Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorJulia CooperUniversity of Colorado Anschutz Medical Campus, Aurora, United States of America
- Senior EditorLynne-Marie PostovitQueens University, Kingston, Canada
Reviewer #1 (Public review):
Summary:
This is an interesting study that addresses whether mitochondrial DNA (mitoDNA) variants impact telomere length (TL), which may be relevant to potential maternal inheritance of TL in offspring. The study addresses this question using a cybrid model approach in which mitochondria from donor platelets from 7 individuals that vary in TL and differ in mitoDNA variants are introduced into 143B cells that lack mitochondria. MitoDNA variants that exhibited reduced complex I activity showed telomere shortening in cybrids and increased telomere dysfunction. Interestingly, these phenotypes could be reduced with NAC antioxidant and NAD+ supplementation, suggesting that ROS and oxidative DNA damage at telomeres contributed to the telomere shortening. They further showed that cybrids with lower levels of ROS correlated with longer TL in the lymphocytes of the mitochondrial donors.
Strengths:
This study provides compelling evidence that mtDNA variants influence TL through a mechanism involving mitochondrial-derived ROS, potentially causing telomeric oxidative damage. The data are robust, and the manuscript is well written. However, the study could be strengthened by addressing the following questions and minor weaknesses below.
Weaknesses:
(1) Introduction. Line 81, the relationship between TL and the risk of lymphoid and myeloid leukemia is not straightforward. POT1 variants associated with long TL increase the risk for lymphoid and myeloproliferative neoplasms (see PMID: 41564438 for example).
(2) Figure 1. Since sex also influences TL, it would be good to know the sex of the selected individuals or explain why this is not necessary.
(3) Please include a description of the 143B cells that were used for cybrid formation in the Results section when introducing the cybrids.
(4) Lines 155-156. The authors note that cybrids from donors 1 and 2 show "pronounced" telomere damage. This result indicates an increase in 53BP1-positive telomeres, which could be indicative of telomere dysfunction or damage. Quantification of the increased chromosome end fusions for cybrids 1 and 2 would strengthen the result. Do the increased fusions correlate with an increase in telomere signal-free ends? These should be apparent in the telomere FISH images of metaphase chromosomes.
(5) Lines 168-169. What is the evidence that the "in vitro metabolic shift" causes acute oxidative stress?
(6) Why did the elevated ROS in cybrid #3 (Figure 4C) not translate to shorter telomeres in the cybrid (Figure 2A)? Perhaps there is a difference between factors that determine TL in the cybrid vs the donor's lymphocytes? In Figure 4B, it appears that the statistical comparisons for mitochondrial superoxide are all relative to Cyb3. If so, why are the comparisons not with the parental 143B rho0 cell line? Please clarify.
(7) Given the heterogeneity in TL and mtDNA variants in the human population, the conclusions could be further strengthened by increasing the number of donors and cybrids analyzed. However, there are admittedly practical factors. Overall, these findings are compelling and provide a solid foundation for expanding this analysis in the future. This is more of a comment than a weakness.
Reviewer #2 (Public review):
Summary:
The authors aim to determine whether mitochondrial genotype influences telomere length. By generating cybrids harboring different mitochondrial backgrounds, the authors seek to establish a mechanistic link between mitochondrial status and telomere biology.
Strengths:
A major strength of the study is the use of cybrid technology, which provides a great approach to investigate the role of mitochondrial DNA independently of the nuclear genome. The authors also employ multiple complementary assays to assess telomere-related phenotypes associated with mitochondrial dysfunction. Together, these experiments generate an interesting dataset that will be of value to researchers interested in the intersection between mitochondrial biology, genome stability, aging, and development. These results also build on previous work supporting roles for ROS/mitochondria in driving telomere shortening.
Weaknesses:
The data support the conclusion that mitochondrial background is associated with differences in telomere length and telomere-related phenotypes. However, some of the mechanistic interpretations would benefit from additional evidence. In particular, the manuscript discusses mitochondrial influences on telomere shortening, yet telomere length in some experiments is assessed at a single time point. Consequently, the current data do not directly address the rate of telomere attrition. Differences observed between cybrid lines could potentially arise from events occurring during cybrid formation, clonal selection, or subsequent cell expansion. Longitudinal analyses across multiple passages, ideally beginning immediately after cybrid generation and controlling for population doublings, would help establish whether mitochondrial function directly affects telomere shortening dynamics. Some experimental results would also benefit from additional quantification, clarification, and some biological replicates are missing.
Overall, this study provides interesting evidence linking mitochondrial background to telomere biology. The cybrid models represent a useful resource for the field, and the work raises important questions regarding mitochondria-telomere communication.
