Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorRafael Fernández-ChaconInstituto de Biomedicina de Sevilla (IBiS), Sevilla, Spain
- Senior EditorLu ChenStanford University, Stanford, United States of America
Reviewer #3 (Public review):
To summarize: The authors' overfilling hypothesis depends crucially on the premise that the very-quickly reverting paired-pulse depression seen after unusually short rest intervals of << 50 ms is caused by depletion of release sites whereas Dobrunz and Stevens (1997) concluded that the cause was some other mechanism that does not involve depletion. The authors now include experiments where switching extracellular Ca2+ from 1.2 to 2.5 mM increases synaptic strength on average, but not by as much as at other synapse types. They contend that the result supports the depletion hypothesis. I didn't agree because the model used to generate the hypothesis had no room for any increase at all, and because a more granular analysis revealed a mixed population with a subset where: (a) synaptic strength increased by as much as at standard synapses; and yet (b) the quickly reverting depression for the subset was the same as the overall population.
The authors raise the possibility of additional experiments, and I do think this could clarify things if they pre-treat with EGTA as I recommended initially. They've already shown they can do this routinely, and it would allow them to elegantly distinguish between pv and pocc explanations for both the increases in synaptic strength and the decreases in the paired pulse ratio upon switching Ca2+ to 2.5 mM. Plus/minus EGTA pre-treatment trials could be interleaved and done blind with minimal additional effort.
Showing reversibility would be a great addition too, because, in our experience, this does not always happen in whole-cell recordings in ex-vivo tissue even when electrical properties do not change. If the goal is to show that L2/3 synapses are less sensitive to changes in Ca2+ compared to other synapse types - which is interesting but a bit off point - then I would additionally include a positive control, done by the same person with the same equipment, at one of those other synapse types using the same kind of presynaptic stimulation (i.e. ChRs).
Specific points (quotations are from the Authors' rebuttal)
(1) Regarding the Author response image 1, I was instead suggesting a plot of PPR in 1.2 mM Ca2+ versus the relative increase in synaptic strength in 2.5 versus in 1.2 mM. This continues to seem relevant.
(2) "Could you explain in detail why two-fold increase implies pv < 0.2?"
a. start with power((2.5/(1 + (2.5/K1) + 1/2.97)),4) = 2*power((1.3/(1 + (1.3/K1) + 1/2.97)),4);
b. solve for K1 (this turns out to be 0.48);
c. then implement the premise that pv -> 1.0 when Ca2+ is high by calculating Max = power((C/(1 + (C/K1) + 1/2.97)),4) where C is [Ca] -> infinity.
d. pv when [Ca] = 1.3. mM must then be power((1.3/(1 + (1.3/K1) + 1/2.97)),4)/Max, which is <0.2.
Note that modern updates of Dodge and Rahamimoff typically include a parameter that prevents pv from approaching 1.0; this is the gamma parameter in the versions from Neher group.
(3) "If so, we can not understand why depletion-dependent PPD should lead to PPF."
When PPD is caused by depletion and pv < 0.2, the number of occupied release sites should not be decreased by more than one-fifth at the second stimulus so, without facilitation, PPR should be > 0.8. The EGTA results then indicate there should be strong facilitation, driving PPR to something like 1.2 with conservative assumptions. And yet, a value of < 0.4 is measured, which is a large miss.
(4) Despite the authors' suggestion to the contrary, I continue to think there is a substantial chance that Ca2+-channel inactivation is the mechanism underlying the very quickly reverting paired-pulse depression. However, this is only one example of a non-depletion mechanism among many, with the main point being that any non-depletion mechanism would undercut the reasoning for overfilling. And, this is what Dobrunz and Stevens claimed to show; that the mechanism - whatever it is - does not involve depletion. The most effective way to address this would be affirmative experiments showing that the quickly reverting depression is caused by depletion after all. Attempting to prove that Ca2+-channel inactivation does not occur does not seem like a worthwhile strategy because it would not address the many other possibilities.
