Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorNicolás UnsainUniversidad Nacional de Córdoba, Cordoba, Argentina
- Senior EditorK VijayRaghavanNational Centre for Biological Sciences, Tata Institute of Fundamental Research, Bangalore, India
Reviewer #1 (Public review):
This paper examines the role of MLCK (myosin light chain kinase) and MLCP (myosin light chain phosphatase) in axon regeneration. Using loss-of-function approaches based on small molecule inhibitors and siRNA knockdown, the authors explore axon regeneration in cell culture and in animal models from central and peripheral nervous systems. Their evidence shows that MLCK activity facilitates axon extension/regeneration, while MLCP prevents it.
Major concerns:
(1) In the title, authors indicate that the observed effects from loss-of-function of MLCK/MLCP take place via F-actin redistribution in the growth cone. However, there are no experiments showing a causal effect between changes in axon growth mediated by MLCK/MLCP and F-actin redistribution.
(2) The author combines MLCK inhibitors with Bleb (Figure 6), trying to verify if both pairs of inhibitors act on the same target/pathway. MLCK may regulate axon growth independent of NMII activity. However, this has very important implications for the understanding not only on how NMII works and affects axon extension, but also in trying to understand what MLCP is doing. One wonders if MLCP actions, which are opposite of MLCK, also independent of NMII activity? The authors, in the discussion section, try to find an explanation for this finding, but I consider it fails since the whole rationale of the manuscript is still around how MLCK and MLCP affect NMII phosphorylation.
What follows is a discussion of the merits and limitations of different claims of the manuscript in light of the evidence presented.
(1) Using western blot and immunohistochemical analyses, authors first show that MLCK expression is increased in DRG sensory neurons following peripheral axotomy, concomitant to an increase in MLC phosphorylation, suggesting a causal effect (Figure 1). The authors claim that it is common that axon growth-promoting genes are upregulated. It would have been interesting at this point to study in this scenario the regulation of MLCP.
(2) Using DRG cultures and sciatic nerve crush in the context of MLCK inhibition (ML-7) and down-regulation, authors conclude that MLCK activity is required for mammalian peripheral axon regeneration both in vitro and in vivo (Figure 2). In parallel, the authors show that these treatments affect as expected the phosphorylation levels of MLC.
The in vitro evidence is of standard methods and convincing. However, here, as well as in all other experiments using siRNAs, no Control siRNAs were used. Authors do show that the target protein is downregulated, and they can follow transfected cells with GFP. Still, it should be noted that the standard control for these experiments has not been done.
(3) The authors then examined the role of the phosphatase MLCP in axon growth during regeneration. The authors first use a known MLCP blocker, phorbol 12,13-dibutyrate (PDBu), to show that is able to increase the levels of p-MLC, with a concomitant increase in the extent of axon regrowth of DRG neurons, both in permissive as well as non-permissive substrates. The authors repeat the experiments using the knockdown of MYPT1, a key component of the MLC-phosphatase, and again can observe a growth-promoting effect (Figure 3).
The authors further show evidence for the growth-enhancing effect in vivo, in nerve crush experiments. The evidence in vivo deserves more evidence and experimental details (see comment 2). A key weakness of the data was mentioned previously: no control siARN was used.
(4) In the next set of experiments (presented in Figure 4) authors extend the previous observations in primary cultures from the CNS. For that, they use cortical and hippocampal cultures, and pharmacological and genetic loss-of-function using the above-mentioned strategies. The expected results were obtained in both CNS neurons: inhibition or knockdown of the kinase decreases axon growth, whereas inhibition or knockdown of the phosphatase increases growth. A main weakness in this set is that drugs were used from the beginning of the experiment, and hence, they would also affect axon specification. As pointed in Materials and Method (lines 143-145) authors counted as "axons" neurites longer than twice the diameter of the cell soma, and hence would not affect the variable measured. In any case, to be sure one is only affecting axon extension in these cells, the drugs should have been used after axon specification and maturation, which occurs at least after 5 DIV.
(5) In Figure 7, the authors a local cytoskeletal action of the drug, but the evidence provided does not differentiate between a localized action of the drugs and a localized cell activity.
