Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorYamini DalalNational Cancer Institute, Bethesda, United States of America
- Senior EditorYamini DalalNational Cancer Institute, Bethesda, United States of America
Reviewer #1 (Public review):
Summary:
This manuscript by Zhao et. al investigates the canonical hedgehog pathway in testis development of Nile tilapia. They used complementary approaches with genetically modified tilapia and transfected TSL cells (a clonal stem Leydig cell line) previously derived from 3-mo old tilapia. The approach is innovative and provides a means to investigate DHH and each downstream component from the ptch receptors to the gli and sf1 transcription factors. They concluded that Dhh binds Ptch2 to stimulate Gli1 to promote an increase in Sf1 expression leading to the onset of 11-ketotesterone synthesis heralding the differentiation of Leydig cells in the developing male tilapia.'
Strengths of the methods and results:
- The use of Nile tilapia is important as it is an important aquaculture species, it shares the genetic pathway for sex determination of mammalian species, and molecular differentiation pathways are highly conserved
- The approach is rigorous and incorporates a novel TSL, clonal stem Leydig cell model that they developed that is relatively faithful in following endogenous developmental steps and can produce the appropriate steroid.
- Tilapia are relatively amenable to CRISPR/Cas9 targeting and, with their accelerated developmental time frame, provide an excellent model system to interrogate specific signaling pathways.
- The stepwise analysis from dhh-gli-sf1 is thoughtful and well done.
Weaknesses of the methods and results:
- Line 162: need to establish and verify the PKH26-labeled TSL cells were unaffected by the dhh-/- environment. No data to support the claim that they were unaffected.
- The rescued phenotype caused by the addition of ptch2-/- to the dhh-/- model is a compelling. To further define potential ptch1 contributions, it would be helpful to examine the expression level of ptch1 in the context of the ptch2-/- and ptch2-/-;dhh-/- mutant animals. Any compensatory increase in ptch1 in either case, without obvious phenotype changes, would support the dominant role for ptch2.
- Activity of individual gli factors need additional reconciliation. The expression profiles for both alternative gli factors should be quantified in each knockout cell line to establish redundancy and/or compensation.
- Figure 5E: An important control is missing that includes evaluation of HEK293 cells transfected with pcDNA3.1-OnGli1 without the addition of pGL3-sf1.
Achieved Aims:
The authors set out to test the hypothesis that the canonical Dhh signaling pathway for Leydig cell differentiation and steroidogenic activity is mediated via ptch2 and gli1 regulation of sf1. The results are strong, there are additional steps needed to verify that redundancy/compensation is not contributing to the outcomes.
This work is important in better understanding of nuanced commonalities and differences in developmental pathways across species. Specific to Leydig cell differentiation and steroidogenesis, their work with tilapia supports conservation of the canonical Dhh pathway; however, there appear to be some differences in downstream mediators compared to mouse. Specifically, they conclude that ptch2/gli1 stimulates sf1 and steroidogenesis in tilapia where gli1 is dispensable in mouse. Instead, Gli3 has recently been shown to play an important role to stimulate Sf1 and support the hedgehog pathway.
Author response:
General Statements
We thank the reviewers for their thoughtful and constructive comments on our manuscript. We have thoroughly considered all points raised and have made extensive revisions to address them. These revisions have significantly strengthened the manuscript.
In summary, the key revisions and clarifications include:
(1) Developmental Time-Course: To address the need for earlier phenotypic analysis, we have performed new immunofluorescence experiments at 30 days after hatching (dah). This new data (Fig. S7) precisely pinpoints the onset of the Leydig cell differentiation defect in dhh-/- mutants, establishing ~30 dah as the critical window for Dhh action.
(2) Role of Ptch1 and Ptch2: We have qualified our conclusions regarding receptor specificity throughout the text to accurately reflect our findings and the limitation posed by the early lethality of ptch1 mutants. The in vivo genetic evidence for Ptch2 (the rescue of dhh-/- by ptch2-/-) is emphasized, while we now explicitly state that a role for Ptch1 cannot be ruled out without future conditional knockout models.
(3) Mechanism between Gli1 and Sf1: In direct response to the reviewers' request for stronger evidence, we have performed a new cold probe competition assay. This experiment provides dose-dependent, biochemical evidence for the specificity of Gli1 binding to the sf1 promoter (New Fig. 5E). Furthermore, we have revised the text throughout the manuscript to use more precise language (e.g., "Gli1 activates sf1 expression") and removed overstated claims of "direct" regulation.
(4) Methodological Rigor and Controls: We have added crucial negative controls for all RNA-FISH experiments using sense probes (New Fig. S9), provided detailed quantification methods for immunofluorescence, clarified the number of biological replicates for transcriptomic analyses, and corrected statistical tests as recommended.
(5) Clarity and Presentation: We have revised the text for clarity, expanded the description of the TSL cell line's validation in the Introduction, added missing details to figure legends and methods, and incorporated suggested key references.
We believe that our detailed responses and the significant new data and textual revisions have fully addressed the reviewers' concerns and have substantially improved the quality and impact of our manuscript.
Point-by-point description of the revisions
Reviewer #1 (Evidence, reproducibility and clarity):
Summary
This manuscript by Zhao et. al investigates the canonical hedgehog pathway in testis development of Nile tilapia. They used complementary approaches with genetically modified tilapia and transfected TSL cells (a clonal stem Leydig cell line) previously derived from 3-mo old tilapia. The approach is innovative and provides a means to investigate DHH and each downstream component from the ptch receptors to the gli and sf1 transcription factors. They concluded that Dhh binds Ptch2 to stimulate Gli1 to promote an increase in Sf1 expression leading to the onset of 11-ketotesterone synthesis heralding the differentiation of Leydig cells in the developing male tilapia.