(5) True that Kusick et al. observed morphological re-docking, but then vesicles would have to re-prime and Mahfooz et al. (2016) showed that re-priming would have to be slower than 110 ms (at least during heavy use at calyx of Held).
Author response:
The following is the authors’ response to the previous reviews
Reviewer #3 (Public review):
The central issue for evaluating the overfilling hypothesis is the identity of the mechanism that causes the very potent (>80% when inter pulse is 20 ms), but very quickly reverting (< 50 ms) paired pulse depression (Fig 1G, I). To summarize: the logic for overfilling at local cortical L2/3 synapses depends critically on the premise that probability of release (pv) for docked and fully primed vesicles is already close to 100%. If so, the reasoning goes, the only way to account for the potent short-term enhancement seen when stimulation is extended beyond 2 pulses would be by concluding that the readily releasable pool overfills. However, the conclusion that pv is close to 100% depends on the premise that the quickly reverting depression is caused by exocytosis dependent depletion of release sites, and the evidence for this is not strong in my opinion. Caution is especially reasonable given that similarly quickly reverting depression at Schaffer collateral synapses, which are morphologically similar, was previously shown to NOT depend on exocytosis (Dobrunz and Stevens 1997). Note that the authors of the 1997 study speculated that Ca2+-channel inactivation might be the cause, but did not rule out a wide variety of other types of mechanisms that have been discovered since, including the transient vesicle undocking/re-docking (and subsequent re-priming) reported by Kusick et al (2020), which seems to have the correct timing.
Thank you for your comments on an alternative possibility besides Ca2+ channel inactivation. Kusick et al. (2020) showed that transient destabilization of docked vesicle pool is recovered within 14 ms after stimulation. This rapid recovery implies that post-stimulation undocking events might be largely resolved before the 20 ms inter-stimulus interval (ISI) used in our paired-pulse ratio (PPR) experiments, arguing against the possibility that post-AP undocking/re-docking events significantly influence PPR measured at 20 ms ISI. Furthermore, Vevea et al. (2021) showed that post-stimulus undocking is facilitated in synaptotagmin-7 (Syt7) knockout synapses. In our study, Syt7 knockdown did not affect PPR at 20 ms ISI, suggesting that the undocking process described in Kusick et al. may not be a major contributor to the paired-pulse depression observed at 20 ms interval in our study. Therefore, it is unlikely that transient vesicle undocking primarily underlies the strong PPD at 20 ms ISI in our experiments. Taken together, the undocking/redocking dynamics reported by Kusick et al. are too rapid to affect PPR at 20 ms ISI, and our Syt7 knockdown data further argue against a significant role of this process in the PPD observed at 20 ms interval.
In an earlier round of review, I suggested raising extracellular Ca2+, to see if this would increase synaptic strength. This is a strong test of the authors' model because there is essentially no room for an increase in synaptic strength. The authors have now done experiments along these lines, but the result is not clear cut. On one hand, the new results suggest an increase in synaptic strength that is not compatible with the authors' model; technically the increase does not reach statistical significance, but, likely, this is only because the data set is small and the variation between experiments is large. Moreover, a more granular analysis of the individual experiments seems to raise more serious problems, even supporting the depletion-independent counter hypothesis to some extent. On the other hand, the increase in synaptic strength that is seen in the newly added experiments does seem to be less at local L2/3 cortical synapses compared to other types of synapses, measured by other groups, which goes in the general direction of supporting the critical premise that pv is unusually high at L2/3 cortical synapses. Overall, I am left wishing that the new data set were larger, and that reversal experiments had been included as explained in the specific points below.
Specific Points:
(1) One of the standard methods for distinguishing between depletion-dependent and depletion-independent depression mechanisms is by analyzing failures during paired pulses of minimal stimulation. The current study includes experiments along these lines showing that pv would have to be extremely close to 1 when Ca2+ is 1.25 mM to preserve the authors' model (Section "High double failure rate ..."). Lower values for pv are not compatible with their model because the k1 parameter already had to be pushed a bit beyond boundaries established by other types of experiments.