References:
(1) Eun-Mi Hur 1, In Hong Yang, Deok-Ho Kim, Justin Byun, Saijilafu, Wen-Lin Xu, Philip R Nicovich, Raymond Cheong, Andre Levchenko, Nitish Thakor, Feng-Quan Zhou. 2011. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A. 2011 Mar 22;108(12):5057-62. doi: 10.1073/pnas.1011258108.
(2) Garrido-Casado M, Asensio-Juárez G, Talayero VC, Vicente-Manzanares M. 2024. Engines of change: Nonmuscle myosin II in mechanobiology. Curr Opin Cell Biol. 2024 Apr;87:102344. doi: 10.1016/j.ceb.2024.102344.
(3) Karen A Newell-Litwa 1, Rick Horwitz 2, Marcelo L Lamers. 2015. Non-muscle myosin II in disease: mechanisms and therapeutic opportunities. Dis Model Mech. 2015 Dec;8(12):1495-515. doi: 10.1242/dmm.022103.
Reviewer #2 (Public review):
Summary:
Saijilafu et al. demonstrate that MLCK/MLCP proteins promote axonal regeneration in both the central nervous system (CNS) and peripheral nervous system (PNS) using primary cultures of adult DRG neurons, hippocampal and cortical neurons, as well as in vivo experiments involving sciatic nerve injury, spinal cord injury, and optic nerve crush. The authors show that axon regrowth is possible across different contexts through genetic and pharmacological manipulation of these proteins. Additionally, they propose that MLCK/MLCP may regulate F-actin reorganization in the growth cone, which is significant as it suggests a novel strategy for promoting axonal regeneration.
Strengths:
This manuscript presents a wide range of experimental models to address its hypothesis and biological question. Notably, the use of multiple in vivo models significantly enhances the overall validity of the study.
Weaknesses:
-The authors previously published that blocking myosin II activity stimulates axonal growth and that MLCK activates myosin II. The present work shows that inhibiting MLCK blocks axonal regeneration while blocking MLCP (the protein that dephosphorylates myosin II) produces the opposite effect. Although this contradiction is discussed, no new evidence has been added to the manuscript to clarify this mechanism or address the remaining questions. Critical unresolved questions include: what happens to myosin II expression when both MLCK and MLCP are inhibited? If MLCK/MLCP are acting through an independent mechanism, what would that mechanism be?
-In the discussion, the authors mention the existence of two myosin II isoforms with opposing effects on axonal growth. Still, there is no evidence in the manuscript to support this point.
-It is also unclear how MLCK/MLCP acts on the actin cytoskeleton. The authors suggest that proteins such as ADF/cofilin, Arp 2/3, Eps8, Profilin, Myosin II, and Myosin V could regulate changes in F-actin dynamics. However, this study provides no experimental evidence to determine which proteins may be involved in the mechanism.
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public review):
This paper examines the role of MLCK (myosin light chain kinase) and MLCP (myosin light chain phosphatase) in axon regeneration. Using loss-of-function approaches based on small molecule inhibitors and siRNA knockdown, the authors explore axon regeneration in cell culture and in animal models. Their evidence shows that MLCK activity facilitates axon extension/regeneration, while MLCP prevents it.
Major concern:
A global inconsistency in the conclusions of the authors is evident when trying to understand the role of NMII in axon growth and to understand the present results in light of previous reports by the authors and many others on the role of NMII in axon extension. The discussion of the matter fails to acknowledge a vast literature on how NMII activity is regulated. The authors study enzymes responsible for the phosphorylation and dephosphorylation of NMII, referring to something that is strongly proven elsewhere, that phosphorylation activates NMII and dephosphorylation deactivates it. The authors mention their own previous evidence using inhibitors of NMII ATPase activity (blebbistatin, Bleb for short) and inhibitors of a kinase that phosphorylates NMII (ROCK), highlighting that Bleb increases axon growth. Since Bleb inhibits the ATPase activity of NMII, it follows that NMII is in itself an inhibitor of axon growth, and hence when NMII is inhibited, the inhibition on axon growth is relieved, and axonal growth takes place (REF1). It is known that NMII exists in an inactive folded state, and ser19 phosphorylation (by MLCK or ROCK) extends the protein, allowing NMII filament formation, ATPase activity, and force generation on actin filaments (REF2). From this, it is derived that if MLCK is inhibited, then there is no NMII phosphorylation, and hence no NMII activity, and, according to their previous work, this should promote axon growth. On the contrary, the authors show the opposite effect: in the lack of phospho-MLC, authors show axon growth inhibition.