Major comments:
(1) Are the key conclusions convincing?
Most results as reported are convincing; however, some conclusions are premature as additional experiments are required to satisfy their claims. For example, the phenotype of the dhh-/- testis is convincing in that Cyp1c1 cells are missing and the addition of ptch2-/- rescues the phenotype indicating a direct path. The link from gli to sf1, however, requires additional study to validate the direct relationship (see item 3 below).
We thank the reviewer for the positive assessment that our principal findings are convincing. Regarding the connection between Gli1 and Sf1, we agree that additional validation was important. We have now performed new experiments and revised our text. As detailed in our response to item 3 below, we have incorporated a cold probe competition assay (new Fig. 5E) which provides dose-dependent evidence for the specificity of Gli1 binding to the sf1 promoter. Furthermore, we have toned down our conclusions in the manuscript.
(2) Should the authors qualify some of their claims as preliminary or speculative, or remove them altogether?
Major: Most significant premature claim is the statement that gli1 directly controls sf1 activity. Additional experiments are required to make this claim (see next statement).
We agree with the reviewer that the claim of "direct" control was premature. We have therefore revised the manuscript accordingly. All statements claiming "direct" regulation of sf1 by Gli1 have been removed or replaced with more accurate descriptions, such as "Gli1 activates sf1 expression" and "Sf1 is a key transcriptional target of Gli1." These changes, coupled with the new functional data from the cold probe competition experiment (Fig. 5E) described in our response to item 3, now provide a robust and appropriately qualified account of our findings.
Minor: As addressed in the discussion section, the ptch1 animals fail to survive limiting the ability to validate both ptch1 and ptch2 roles. Thus, the conclusion that only ptch2 is required should be qualified.
We thank the reviewer for this rigorous comment. We fully acknowledge the limitation imposed by the early lethality of ptch1 mutants, which precludes a definitive in vivo assessment of its potential role in postnatal testis development. In direct response to this point, we have revised the text throughout the manuscript to more accurately reflect the strength of our conclusions. Specifically, in the Results section, we now state that “This differential receptor requirement implies that Ptch2 likely acts as the functional receptor for transducing Dhh signals in TSL cells” (lines 174–176). Furthermore, we have strengthened the Discussion by explicitly stating: “Therefore, while our findings strongly nominate Ptch2 as the principal receptor for Dhh in SLCs, a definitive exclusion of a role for Ptch1 will require future studies employing Leydig cell–specific conditional knockout models” (lines 265–268). We believe these revisions provide a appropriately qualified interpretation of our data while maintaining the compelling narrative of Ptch2's primary role.
Major: There are a couple of key references missing however, please consider including:
- Kothandapani A, Lewis SR, Noel JL, Zacharski A, Krellwitz K, Baines A, Winske S, Vezina CM, Kaftanovskaya EM, Agoulnik AI, Merton EM, Cohn MJ, Jorgensen JS.PLoS Genet. 2020 Jun 4;16(6):e1008810. doi: 10.1371/journal.pgen.1008810. eCollection 2020 Jun.PMID: 32497091
- Park SY, Tong M, Jameson JL.Endocrinology. 2007 Aug;148(8):3704-10. doi: 10.1210/en.2006-1731. Epub 2007 May 10.PMID: 17495005
We have included the key references: Kothandapani A, et al. (2020). PLoS Genet. and Park SY, et al. (2007). Endocrinology.
(3) Would additional experiments be essential to support the claims of the paper? Request additional experiments only where necessary for the paper as it is, and do not ask authors to open new lines of experimentation. Additional experiments are suggested to strengthen the direct connection between gli1 and sf1:
Major: Figure 5F shows evidence for increased sf1-luc activity upon co-transfection of OnGli1 in TSL cells. These data would be strengthened with evaluation of the same sf1 promoter that has each/both putative GLI binding sites mutated.
We thank the reviewer for this insightful suggestion. To further strengthen the evidence for the functional connection between Gli1 and the sf1 promoter, we have performed a new cold probe competition experiment. Given the potential presence of other unpredicted Gli-binding motifs within the 5-kb sf1 promoter region and the practical constraints, we employed an alternative, robust biochemical approach. This assay used a wild-type oligonucleotide containing the canonical Gli-binding motif (GACCACCCA) as a specific competitor. As shown in the new Fig. 5E, this cold probe caused a significant, dose-dependent reduction in Gli1-induced sf1-luc activity, while a mutated control probe (TTAATTAAA) had no effect. This result provides strong evidence that Gli1-mediated transactivation of the sf1 promoter is dependent on its specific binding to this consensus motif.
Furthermore, in response to the reviewer's comment, we have revised the manuscript text to use more precise language, such as "Gli1 activates sf1 expression" and "Sf1 is a key transcriptional target of Gli1," toning down any overstated claims of direct regulation. Together with the existing data-which includes the original luciferase assay, the new competition experiment, and key loss-of-function/gain-of-function genetic evidence from SLCs transplantation-we believe our study now provides a compelling and multi-faceted case for Gli1 being the key regulator of sf1 within this pathway. We are confident that these revisions have satisfactorily addressed the point raised.
Major: All 8xGli-luciferase assays should include evaluation of the mutant 8xGli-luciferase plasmid as a negative control.