It should be noted that we did not arbitrarily pushed the k1 parameter beyond boundaries, but estimated the range of k1 based on the fast time constant for recovery from paired pulse depression as shown in Fig. 3-S2-Ab.
The authors now report a mean increase in synaptic strength of 23% after raising Ca to 2.5 mM. The mean increase is not quite statistically significant, but this is likely because of the small sample size. I extracted a 95% confidence interval of [-4%, +60%] from their numbers, with a 92% probability that the mean value of the increase in the full population is > 5%. I used the 5% value as the greatest increase that the model could bear because 5% implies pv < 0.9 using the equation from Dodge and Rahamimoff referenced in the rebuttal. My conclusion from this is that the mean result, rather than supporting the model, actually undermines it to some extent. It would have likely taken 1 or 2 more experiments to get above the 95% confidence threshold for statistical significance, but this is ultimately an arbitrary cut off.
Our key claim in Fig. 3-S3 is not the statistical non-significance of EPSC changes, but the small magnitude of the change (1.23-fold). This small increase is far less than the 3.24-fold increase predicted by the fourth-power relationship (D&R equation, Dodge & Rahamimoff, 1967), which would be valid under the conditions that the fusion probability of docked vesicles (pv) is not saturated. We do not believe that addition of new experiments would increase the magnitude of EPSC change as high as the Dodge & Rahamimoff equation predicts, even if more experiments (n) yielded a statistical significance. In other words, even a small but statistically significant EPSC changes would still contradict with what we expect from low pv synapses. It should be noted that our main point is the extent of EPSC increase induced by high external [Ca2+], not a p-value. In this regard, it is hard for us to accept the Reviewer’s request for larger sample size expecting lower p-value.
Although we agree to Reviewer’s assertion that our data may indicate a 92% probability for the high Ca2+ -induced EPSC increases by more than 5%, we do not agree to the Reviewer’s interpretation that the EPSC increase necessarily implies an increase in pv. We are sorry that we could not clearly understand the Reviewer’s inference that the 5% increase of EPSCs implies pv < 0.9. Please note that release probability (pr) is the product of pv and the occupancy of docked vesicles in an active zone (pocc). We imagine that this inference might be under the premise that pocc is constant irrespective of external [Ca2+]. Contrary to the Reviewer’s premise, Figure 2c in Kusick et al. (2020) showed that the number of docked SVs increased by c. a. 20% upon increasing external [Ca2+] to 2 mM. Moreover, Figure 7F in Lin et al. (2025) demonstrated that the number of TS vesicles, equivalent to pocc increased by 23% at high external [Ca2+]. These extents of pocc increases are similar to our magnitude of high external Ca2+ -induced increase in EPSC (1.23-fold). Of course, it is possible that both increase of pocc and pv contributed to the high [Ca2+]o-induced increase in EPSC. The low PPR and failure rate analysis, however, suggest that pv is already saturated in baseline conditions of 1.3 mM [Ca2+]o and thus it is more likely that an increase in pocc is primarily responsible for the 1.23-fold increase. Moreover, the 1.23-fold increase, does not match to the prediction of the D&R equation, which would be valid at synapses with low pv. Therefore, interpreting our observation (1.23-fold increase) as a slight increase in pocc is rather consistent with recent papers (Kusick et al.,2020; Lin et al., 2025) as well as our other results supporting the baseline saturation of pv as shown in Figure 2 and associated supplement figures (Fig. 2-S1 and Fig. 2-S2).
(2) The variation between experiments seems to be even more problematic, at least as currently reported. The plot in Figure 3-figure supplement 3 (left) suggests that the variation reflects true variation between synapses, not measurement error.