We thank the Reviewer for taking time to review our manuscript, and we really appreciated the comments from the reviewer. We have tried our best to revise the manuscript to address all the comments raised by the Reviewer.
Reporting evidence challenging previous conclusions is common business in scientific endeavors, but the problem with the current manuscript is that it fails to point to and appropriately discuss this contradiction. Instead, the authors refer to the fact that MLCK and Bleb inhibit NMII in different steps of the activation process. While this is true, this explanation does not solve the contradiction. There are many options to accommodate the information, but it is not the purpose of this revision to provide them. Since the manuscript is focused solely on phosphorylation states of MLC and axon extension, the claims are simply at odds with the current literature, and this important finding, if true, is not properly discussed.
Thank you for reviewer's very good comments. As suggested by Reviewer, we discuss more detail it in our revised manuscripts (line 357-368; line 373-374).
What follows is a discussion of the merits and limitations of different claims of the manuscript in light of the evidence presented.
(1) Using western blot and immunohistochemical analyses, authors first show that MLCK expression is increased in DRG sensory neurons following peripheral axotomy, concomitant to an increase in MLC phosphorylation, suggesting a causal effect (Figure 1). The authors claim that it is common that axon growth-promoting genes are upregulated. It would have been interesting at this point to study in this scenario the regulation of MLCP, which is a main subject in this work, and expect its downregulation.
We thank the Reviewer for taking time to review our manuscript, and we really appreciated the positive comments from the Reviewer.
(2) Using DRG cultures and sciatic nerve crush in the context of MLCK inhibition and down-regulation, authors conclude that MLCK activity is required for mammalian peripheral axon regeneration both in vitro and in vivo (Figure 2).
The in vitro evidence is of standard methods and convincing. However, here, as well as in all other experiments using siRNAs, it is not clear what the control is about (the identity of the plasmids and sequences, if any).
We used the pCMV–EGFP–N3 as control, and the pCMV–EGFP–N3 plasmid was from Clontech, Inc. (line 114-115).
Related to this, it is not helpful to show the same exact picture as a control example in Figures 2 and 3 (panels J and E, respectively). Either because they should not have received the same control treatment, or simply because it raises concern that there are no other control examples worth showing. In these images, it is not also clear where and how the crush site is determined in the GFP channel. This is of major importance since the axonal length is measured from the presumed crush site. Apart from providing further details in the text, the authors should include convincing images.
Thank you so much for your comments. We changed the control example in Figure 3J. For sciatic nerve regeneration experiments, the sciatic nerve was exposed at the sciatic notch by a small incision 2 days after the in vivo electroporation. The nerve was then crushed, and the crush site was marked with a 11-0 nylon epineural suture. After surgeries, the wound was closed, and the mice were allowed to recover. Three days after the sciatic nerve crush, the whole sciatic nerves from the perfused animals were dissected out and postfixed overnight in 4% PFA at 4°C. Before whole-mount flattening, it was confirmed that the place of epineural suture matched the injury site, and experiments were included in the analysis only when the crush site was clearly identifiable. Using whole mounted tissue, all identifiable EGFP-labeled axons in the sciatic nerve were manually traced from the crush site to the distal growth cone to measure the length of axon regeneration. (line 159-164).
(3) The authors then examined the role of the phosphatase MLCP in axon growth during regeneration. The authors first use a known MLCP blocker, phorbol 12,13-dibutyrate (PDBu), to show that is able to increase the levels of p-MLC, with a concomitant increase in the extent of axon regrowth of DRG neurons, both in permissive as well as non-permissive. The authors repeat the experiments using the knockdown of MYPT1, a key component of the MLC-phosphatase, and again can observe a growth-promoting effect (Figure 3).