We thank the reviewer for highlighting the importance of reporter assay controls. In our study, we included the empty vector pGL4.23, which lacks any Gli-binding sites, as the fundamental negative control. As shown in Fig. 4C, this vector showed minimal background activity that was unresponsive to Dhh, confirming that the strong luciferase induction in the 8xGli-reporter is entirely dependent on functional Gli-binding sites. While a mutated 8xGli construct is one valid approach, we think that the use of an empty vector is functionally equivalent and equally rigorous for establishing specificity. We are confident that our current data unambiguously demonstrate Gli-dependent activation. For clarity, we have explicitly stated in the figure legend and methods that pGL4.23 served as the negative control.
Minor: Figure 5D experiment that includes TSL-gli1(also 2,3) +/- OnDhh; please examine whether the absence of Gli affects expression of sf1 in each condition. In other words, provide a loss-of-function of Gli connection to regulation of sf1.
We measured the mRNA expression levels of sf1 in TSL-WT, TSL-gli1-/-, TSL-gli2-/-, and TSL-gli3-/- cells using qRT-PCR. The results are presented in the new Supplementary Figure S8A. The results show that the loss of gli1 leads to a significant reduction in the expression of sf1. In contrast, the knockout of gli2 or gli3 had no significant effect on sf1 expression levels.
(4) Are the suggested experiments realistic in terms of time and resources? It would help if you could add an estimated cost and time investment for substantial experiments.
Given the expertise, it is not anticipated that the suggested experiments would be a significant burden to this group.
We appreciate the reviewer's considerations. Now, we have performed the additional key experiments, which have been incorporated into the revised manuscript. We believe these new data have fully addressed the points raised.
(5) Are the data and the methods presented in such a way that they can be reproduced?
Most methods are adequately described or referenced to previous detailed description. There were, however, some methods that could benefit from additional details:
Major: IF quantification data: please provide details on how the number of positive cells were quantified and presented, for example, how many cells from how many sections for each genotype were included for the analysis?
We have added relevant information in the "Materials and Methods" section in line 369-373: “For each biological replicate (n\=5-6 fish per genotype), three non-serial, non-adjacent testis sections were analyzed. From each section, three representative fields of view were captured to ensure non-overlapping sampling. All positive cells number of Vasa, Sycp3 and Cyp11c1 was quantified by Image J Pro 1.51 software using default parameters.”
Major: FISH: No controls are present, for example, scrambled RNA probes. Further, please clarify or address the significant presence of message in the nucleus.
As suggested, we have now included negative control experiments using sense RNA probes for all genes (ptch1, ptch2, gli1, gli2, gli3). These controls showed no specific signal, confirming the specificity of our antisense probe hybridization. These data are now presented in the new Supplementary Figure S9.
Major: TSL cells: TSL-onDhh, -onSf1: provide evidence for increase in expression
We measured the mRNA expression levels of dhh in TSL-WT and TSL-OnDhh, and sf1 in TSL-WT and TSL-OnSf1 using qRT-PCR. The results are presented in the new Supplementary Figure S8B. The results show that overexpression of Dhh and Sf1 significantly increased the mRNA expression levels of dhh and sf1, respectively.
Major: TSL + SAG cells and other treatments in general: how long were they treated before transplantation?
Response: We have added relevant information in the "Materials and Methods" section in line 398-399: “For the SAG treatment experiment, TSL cells were incubated with 0.5 μM SAG for 48 hours before transplantation.”
Major: Transcriptome analyses: how many replicates were used for each cell line? Please clarify-the results presented in Fig 5E: how was this plot generated, it is interpreted that all three cell lines were combined and compared to the WT line. It is not clear how this was achieved.
We have added relevant information in the "Materials and Methods" section in line 445-447: “For the SAG treatment experiment, TSL cells were incubated with 0.5 μM SAG for 48 hours before collection. For each genotype, cells from three independent culture wells were pooled.
Added relevant information in the "Results" section in line 198-202: “…we performed transcriptomic profiling of TSL cells under conditions of pathway activation: Dhh overexpression (TSL-OnDhh), Gli1 overexpression (TSL-OnGli1), and SAG treatment (TSL+SAG). Comparative RNA-seq analysis identified a core set of 33 genes consistently upregulated across all three conditions.”
(6) Are the experiments adequately replicated and statistical analysis adequate?
Most are adequate and appropriate, some questions remain:
- Transcriptomes-how many replicates (see above)?
- IF quantification-how were cells identified/how many sections (see above)?
Minor: Statistics: methods indicate that a student's t-test was used, but ANOVA's are also used, which is appropriate. There are data presented that should be reevaluated via an ANOVA: Figure 4D, 4N-R; Figure 5G-no stats indicated in figure legend.
We sincerely thank the reviewer for highlighting the inappropriate use of statistical tests in our original submission. We have re-analyzed all data using the ANOVA-based methods as suggested in the specific detail. We confirm that these changes do not alter the overall interpretation of our results but provide a more robust and statistically sound foundation for our conclusions. We changed “Differences were determined by two-tailed independent Student's t-test” to “Statistical significance was determined by one-way ANOVA followed by Tukey's test (C, Q-U, different letters above the error bar indicate statistical differences at P < 0.05) or Student's t-test (D) (*, P < 0.05; **, P < 0.01; NS, no significant difference).”
In lines 719-721 we added “Statistical significance was determined by one-way ANOVA followed by Tukey's test (E, different letters above the error bar indicate statistical differences at P < 0.05) or Student's t-test (B, H) (*, P < 0.05; **, P < 0.01; NS, no significant difference).” in line 745-747.
Reviewer #1 (Significance):
The data presented in this manuscript provides important context towards the connection between the DHH pathway, Sf1, and steroidogenesis.