Note that there was a substantial variance in the number of docked or TS vesicles at baseline and its fold changes at high external Ca2+ condition in previous studies too (Lin et al., 2025; Kusick et al., 2020). Our study did not focus on the heterogeneity but on the mean dynamics of short-term plasticity at L2/3 recurrent synapses. Acknowledging this, the short-term plasticity of these synapses could be best explained by assuming that vesicular fusion probability (pv) is near to unity, and that release probability is regulated by pocc. In other words, even though pv is near to unity, synaptic strength can increase upon high external [Ca2+], if the baseline occupancy of release sites (pocc) is low and pocc is increased by high [Ca2+]. Lin et al. (2025) showed that high external [Ca2+] induces an increase in the number of TS vesicles (equivalent to pocc) by 23% at the calyx synapses. Different from our synapses, the baseline pv (denoted as pfusion in Lin et al., 2025) of the calyx synapse is not saturated (= 0.22) at 1.5 mM external [Ca2+], and thus the calyx synapses displayed 2.36-fold increase of EPSC at 2 mM external [Ca2+], to which increases in pocc as well as in pv (from 0.22 to 0.42) contributed. Therefore, the small increase in EPSC (= 23%) supports that pv is already saturated at L2/3 recurrent synapses.
And yet, synaptic strength increased almost 2-fold in 2 of the 8 experiments, which back extrapolates to pv < 0.2.
We are sorry that we could not understand the first comment in this paragraph. Could you explain in detail why two-fold increase implies pv < 0.2?
If all of the depression is caused by depletion as assumed, these individuals would exhibit paired pulse facilitation, not depression. And yet, from what I can tell, the individuals depressed, possibly as much as the synapses with low sensitivity to Ca2+, arguing against the critical premise that depression equals depletion, and even arguing - to some extent - for the counter hypothesis that a component of the depression is caused by a mechanism that is independent of depletion.
For the first statement in this paragraph, we imagine that ‘the depression’ means paired pulse depression (PPD). If so, we can not understand why depletion-dependent PPD should lead to PPF. If the paired pulse interval is too short for docked vesicles to be replenished, the first pulse-induced vesicle depletion would result in PPD. We are very sorry that we could not understand Reviewer’s subsequent inference, because we could not understand the first statement.
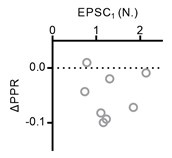
I would strongly recommend adding an additional plot that documents the relationship between the amount of increase in synaptic strength after increasing extracellular Ca2+ and the paired pulse ratio as this seems central.
We found no clear correlation of EPSC1 with PPR changes (ΔPPR) as shown in the figure below.
Author response image 1.
Plot of PPR changes as a function of EPSC1.
(3) Decrease in PPR. The authors recognize that the decrease in the paired-pulse ratio after increasing Ca2+ seems problematic for the overfilling hypothesis by stating: "Although a reduction in PPR is often interpreted as an increase in pv, under conditions where pv is already high, it more likely reflects a slight increase in pocc or in the number of TS vesicles, consistent with the previous estimates (Lin et al., 2025)."
We admit that there is a logical jump in our statement you mentioned here. We appreciate your comment. We re-wrote that part in the revised manuscript (line 285) as follows:
“Recent morphological and functional studies revealed that elevation of [Ca2+]o induces an increase in the number of TS or docked vesicles to a similar extent as our observation (Kusick et al., 2020; Lin et al., 2025), raising a possibility that an increase in pocc is responsible for the 1.23-fold increase in EPSC at high [Ca2+]o . A slight but significant reduction in PPR was observed under high [Ca2+]o too. An increase in pocc is thought to be associated with that in the baseline vesicle refilling rate. While PPR is always reduced by an increase in pv, the effects of refilling rate to PPR is complicated. For example, PPR can be reduced by both a decrease (Figure 2—figure supplement 1) and an increase (Lin et al., 2025) in the refilling rate induced by EGTA-AM and PDBu, respectively. Thus, the slight reduction in PPR is not contradictory to the possible contribution of pocc to the high [Ca2+]o effects.”
I looked quickly, but did not immediately find an explanation in Lin et al 2025 involving an increase in pocc or number of TS vesicles, much less a reason to prefer this over the standard explanation that reduced PPR indicates an increase in pv.