The authors further show evidence for the growth-enhancing effect in vivo, in nerve crush experiments. The evidence in vivo deserves more evidence and experimental details (see comment 2). Some key weaknesses of the data were mentioned previously (unclear RNAi controls and duplication of shown images), but in this case, it is also not clear if there is a change only in the extent of growth, or also in the number of axons that are able to regenerate.
Thank you so much for your comments. We used same control as in vitro experiments (the pCMV– EGFP–N3 plasmid was from Clontech, Inc), and we also changed the control image in Figure 3J. For in vivo axon regeneration experiments, we measured the lengths of all identifiable EGFP-labelled axons in the sciatic nerve from the crush site to the distal axonal ends. The number of EGFP labeled regenerating axons were actually determined by the electroporation rate of EGFP, which is similar, but not identical, in different mice. Thus, our data only can show the differences in axon lengths among different experimental conditions. Such approach has been used in many of our previously published papers (e.g. Saijilafu et al. Nature Communications, 2011, Saijilafu et al. Nature Communications, 2013). (line 152-153).
(4) In the next set of experiments (presented in Figure 4) authors extend the previous observations in primary cultures from the CNS. For that, they use cortical and hippocampal cultures, and pharmacological and genetic loss-of-function using the above-mentioned strategies. The expected results were obtained in both CNS neurons: inhibition or knockdown of the kinase decreases axon growth, whereas inhibition or knockdown of the phosphatase increases growth. A main weakness in this set is that it is not indicated when (at what day in vitro, DIV) the treatments are performed. This is important to correctly interpret the results, since in the first days in vitro these neurons follow well-characterized stages of development, with characteristic cellular events with relevance to what is being evaluated. Importantly, this would be of value to understand whether the treatments affect axonal specification and/or axonal extension. Although these events are correlated, they imply a different set of molecular events.
The treatments were started from the initial of cell culture period, and this procedure may affect axon specification as the Reviewer point out. However, we mainly focused on axon length in our experiments, thus, for quantification of axon length, neurons with processes longer than twice the diameter of cell bodies were photographed, and the longest axon of each neuron was measured. We revised the manuscript as suggested by the reviewer (line 143-145).
The title of this section is misleading: line 241 "MLCK/MLCP activity regulated axon growth in the embryonic CNS"... the title (and the conclusion) implies that the experiments were performed in situ, looking at axons in the developing brain. The most accurate title and conclusion should mention that the evidence was collected in CNS primary cultures derived from embryos.
We have revised the manuscript as suggested by the reviewer (line 251).
(5) Performing nerve crush injury in CNS nerves (optic nerve and spinal cord), and the local application of PBDu, the author shows contrasting results (Figure 5). In the ON nerve, they can see axons extending beyond the lesion site due to PBDu. On the contrary, the authors fail to observe so in the corticospinal tract present in the spinal cord. The authors fail to discuss this matter in detail. Also, they accommodate the interpretation of the evidence in light of a process known as axon retraction, and its prevention by MLCP inhibition. Since the whole paper is on axon extension, and it is known that mechanistically axon retraction is not merely the opposite of axon extension, the claim needs far more evidence.
Thank you so much for your comments. Compared to optic nerve axons, corticospinal tract axons exhibit a reduced intrinsic axon growth capability. Consequently, we observed that PBDu stimulates optic nerve axon regeneration. However, unfortunately, we did not detect any enhancement in corticospinal tract axons beyond the injury site in SCI following the inhibition of myosin light chain phosphatase (MLCP) with PBDu.
In panel 5F and the supplementary data, the authors mention the occurrence of retraction bulbs, but the images are too small to support the claim, and it is not clear how these numbers were normalized to the number of axons labeled in each condition.
Thank you so much for your comments. In this study, we used a similar method from Ertürk et al. (2007) to quantify the retraction bulb. Both maximum width of the enlarged distal tip of the axon and the width of its immediately adjacent axon shaft was measured. Then, the ratio of these two widths was then calculated. An axonal tip was considered as a retraction bulb if its tip/shaft ratio exceeded 4. Averages number of retraction bulb were calculated from 3 sections in every mice for each group (n=5). (line 187-191).
[Ref] Ertürk A, Hellal F, Enes J, and Bradke F (2007). Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci 27, 9169–9180. [PubMed:17715353].