The audience would likely include developmental biologists, including those related to differentiation of any hormone producing cell type and especially those focused on steroidogenesis onset. Clinical interests will be related to sex determination and differentiation, especially related to male sex phenotype differentiation. Basic scientists will be especially interested.
Expertise: mouse fetal testis differentiation and maturation, steroidogenesis, hedgehog, sf1. Good fit except for the animal model, but they are surprisingly similar.
Reviewer #2 (Evidence, reproducibility and clarity):
In this work, Zhao et al., investigated the role of Dhh signaling pathway in the proliferation and differentiation of leydig lineage cells in the testes of Nile tilapia, an economic important farmed fish. By generating dhh mutants, the authors showed that loss of Dhh in tilapia recapitulated mammalian phenotypes, characterized by testicular hypoplasia and androgen insufficiency. A previous established TSL line was used to rescue the deficits in dhh-/- testes, which demonstrated that Dhh regulates the differentiation of SLCs rather than their survival. By generating mutant TSL lines, the authors aimed to identify the downstream players under Dhh in tilapia. Based on the data, the authors propose that a dhh-ptch2-gli1-sf1 axis exists in leydig cell lineage development.
How secreted dhh from Sertoli cells affect the Leydig cells remains elusive. While previous studies have revealed the paracrine role of Sertoli cell secreted Dhh in the regulation of Leydig cell development and maturation, the authors provided some new insights into the issue using tilapia as a model. Unfortunately, this work is not well performed, and the conclusions are not well supported by the current data. And to reach logic conclusions, more meaningful experiments should be performed, and more convincing data should be provided.
Strength:
The authors used genetic mutants, TSL lines, and cell transplantation techniques to address the questions. The manuscript is technically sound, and overall is well-written.
Limitations:
Experimental design should be optimized, and more convincing data should be provided to reach solid conclusion.
(1) The SLCs (stem leydig cells) used in this work. The SLC line was established from 3-month-old immature XY tilapia. The authors claimed that this line is a SLC line only because they express a few Leydig markers such as pdgfra and nestin. However, in my opinion, the identity of the cell line is not clear. It is suggested to perform more experiments, including flow cytometry assay or single cell RNA sequencing analysis, to further characterize this line, to demonstrate that this line is a real SLCs that are equivalent to the SLCs in 3-month testes of tilapia. According to the previous publication (2020), the information about the line was not well presented.
We thank the reviewer for this comment regarding the characterization of the TSL cell line. The identity of TSL as a stem Leydig cell line was rigorously established in our previous publication (Huang et al., 2020), which provided comprehensive molecular, in vitro, and in vivo functional evidence that meets the definitive criteria for an SLC. This includes its stable expression of established SLC markers (pdgfrα, nestin, coup-tfii), its capacity to differentiate into steroidogenic cells producing 11-KT in vitro, and most critically, its ability to colonize the testicular interstitium, differentiate into Leydig cells, and restore androgen production upon transplantation in vivo.
In direct response to the reviewer's point, we have revised the Introduction of our manuscript to provide a more detailed and clear description of the TSL line's origin and validation (lines 95-105) as “Furthermore, a stem Leydig cell line (TSL) has been established from the testis of a 3-month-old Nile tilapia. TSL expresses platelet-derived growth factor receptor α (pdgfrα), nestin, and chicken ovalbumin upstream promoter transcription factor II (coup-flla), which are usually considered as SLC-related markers in several other species. Notably, this cell line exhibits the capacity to differentiate into 11-ketotestosterone (11-KT)-producing Leydig cells both in vitro and in vivo. When cultured in a defined induction medium, TSL cells differentiate into a steroidogenic phenotype, expressing key steroidogenic genes including star1, star2, and cyp11c1, and producing 11-KT; upon transplantation into recipient testes, TSL cells successfully colonize the interstitial compartment, activate the expression of steroidogenic genes, and restore 11-KT production”, ensuring that readers can fully appreciate its well-founded identity as a SLC model without needing to consult the original publication. We are confident that the existing body of evidence solidly supports all conclusions drawn from its use in this study.
(2) How loss of dhh affects testicular and the leydig cell lineage development are not clearly investigated. In the current manuscript, the characterization of dhh mutant was not enough and lack of in-depth investigation. The authors primarily looked at testes at 90 dph when Leydig cell lineage was well developed. In my opinion, this time was too late. To investigate the earlier events that are affected by loss of dhh, I suggested to perform experiments at earlier time points, in particular around the initiation stages of the sex differentiation and Lyedig cell specification/maturation.
We thank the reviewer for this insightful comment. We agree that a thorough developmental analysis is crucial. In response to this point, we have now performed an in-depth investigation at earlier stages to precisely define the phenotype onset.
Our revised manuscript includes new data from a developmental time-course analysis. While our initial characterization included 5, 10, and 20 dah, we now identified 30 dah as the critical window for Leydig cell differentiation onset, which was also supported by prior work (Zheng et al.). Our new immunofluorescence data at 30 dah now clearly show that Cyp11c1-positive cells are present in wild-type testes but are entirely absent in dhh-/- mutants (Fig. S7). This finding pinpoints the initial failure of SLC differentiation.
We have integrated this key finding into the Discussion (lines 234-239) as “To define the onset of Leydig cell differentiation, we performed a developmental time-course analysis. This revealed that Cyp11c1-positive steroidogenic cells first appear in wild-type testes at 30 dah, while being conspicuously absent in dhh-/- mutants at this same stage (Fig. S7). This clear temporal pattern establishes ~30 dah as the developmental window when SLCs initiate their differentiation program in the Nile tilapia.”