Fig. 7F of Lin et al. (2025) shows an 1.23-fold increase in the number of TS vesicles by high external [Ca2+]. The same figure (Fig. 7E) in Lin et al. (2025) also shows a two-fold increase of pfusion (equivalent to pv in our study) by high external [Ca2+] (from 0.22 to 0.42,). Because pocc is the occupancy of TS vesicles in a limited number of slots in an active zone, the fold change in the number of TS vesicles should be similar to that of pocc.
The authors should explain why the most straightforward interpretation is not the correct one in this particular case to avoid the appearance of cherry picking explanations to fit the hypothesis.
The results of Lin et al. (2025) indicate that high external [Ca2+] induces a milder increase in pocc (23%) compared to pv (190%) at the calyx synapses. Because the extent of pocc increase is much smaller than that of pv and multiple lines of evidence in our study support that the baseline pv is already saturated, we raised a possibility that an increase in pocc would primarily contribute to the unexpectedly low increase of EPSC at 2.5 mM [Ca2+]o. As mentioned above, our interpretation is also consistent with the EM study of Kusick et al. (2020). Nevertheless, the reduction of PPR at 2.5 mM Ca2+ seems to support an increase in pv, arguing against this possibility. On the other hand, because pocc = k1/(k1+b1) under the simple vesicle refilling model (Fig. 3-S2Aa), a change in pocc should associate with changes in k1 and/or b1. While PPR is always reduced by an increase in pv, the effects of refilling rate to PPR is complicated. For example, despite that EGTA-AM would not increase pv, it reduced PPR probably through reducing refilling rate (Fig. 2-S1). On the contrary, PDBu is thought to increase k1 because it induces two-fold increase of pocc (Fig. 7L of Lin et al., 2025). Such a marked increase of pocc, rather than pv, seems to be responsible for the PDBu-induced marked reduction of PPR (Fig. 7I of Lin et al., 2025), because PDBu induced only a slight increase in pv (Fig. 7K of Lin et al., 2025). Therefore, the slight reduction of PPR is not contradictory to our interpretation that an increase in pocc might be responsible for the slight increase in EPSC induced by high [Ca2+]o.
(4) The authors concede in the rebuttal that mean pv must be < 0.7, but I couldn't find any mention of this within the manuscript itself, nor any explanation for how the new estimate could be compatible with the value of > 0.99 in the section about failures.
We have never stated in the rebuttal or elsewhere that the mean pv must be < 0.7. On the contrary, both of our manuscript and previous rebuttals consistently argued that the baseline pv is already saturated, based on our observations including low PPR, tight coupling, high double failure rate and the minimal effect of external Ca2+ elevation.
(5) Although not the main point, comparisons to synapses in other brain regions reported in other studies might not be accurate without directly matching experiments.
Please understand that it not trivial to establish optimal experimental settings for studying other synapses using the same methods employed in the study. We think that it should be performed in a separate study. Furthermore, we have already shown in the manuscript that action potentials (APs) evoked by oChIEF activation occur in a physiologically natural manner, and the STP induced by these oChIEF-evoked APs is indistinguishable from the STP elicited by APs evoked by dual-patch electrical stimulation. Therefore, we believe that our use of optogenetic stimulation did not introduce any artificial bias in measuring STP.
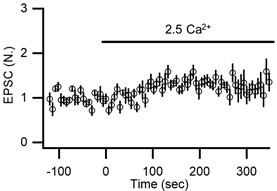
As it is, 2 of 8 synapses got weaker instead of stronger, hinting at possible rundown, but this cannot be assessed because reversibility was not evaluated. In addition, comparing axons with and without channel rhodopsins might be problematic because the channel rhodopsins might widen action potentials.