(6) The author combines MLCK and MLCP inhibitors with Bleb, trying to verify if both pairs of inhibitors act on the same target/pathway (Figure 6). The rationale is wrong for at least two reasons.
a- Because both lines of evidence point to contrasting actions of NMII on axon growth, one approach could never "rescue" the other.
If MLCK regulates axon growth through the activation of Myosin, the inhibitory effect of ML-7 (an MLCK inhibitor) on axon growth might be influenced by Bleb, a NMII inhibitor. However, our findings reveal that the combination of Bleb and ML-7 does not alter the rate of axon outgrowth compared to ML-7 alone. This suggests that the roles of ML-7 and Bleb in axon growth are independent. It means MLCK may regulates axon growth independent of NMII activity.
b. Because the approaches target different steps on NMII activation, one could never "prevent" or rescue the other. For example, for Bleb to provide a phenotype, it should find any p-MLC, because it is only that form of MLC that is capable of inhibiting its ATPase site. In light of this, it is not surprising that Bleb is unable to exert any action in a situation where there is no p-MLC (ML-7, which by inhibiting the kinase drives the levels of p-MLC to zero, Figure 4A). Hence, the results are not possible to validate in the current general interpretation of the authors. (See 'major concern').
The reported mechanism of blebbistatin is not through competition with the ATP binding site of myosin. Instead, it selectively binds to the ATPase intermediate state associated with ADP and inorganic phosphate, which decelerates the phosphate release. Importantly, blebbistatin does not impede myosin's interaction with actin or the ATP-triggered disassociation of actomyosin. It rather inhibits the myosin head when it forms a product complex with a reduced affinity for actin. This indicates that blebbistatin functions by stabilizing a particular myosin intermediate state that is independent of the phosphorylation status of myosin light chain (MLC).
[Ref] Kovács M, Tóth J et al. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem. 2004 Aug 20;279(34):35557-63. doi: 10.1074/jbc.M405319200.
(7) In Figure 7, the authors argue that the scheme of replating and using ML7 before or after replating is evidence for a local cytoskeletal action of the drug. However, an alternative simpler explanation is that the drug acts acutely on its target, and that, as such, does not "survive" the replating procedure. Hence, the conclusion raised by the evidence shown is not supported.
In our study, we meticulously assessed the neuronal survival rates across various experimental groups. The findings indicate no significant variation in survival rates among the groups. This suggests that the drug treatment exerts no discernible influence on cell viability but primarily modulates axonal elongation."
Author response image 1.
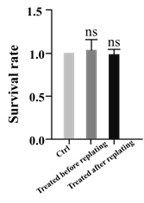
(8) In Figure 8, the authors show that the inhibitory treatments on MLCK and MLCP (ML7 and PRBu) alter the morphology of growth cones. However, it is not clear how this is correlated with axon growth. The authors also mention in various parts of the text that a local change in the growth cone is evidence for a local action/activity of the drug or enzyme. However, the local change<->local action is not a logical truth. It can well be that MLCK and MLCP activity trigger molecular events that ultimately have an effect elsewhere, and by looking at "elsewhere" one observes of course a local effect but is not because the direct action of MLCK or MLCP are localized. To prove true localized effects there are numerous efforts that can be made, starting from live imaging, fluorescent sensors, and compartmentalized cultures, just to mention a few.
About the relationship between growth cone size and its growth rate, the previous published literatures found that a fast-growing axon tended to have small growth cones (Mason C. et al. 1997). A recent study on Aplysia further supports this by noting that growth cones enlarge significantly when axonal elongation halts (Miller and Suter, 2018). Consistent with these findings, our data indicate that inhibiting MLCP with PDBu treatment leads to a reduction in growth cone size, which in turn promotes axon regeneration.
[Ref] Mason CA, Wang LC. Growth cone form is behavior-specific and, consequently, position-specific along the retinal axon pathway. J Neurosci. 1997; 13:1086–1100. [PubMed: 8994063]
[Ref] Miller KE, Suter DM. An Integrated Cytoskeletal Model of Neurite Outgrowth. Front Cell Neurosci. 2018 Nov 26;12:447. doi: 10.3389/fncel.2018.00447. eCollection 2018.