Concurrently, our analysis of the 90 dah timepoint remains vital, as it represents a mature stage with robust spermatogenesis and a stabilized somatic niche. This allows for a comprehensive assessment of the ultimate functional consequences of the early differentiation block, including its impact on germ cell support and overall testicular architecture.
Thus, our study now provides a complete developmental perspective: the 30 dah timepoint identifies the initiation of the Dhh-dependent defect, while the 90-dah analysis reveals the mature, functional outcomes within the intact testicular niche.
(3) The authors claimed that there was a ptch2-gli1-sf1 axis. The conclusion was drawn largely based on data that generated from the in vitro cultured TSL line. More data from genetic mutant tilapia are required to support the conclusion.
We thank the reviewer’s insightful comments regarding the need for robust in vivo validation. In fact, our conclusion of a Dhh-Ptch2-Gli1-Sf1 axis is supported by an integrated experimental strategy, combining key in vivo evidence with targeted in vitro analyses to build a coherent model.
(1) Evidence for Ptch2 as the key receptor: The role of Ptch2 is supported by a pivotal in vivo genetic experiment. The observation that the dhh-/- testicular phenotype is fully rescued in dhh-/-;ptch2-/- double mutants provides compelling genetic evidence that Ptch2 is the essential receptor for Dhh in vivo (Fig. 4E-U). We acknowledge that the early embryonic lethality of global ptch1 mutation precludes its functional analysis in postnatal testis development. Therefore, while our data strongly nominate Ptch2 as the principal receptor, we have qualified our conclusions in the revised manuscript to reflect that a role for Ptch1 cannot be definitively excluded without Leydig cell-specific conditional knockout models.
(2) Evidence for Gli1 and its regulation of Sf1: The role of Gli1 as the key transcriptional effector was efficiently identified using our well-characterized TSL system, a valid approach for dissecting this highly conserved signaling cascade. The functional connection between Gli1 and Sf1 is supported by multiple lines of evidence: transcriptomic profiling, promoter analysis, luciferase reporter assays (including a new cold probe competition experiment), and most importantly, in vivo functional validation via SLC transplantation. The latter demonstrated that Sf1 is both necessary and sufficient for SLC differentiation within the testicular niche (Fig. 5).
In direct response to the reviewer's points, we have thoroughly revised the manuscript text to ensure all claims are accurately stated, particularly regarding the receptor specificity and the nature of the Gli1-Sf1 regulatory relationship. We believe our study provides a solid foundation for the proposed signaling axis.
Overall, better experimental design should be planned, including the rescue experiments. Some key information was missed. For instance, the identity of the stem Leydig cells was not clearly presented.
We have explained it in point #1.
Figures:
Figure 1: The authors described the phenotypes at 90 dph. Loss of dhh led to severe phenotypes in testicular formation, as evidenced by defective formation of Vasa, a germline stem cell marker; loss of expression of cyp11c1, a leydig cell marker; and loss of sycp3, a marker of meiosis of spermatogonia.
However, in my opinion, 90 dph was too late. To investigate the role of dhh in Leydig cell lineage, the authors are suggested to focus on earlier developmental stages when the sex differentiation and maturation of leydig cells occur. This work is actually a development biology one that investigates how dhh loss in Sertoli cells affects the development of Leydig cells. The careful characterization of earliest testicular phenotypes of dhh mutant is very important.
We have explained it in point #2.
Figure 2: Please clarify the logic for performing rescue experiments using 11-KT. Provided the critical role of 11-KT in the testis development and spermatogenesis, it was not unexpected that 11-KT treatment can rescue most of the cell types in testes. If dhh is absolutely required for LC lineage development maturation, adding 11-KT at 30 dph will not have an effect. Why not perform rescue experiments using Dhh protein?
We thank the reviewer for this insightful comment, which allows us to clarify the logical progression of our experimental design, a process central to genetic discovery.
When we first characterized the dhh-/- mutant, we observed a complex suite of phenotypes: testicular hypoplasia, arrested germ cell development, a profound deficiency of Leydig cells, and drastically low androgen levels. A primary challenge was to distinguish which defects were direct consequences of losing Dhh signaling and which were secondary effects of the overall testicular failure.
We therefore employed a classic genetic strategy: phenotypic dissection through targeted rescue. The 11-KT rescue experiment was designed to test a foundational hypothesis: Are the severe testicular defects in dhh-/- mutants primarily a consequence of the systemic androgen deficiency? The results provided a pivotal and clear answer: while 11-KT treatment partially rescued germ cell development and testicular structure, it completely failed to restore the population of Cyp11c1-positive Leydig cells. This critical finding allowed us to dissociate the phenotypes, demonstrating that the Leydig cell defect is a primary, cell-autonomous consequence of Dhh loss, not a secondary effect of low androgen.
This conclusion logically propelled the next phase of our research: to shift focus from systemic hormone action to the local, niche role of Dhh in regulating the Leydig lineage directly. This led directly to the TSL transplantation experiments and the mechanistic dissection of the Ptch2-Gli1-Sf1 axis within SLCs.
Regarding the use of Dhh protein, we agree it is a complementary approach. However, producing biologically active, recombinant Hedgehog ligand is challenging due to its essential dual lipid modification, which is required for solubility and activity. Our transplantation experiments with TSL-OnDhh cells (Fig. 3) functionally demonstrate that providing Dhh signaling in a cell-autonomous manner is sufficient to rescue differentiation, thereby directly addressing the core question without the need for recombinant protein.