We continuously monitored series resistance and baseline EPSC amplitude throughout the experiments. The figure below shows the mean time course of EPSCs at two different [Ca2+]o. As it shows, we observed no tendency for run-down of EPSCs during experiments. If any, such recordings were discarded from analysis. In addition, please understand that there is a substantial variance in the number of docked vesicles at both baseline and high external Ca2+ (Lin et al., 2025; Kusick et al., 2020) as well as short-term dynamics of EPSCs at our synapses.
Author response image 2.
Time course of normalized amplitudes of the first EPSCs during paired-pulse stimulation at 20 ms ISI in control and in the elevated external Ca2+ (n = 8).
(6) Perhaps authors could double check with Schotten et al about whether PDBu does/does not decrease the latency between osmotic shock and transmitter release. This might be an interesting discrepancy, but my understanding is that Schotten et al didn't acquire information about latency because of how the experiments were designed.
Schotten et al. (2015) directly compared experimental and simulation data for hypertonicity-induced vesicle release. They showed a pronounced acceleration of the latency as the tonicity increases (Fig. 2-S2), but this tonicity-dependent acceleration was not reproduced by reducing the activation energy barrier for fusion (ΔEa) in their simulations (Fig. 2-S1). Thus, the authors mentioned that an unknown compensatory mechanism counteracting the osmotic perturbation might be responsible for the tonicity-dependent changes in the latency. Importantly, their modeling demonstrated that reducing ΔEa, which would correspond to increasing pv results in larger peak amplitudes and shorter time-to-peak, but did not accelerate the latency. Therefore, there is currently no direct explanation for the notion that PDBu or similar manipulations shorten latency via an increase in pv.
(7) The authors state: "These data are difficult to reconcile with a model in which facilitation is mediated by Ca2+-dependent increases in pv." However, I believe that discarding the premise that depression is always caused by depletion would open up wide range of viable possibilities.
We hope that Reviewer understands the reasons why we reached the conclusion that the baseline pv is saturated at our synapses. First of all, strong paired pulse depression (PPD) cannot be attributed to Ca2+ channel inactivation because Ca2+ influx at the axon terminal remained constant during 40 Hz train stimulation (Fig.2 -S2). Moreover, even if Ca2+ channel inactivation is responsible for the strong PPD, this view cannot explain the delayed facilitation that emerges subsequent pulses (third EPSC and so on) in the 40 Hz train stimulation (Fig. 1-4), because Ca2+ channel inactivation gradually accumulates during train stimulations as directly shown by Wykes et al. (2007) in chromaffin cells. Secondly, the strong PPD and very fast recovery from PPD indicates very fast refilling rate constant (k1). Under this high k1, the failure rates were best explained by pv close to unity. Thirdly, the extent of EPSC increase induced by high external Ca2+ was much smaller than other synapses such as calyx synapses at which pv is not saturated (Lin et al., 2025), and rather similar to the increases in pocc estimated at calyx synapses or the EM study (Kusick et al., 2020; Lin et al., 2025).
Reference
Wykes et al. (2007). Differential regulation of endogenous N-and P/Q-type Ca2+ channel inactivation by Ca2+/calmodulin impacts on their ability to support exocytosis in chromaffin cells. Journal of Neuroscience, 27(19), 5236-5248.
Reviewer #3 (Recommendations for the authors):
I continue to think that measuring changes in synaptic strength when raising extracellular Ca2+ is a good experiment for evaluating the overfilling hypothesis. Future experiments would be better if the authors would include reversibility criteria to rule out rundown, etc. Also, comparisons to other types of synapses would be stronger if the same experimenter did the experiments at both types of synapses.
We observed no systemic tendency for run-down of EPSCs during these experiments (Author response image 2). Furthermore, the observed variability is well within the expected variance range in the number of docked vesicles at both baseline and high external Ca²⁺ (Lin et al., 2025; Kusick et al., 2020) and reflects biological variability rather than experimental artifact. Therefore, we believe that additional reversibility experiments are not warranted. However, we are open to further discussion if the Reviewer has specific methodological concerns not resolved by our present data.
For the second issue, as mentioned above, we think that studying at other synapse types should be done in a separate study.