References:
(1) Eun-Mi Hur 1, In Hong Yang, Deok-Ho Kim, Justin Byun, Saijilafu, Wen-Lin Xu, Philip R Nicovich, Raymond Cheong, Andre Levchenko, Nitish Thakor, Feng-Quan Zhou. 2011. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A. 2011 Mar 22;108(12):5057-62. doi: 10.1073/pnas.1011258108.
(2) Garrido-Casado M, Asensio-Juárez G, Talayero VC, Vicente-Manzanares M. 2024. Engines of change: Nonmuscle myosin II in mechanobiology. Curr Opin Cell Biol. 2024 Apr;87:102344. doi: 10.1016/j.ceb.2024.102344.
(3) Karen A Newell-Litwa 1, Rick Horwitz 2, Marcelo L Lamers. 2015. Non-muscle myosin II in disease: mechanisms and therapeutic opportunities. Dis Model Mech. 2015 Dec;8(12):1495-515. doi: 10.1242/dmm.022103.
Reviewer #2 (Public review):
Summary:
Saijilafu et al. demonstrate that MLCK/MLCP proteins promote axonal regeneration in both the central nervous system (CNS) and peripheral nervous system (PNS) using primary cultures of adult DRG neurons, hippocampal and cortical neurons, as well as in vivo experiments involving sciatic nerve injury, spinal cord injury, and optic nerve crush. The authors show that axon regrowth is possible across different contexts through genetic and pharmacological manipulation of these proteins. Additionally, they propose that MLCK/MLCP may regulate F-actin reorganization in the growth cone, which is significant as it suggests a novel strategy for promoting axonal regeneration.
Strengths:
This manuscript presents a comprehensive array of experimental models, addressing the biological question in a broad manner. Particularly noteworthy is the use of multiple in vivo models, which significantly strengthens the overall validity of the study.
We thank the Reviewer for taking time to review our manuscript, and we really appreciated the positive comments from the Reviewer.
Weaknesses:
The following aspects apply:
(1) The manuscript initially references prior research by the authors suggesting that NMII inhibition enhances axonal growth and that MLCK activates NMII. However, the study introduces a contradiction by demonstrating that MLCK inhibition (via ML-7 or siMLCK) inhibits axonal growth. This inconsistency is not adequately addressed or discussed in the manuscript.
Thank you for reviewer's very good comments. As suggested by Reviewer, we discuss more detail it in our revised manuscripts (line 357-368; line373-374).
(2) While the study proposes that MLCK/MLCP regulates F-actin redistribution in the growth cone, the mechanism is not explored in depth. The only figure showing how pharmacological manipulation affects the growth cone suggests that not only F-actin but also the microtubule cytoskeleton might be affected, indicating that the mechanism may not be specific. A deeper exploration of this relationship in DRG neurons, in addition to cortical neurons, as shown in the study, would be beneficial.
Thank you for your insightful suggestion. However, our study primarily focuses on actin and myosin dynamics in the context of axonal elongation, as indicated by our direct observations in growing dorsal root ganglia (DRGs). Athamneh et al. (2017) elegantly demonstrated that the bulk movement of microtubules (MTs), rather than their assembly, predominantly drives MT advance during axonal elongation. Consequently, our manuscript concentrates on the actomyosin system, which is central to our findings. While the role of MTs in axonal growth is indeed significant and fascinating, the data we present is predominantly concerned with the actomyosin mechanism.
[Ref] Athamneh, A. I. M. et al. Neurite elongation is highly correlated with bulk forward translocation of microtubules. Scientific Reports 7, (2017).
(3) In the sciatic nerve injury experiments, it would be crucial to include additional controls that clearly demonstrate that siMYPT1 treatment increases MLCP in the L4-L5 ganglia. Additionally, although the manuscript mentions quantifying axons expressing EGFP, the Materials and Methods section only discusses siMYPT1 electroporation, which could lead to confusion.
Thank you for your suggestion. However, due to the unavailability of a suitable commercial MLCP antibody, we were unable to directly detect MLCP expression. Instead, we assessed the phosphorylation level of myosin light chain (MLC) as a proxy to indicate that siMYPT1 transfection effectively downregulates MLCP activity in L4/5 dorsal root ganglia (DRG). This approach was taken to ensure the integrity of our findings despite the limitations in antibody availability.