Figure 3. The authors showed that in dhh-/- testes, TSL engrafted equivalently but failed to express Cyp11c1. This result was strange which raised a question about the identity of the TSLs, as I have mentioned above. The authors claimed that the TSLs are stem Leydig cells, which I doubt. Additional data should provided to support the statement.
In the testicular environment, the transplanted TSLs should be able to colonize and differentiate into more mature leydig cells. Only a small portion of the PKH26-labled TSLs became Cyp11c1 positive after transplantation, can the authors comment this observation?
To address "Mutation of dhh blocks SLC differentiation", the authors should first carefully examine the TSL lineage development using dhh mutant. Then, investigate how loss of dhh disrupts the cross talk between Sertoli cells and Leydig cells. why bother performing transplanted TSLs? Please clarify. Why not perform rescue experiments using Dhh protein at appropriate developmental stages?
We thank the reviewer for these comments, which allow us to clarify the rationale and interpretation of our key experiments.
(1) We have provided comprehensive evidence establishing the TSL line as a SLC line (Response to Point #1). The observation that WT TSL cells engraft but fail to differentiate in the dhh-/- testicular environment is not strange; it is, in fact, the core and most crucial finding of this experiment. It provides direct functional evidence that the dhh-/- niche lacks the essential signals required to initiate SLC differentiation, consistent with the severe deficiency of endogenous Cyp11c1+ cells in these mutants (Fig. 1I-J', N).
(2) The reviewer's concern about "only a small portion" of cells differentiating is based on a misunderstanding. Our quantitative data (Fig. 3F) show that approximately 78% of the transplanted PKH26+ TSL cells successfully differentiated into Cyp11c1+ cells in WT hosts. This high efficiency robustly demonstrates the differentiation potential of TSL cells and the permissiveness of the WT niche. The near-zero differentiation rate in the dhh-/- host (Fig. 3F) starkly highlights the specific and severe defect in the mutant microenvironment.
(3) The TSL transplantation experiment was the most direct strategy to test why Cyp11c1+ cells are absent in dhh-/- testes. It allowed us to distinguish between a failure in SLC differentiation and other possibilities (e.g., cell death). The finding that functional SLCs cannot differentiate in the mutant niche logically directed our subsequent focus onto the cell-intrinsic molecular mechanism (the Ptch2-Gli1-Sf1 axis) within the Leydig lineage. While Sertoli-Leydig crosstalk is an important area, it was beyond the scope of this study aimed at defining the intrinsic differentiation pathway.
(4) Regarding Dhh protein rescue, generating bioactive, lipid-modified recombinant Hh protein is technically challenging. Our transplantation of TSL-OnDhh cells (Fig. 3) functionally demonstrates that providing Dhh signaling in a cell-autonomous manner is sufficient to rescue differentiation, effectively addressing this question without the need for recombinant protein.
Figure S3. “To assess whether dhh mutation affects androgen-producing cells outside Leydig cells, 11-KT levels were analyzed during early testicular development before SLCs differentiation. IF analyses revealed that no Cyp11c1 positive cells were present in the testes of XY WT fish at 5, 10, and 20 dah, indicating that SLCs had not yet differentiated at these stages (Fig. S3A-C). Tissue fluid 11-KT levels showed no significant differences between WT and dhh-/- XY fish at 5, 10, and 20 dah (Fig. S3D)”. These observations suggested that loss of dhh does not affect the specification of SLCs, but affect its differentiation into mature LCs. The differentiation of Cyp11c1 should be later than 20 dah. So when is the earliest time point for formation of Cyp11c1 positive cells, and how loss of dhh affect this? These are important questions to answer.
We agree with the reviewer's interpretation that our data suggest dhh loss affects SLC differentiation rather than initial specification. In direct response to the need for earlier timepoints, we have now performed and included an analysis at 30 dah, which we identified as the critical window for Leydig cell differentiation onset. Our new data (Fig. S7) show that Cyp11c1+ cells are present in WT testes but are entirely absent in dhh-/- mutants at this stage. This precisely pinpoints the initiation of the phenotypic divergence and establishes ~30 dah as the developmental window when Dhh signaling is required to drive SLC differentiation. Our study therefore now provides a complete developmental perspective, from the initial failure at 30 dah to the mature functional outcomes at 90 dah.
Figure 4. The authors generated ptch1/2 mutant TSL lines, and luciferase assay was performed, and based on the results, the authors concluded that Ptch2, but not Ptch1, is specifically required for transducing Dhh signals in TSLs. The conclusion was only based on luciferase assay using TSLs. Whether this was the case in testes at animal level is not clear. Clearly, more genetic experiments, using ptch mutants, should performed to substantiate this.
The authors stated “Ptch2 acts as the obligate receptor for Dhh signaling during testis development”. If ptch2 is required for TSL lineage, why ptch2-/- testes exhibited no significant differences in testicular histology and Leydig cell (Cyp11c1+) populations and serum 11-KT levels? This contradictory statement need to be addressed.
We thank the reviewer for these critical comments, which allow us to clarify the logic underlying our conclusions regarding Ptch2.
(1) In Vivo Genetic Evidence for Ptch2: Our conclusion that Ptch2 is the primary receptor for Dhh is not based solely on the TSL luciferase assays. It is definitively supported by a key in vivo genetic experiment: the complete phenotypic rescue in the dhh-/-;ptch2-/- double mutants (Fig. 4F-R). In genetic terms, the loss of the receptor (ptch2) suppressing the phenotype caused by the loss of the ligand (dhh) is classic evidence for a ligand-receptor relationship within a linear pathway. This in vivo evidence strongly substantiates Ptch2's role at the animal level. The early embryonic lethality of ptch1 mutants precludes a similar in vivo test for Ptch1 in postnatal testis development.
(2) Addressing the Apparent Contradiction of the ptch2-/- Phenotype: The reviewer raises an excellent point, which stems from the fundamental biology of the Hh pathway as shown in Author response image 1. Ptch receptors are inhibitory. In the absence of ligand, Ptch suppresses pathway activity.
Author response image 1.
The canonical Hh signaling pathway. In the dhh-/- mutant, the pathway is suppressed due to unopposed Ptch activity, leading to a failure in SLC differentiation. In the ptch2-/- mutant, this key inhibitory brake is removed, leading to constitutive activation of the pathway. The fact that ptch2-/- testes are normally indicates that this level of pathway activation is not detrimental and, crucially, is sufficient to support wild-type levels of Leydig cell development and steroidogenesis. This lack of a phenotype in the receptor mutant, contrasted with the severe ligand mutant phenotype, is a common and expected observation in signaling pathways where the receptor acts as a tonic inhibitor.
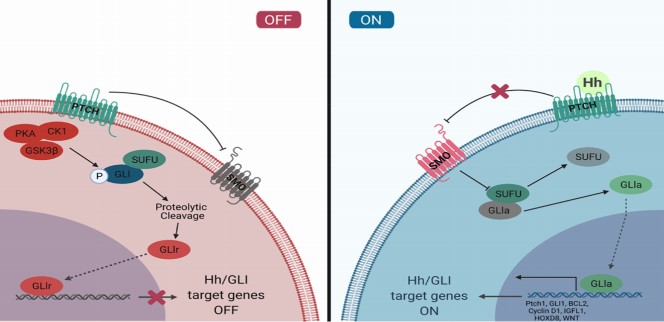
In summary, the normal development of ptch2-/- testes is not contradictory but is entirely consistent with its role as the inhibitory receptor for Dhh. The severe phenotype in dhh-/- mutants and its specific rescue by removing ptch2 provides compelling genetic evidence for their functional relationship. We have revised the text throughout the manuscript to ensure these conclusions are accurately stated.
Figure 5. The authors generated gli1/2/3 mutant TSL lines, and luciferase assay was performed, and based on the results, the authors concluded that Gli1, but not Gli2/3, was specifically required for transducing Dhh signals in TSL cells. The conclusion is drawn, only based on luciferase assay using TSLs. Whether this was the case in testes at animal level is not clear. Clearly, more genetic experiments should performed to substantiate this, using the gli mutant fish.
To identify Gli1-dependent targets in SLCs, the authors compared transcriptomes of TSLWT, Dhh-overexpressing (TSL-OnDhh), Gli1-overexpressing (TSL-OnGli1), and SAG-treated (TSL+ SAG) TSL cells. While this experiments can be used to identify dhh target genes, it is better to use gli mutant cell lines. Since the authors have generate gli1/2/3 mutants, why not using these mutant fish to identify/confirm the Gli targets?
We thank the reviewer for these comments.
(1) We acknowledge that Gli1 as the key transcriptional effector is primarily based on our in vitro evidence using the TSL cell line. We have revised the manuscript accordingly to ensure this is stated precisely, avoiding overstatement.
(2) Concerning the transcriptomic analysis, the reviewer suggests using glis mutant cell lines. While this is a valid approach, our strategy of profiling pathway activation (via Dhh/Gli1 overexpression or SAG treatment) was deliberately chosen to provide a high signal-to-noise ratio for identifying genes that are positively upregulated during the differentiation process. Analyzing loss-of-function mutants under basal conditions can be confounded by potential compensatory mechanisms among the Gli family members, potentially masking the specific transcriptional signature of pathway activation we sought to capture.
By the way, we have generated gli1/2/3 mutant TSL cell lines for the functional luciferase assays, but we have not generated the corresponding glis mutant fish lines, which would represent a substantial new line of investigation.
Reviewer #2 (Significance):
While previous studies have revealed the paracrine role of Sertoli cell secreted Dhh in the regulation of Leydig cell development and maturation, the authors provided some new insights into the issue using tilapia as a model.
Reviewer #3 (Evidence, reproducibility and clarity):
Summary
The authors investigate the Dhh signaling pathway in Leydig cell differentiation in the tilapia model. They generated multiple mutant lines in different hedgehog pathway components and utilized a Leydig stem cell line to interrogate Leydig cell differentiation. Through this analysis, the authors demonstrate that Dhh regulates Leydig differentiation rather than survival. They also found that Ptch2 is the specific receptor that mediates signaling to promote Leydig differentiation and that Gli1 is the primary Gli involved. Furthermore, they show that a known regulator of Leydig cell development and function, SF1, is a downstream transcriptional target. Overall, the study identifies previously unknown information as to how Dhh signaling regulates Leydig cell development, which is necessary for testosterone production by the testis.
Major Comments
(1) In the RNAseq analysis is not clear exactly how the 33 "up-regulated" genes were identified. What was the methodology for identification of these genes? Some of the genes were down-regulated or not different in the OnGli condition and some in the OnDhh condition were not differentially expressed, as shown in Fig S8B. Therefore, it is unclear why all 33 genes are classified as upregulated "across all three conditions".