About the electroporation method section, we have now included detailed information about the control plasmid used in our experiments to ensure a clear understanding of our experimental setup and to validate our results. A 1 μl solution containing indicated siRNAs together with the plasmid encoding EGFP (pCMV–EGFP–N3) was then microinjected into the L4–L5 DRG….. (line 152-153).
(4) In some panels, it is difficult to differentiate the somas from the background (Figures 3, 4, 7). In conditions where images with shorter axonal lengths are represented, it is unclear whether this is due to fewer cells or reduced axonal growth (Figures 2, 4, 6).
In the original submission, there was some loss of image quality while converting the TIFF to PDF. We improved the quality of images in our revised manuscripts.
Recommendations for the authors:
Reviewer #1 (Recommendations for the authors):
There are a number of typos and language errors that should be thoroughly revised. For example, line 219: "It is well known that the opposite role of MLCK and MLCP to regulate the MLC phosphorylation status". The term "opposite role" is vague. Using "opposite roles" and specifying that they are in regulating MLC phosphorylation status clarifies the relationship between MLCK and MLCP. Also, the original phrase "to regulate" was not correctly integrated into the sentence. Rephrasing it to "in regulating" makes the role of MLCK and MLCP clearer.
We have revised the manuscript as suggested by the reviewer (line 229).
In the same line, there is a high number of panels that are not referred to in the text or references for panels that have another letter. Just to mention a few:
- line 199: "(Figure 1F, G)", → BUT figure 1 contains no G panel.
We have revised the manuscript as suggested by the reviewer (line 209).
- line 203: "The results showed that ML-7 administration led to a significant reduction in MLC phosphorylation levels (Figure 2A, B) and impaired axonal growth in sensory neurons (Figure 2C, D). → BUT panel C is related to A and B, and only D and E show impaired axonal growth.
We have revised the manuscript as suggested by the reviewer (line 214; line 215; line 217; line 219 ).
Reviewer #2 (Recommendations for the authors):
(1) Improving the quality of the images would significantly strengthen the results presented.
In the original submission, there was some loss of image quality while converting the TIFF to PDF. We improved the quality of images in our revised manuscripts.
(2) The representative images of controls do not always show the same number of cells or axonal growth (e.g., Figure 4).
We have changed some images as suggested by the reviewer.
(3) The text has citation errors when referring to the figure labels.
Upon thorough review, we have carefully examined our manuscript and have made the necessary corrections to address the identified errors. We appreciate the opportunity to enhance the quality of our work and believe that these revisions have significantly improved the clarity of our manuscript.
(4) What happens to MLCK levels when MLCP activity is inhibited in the optic nerve?
Upon analyzing our experimental data, we observed no significant alterations in the protein levels of MLCK when the activity of MLCP was inhibited. This finding suggests that the regulatory mechanisms governing MLCK expression may not be directly influenced by short-term MLCP inhibition. It is plausible that the duration of the inhibition period was insufficient to elicit a detectable change in MLCK expression levels.
(5) The text in line 266: "In contrast, local PBS administration at the injury site or intravitreal PDBu injection induced little axon regeneration beyond the injury site (Figure 5 A-C)." However, this is not reflected in the figure.
In our revised manuscript, we have provided a more precise description of our findings: In contrast, local PBS administration at the injury site or intravitreal PDBu injection did not significantly enhance axon regeneration beyond the injury site (Figure 5 A-C). This observation suggests that the only treatment employed in the injury site (the inhibition of MLCP activity within the growth cone) effective promote axonal growth. (line 276-279).
(6) Line 287: The phrase "Consistent with our previous study" requires a citation to support it.
We added the reference paper; Consistent with our previous study 1, the inhibition of myosin II activity with 25 μM blebbistatin markedly promoted axonal growth (Figure 6A, B). (line 298)
(7) Line 333: The paper cited by Yu P et al. (2012) does not mention MLCK or p-MLC, so it appears to be misquoted.
Thank you for comments. We rechecked this cited paper and confirmed that the author provided the western data C in the supplementary figure 1, it showed that Bleb did not alter the phosphorylation status of MLC.