We have clarified this methodology in the Materials and Methods section in line 452-454: “Differentially expressed genes (DEGs) were identified for each condition (TSL-OnDhh, TSL-OnGli1, TSL+SAG) compared to TSL-WT controls using edgeR (threshold: FDR < 0.05, |log2(foldchange)| ≥ 1.5). And we Added relevant information in the Results section in line 198-202: we performed transcriptomic profiling of TSL cells under conditions of pathway activation: Dhh overexpression (TSL-OnDhh), Gli1 overexpression (TSL-OnGli1), and SAG treatment (TSL+SAG). Comparative RNA-seq analysis identified a core set of 33 genes consistently upregulated across all three conditions (Fig. 5C, S6A).”
We have also updated Fig. S8B to include a clear value and to better visualize the FPKM value levels of these 33 genes across the conditions.
(2) In figure 4A (and possibly B), it appears that ptch RNA is in the nucleus of the cell. Why would the RNA be primarily in the nucleus? Is the RNA detection accurate? Were controls done? The methods state that sense probes were made but no how they compared to the antisense probes. This comment can also be applied to the gli FISH, particularly gli3 (Figure 5).
This is an excellent observation. We speculate that the apparent nuclear signal may be due to strong transcriptional activity in the nucleus. To confirm the specificity of our FISH experiment, we performed FISH with sense RNA probes as negative controls for all genes (ptch1, ptch2, gli1, gli2, gli3), and no specific signals were observed (see New Fig. S9).
Minor comments
(1) In the introduction, please include information as to when tilapia reach sexual maturity
We have added this information to the Introduction in line 91-92: early sexual maturity (approximately 3 months after hatching for males and 6 months after hatching for females).
(2) When first mentioning experiments that use the PKH26 dye, please give a brief description of the dye in the text of the results. This is described in the methods but it would be helpful to have some information about what PKH26 is in the results to more easily understand the figure and experimental design.
We have added a brief description in the Results section in line 151-152: “To dissect Leydig cell lineage impairment in dhh-/- testes, we transplanted the TSL labeled with PKH26 (a fluorescent red hydrophobic membrane dye that enables tracking of transplanted cells) into WT and dhh-/- testes (Fig. 3A).”
(3) In the statistical analysis section of the methods, the authors state that two-tailed t-tests were performed however in the figure legends it states that ANOVA was done for some of the statistical analysis. Please clarify this.
We have updated the Statistical Analyses section in Methods to clarify in line 472-476: “A two-tailed independent Student’s t-test was used to determine the differences between the two groups. One-way ANOVA, followed by Tukey multiple comparison, was used to determine the significance of differences in more than two groups. P < 0.05 was used as a threshold for statistically significant differences.”
(4) Figures - in figures that have charts with the Y-axis labeled as "relative positive cells", or similar, please explain what exactly is meant by "relative". What is it relative to?
We have revised all relevant Y-axis labels and figure legends to explicitly state the quantification method. For example, we now use: "Vasa+ / DAPI+ (%), Sycp3+ / DAPI+ (%) or Cyp11c1+ / DAPI+ (%).
(5) Figure 1: please point out the testes in panels A and B
We have indicated the position of the testes with arrows in Figures 1A and B.
(6) In figure 4, it would be helpful for the WT images from S7 moved to fig 4.
We have moved representative WT images from Fig. S7 into Fig. 4 for easier comparison with the mutant phenotypes.
(7) Figure 4E: Are the yellow bars comparable to each other. Is there any significance to the increased luciferase with 8xGli in ptch2-/- as compared to the other genotypes?
We thank the reviewer for this astute observation. Yes, the yellow bars are directly comparable, and the elevated basal luciferase activity of the 8xGli reporter in the ptch2-/- TSL cells is indeed significant and expected. The genetic ablation of ptch2 removes this inhibition, leading to ligand-independent, constitutive activation of the downstream signaling cascade. The observed increase in basal reporter activity in the ptch2-/- cells is a classic manifestation of this mechanism.
The primary objective of this experiment was to test the cells' responsiveness to Dhh stimulation across genotypes. The key finding is that while wild-type and ptch1-/- cells showed a significant response to Dhh, the ptch2-/- cells-which already exhibited high basal activity-were completely unresponsive. This combination of constitutive activation and ligand insensitivity in the ptch2-/- genotype provides particularly strong genetic evidence that Ptch2 is the essential receptor mediating Dhh signal transduction in this system.
(8) Figure 5G: please include what exactly what each construct name stands for in the figure legend
We have expanded the legend for Fig. 5G to define each construct.
(9) Figure S8B: please include what the values in the table are (eg are these the significance values?)
We have updated the caption for Figure S8B (now Figure S6B): “The FPKM value for each gene in each sample is indicated within the squares. The color gradient from blue to red reflects low to high expression levels per row (gene).”
Reviewer #3 (Significance):
Strengths and limitations:
The genetics of the tilapia system and the availability of the tilapia Leydig stem cell lines were particular strengths of this study. The study utilizes fish genetics to genetically interrogate the Dhh signaling pathway in Leydig cell development through generation and analysis of mutant lines. The tilapia Leydig stem cell line was an integral part of this study as it allowed for genetic and chemical manipulation of Dhh signaling in undifferentiated Leydig cells and, through transplantation into testes, allowed for analysis of how Leydig cell differentiation was affected.
Advance:
The study makes significant advances as to how Dhh signaling instructs Leydig cell differentiation, including identification of the Ptch receptor and Gli transcription factor that function downstream of Dhh in this process. Furthermore, they identify a direct link between Dhh signaling and Sf1 expression, which is known to important for Leydig cell function.
Audience:
This study will be of particular interest to reproductive biologists, endocrinologists, and developmental biologists. The study may also be of interest to researchers and physicians investigating cancers that are promoted by androgens produced by Leydig cells of the testis.