Reviewer #3 (Public review):
Strengths:
Mahieu and colleagues address an interesting and underexplored question: whether non-pathogenic variation in the mitochondrial genome contributes to the inter-individual variability of human telomere length (TL). Using a Belgian Flow-FISH reference cohort (n=491) to identify donors at TL extremes, they generate transmitochondrial cybrids from platelets of seven donors of distinct mtDNA subhaplogroups and characterize the resulting cells with a broad and well-executed toolkit (TRF, TeSLA, ddTRAP, EPR-based mitoROS, Seahorse with permeabilized-cell ETC dissection, LC-MS metabolomics, telomeric PAR-FISH). The most compelling finding is that cybrids derived from donors with low complex I (CI) activity undergo rapid telomere shortening during the glycolysis-to-OXPHOS transition of cybrid formation, and that this is largely prevented by co-treatment with NAC and the NAD⁺ precursor nicotinamide riboside, supporting a model in which CI sustains the NAD⁺ pool required for PARP1-mediated repair of oxidative damage at telomeres. The authors further report an inverse correlation between donor lymphocyte TL and mitoROS in the corresponding cybrids, and provide preliminary evidence that the K1a-defining ATP6 A177T variant (m.G9055>A) may be enriched in long-telomere individuals.
Weaknesses:
(1) Statistical support and donor sampling for the central in vivo correlation (Figure 4C).
The inverse correlation between donor lymphocyte TL and cybrid mitoROS (R²=0.794, p=0.007) is the principal in vivo claim of the paper, but it is built on seven donors deliberately selected from the extremes of the Flow-FISH distribution. Sampling at the tails of the outcome variable can substantially inflate apparent correlation strength and significance. I would encourage the authors to (i) explicitly state this sampling structure where the correlation is introduced, (ii) report a leave-one-out sensitivity analysis to confirm the relationship is not driven by one or two donors (Cyb3 and Cyb6 appear to anchor the line), and (iii) where feasible, extend the analysis to additional donors with intermediate TL to test whether the relationship holds across the full distribution. Even a modest expansion (e.g., 4 to 5 additional donors at P25 to P75) would substantially strengthen this central claim.
(2) Reconciling the cybrid CI / TL relationship (Fig 3B) with the absence of a CI / TL relationship in donor lymphocytes (Figure 4A).
Figure 3B shows a strong correlation between CI activity and TL in cybrids (R²=0.87), while Figure 4A shows no correlation between donor CI activity (measured in the same cybrids) and donor lymphocyte TL. The authors acknowledge this, but the manuscript subsequently builds toward a CI-centric model of in vivo TL regulation, which seems to outrun the data. The most internally consistent interpretation is that the cybrid CI phenotype reports a sensitized in vitro response to the acute oxidative stress of the metabolic shift, rather than a steady-state determinant of leukocyte TL. I would suggest reframing the abstract, significance statement, and Discussion to make this distinction clearer. The in vitro CI / NAD⁺ / PARP1 axis is a strong finding on its own, while the in vivo role of CI activity (as opposed to ROS more broadly) is not yet established here. Donor #1's profile (very long lymphocyte TL, low CI activity, severe shortening in cybrids, no telomere inheritance in offspring) is informative in this regard and could be discussed more directly as a case that helps delineate where the cybrid model does and does not recapitulate in vivo biology.
(3) The K1a / ATP6 A177T inheritance claim.
The proposal that K1a (and specifically ATP6 A177T) contributes to maternal inheritance of long telomeres is intriguing but currently rests on three pedigrees (one of which, donor #1, does not support the hypothesis) and a chi-square test that does not reach significance (p=0.153, Figure 4F). The supporting evidence is also limited by the fact that platelet-mediated mitochondrial transfer delivers donor mitochondrial proteins, lipids, and residual mtRNA in addition to mtDNA, making it difficult to attribute the cybrid phenotype of donor #6 specifically to the ATP6 A177T variant. I would recommend either: (a) extending the genotyping screen to additional unrelated donors and, if feasible, confirming the effect of ATP6 A177T through an isogenic approach (e.g., mtDNA base editing in a clean background), or (b) softening the relevant statements to "suggestive trend warranting larger studies," and presenting the K1a observation as hypothesis-generating rather than supportive. The Ashkenazi-centenarian connection raised in the Discussion is an excellent direction for follow-up and could be framed accordingly.
Author response:
Public Reviews:
Reviewer #1 (Public review):
Summary:
This is an interesting study that addresses whether mitochondrial DNA (mitoDNA) variants impact telomere length (TL), which may be relevant to potential maternal inheritance of TL in offspring. The study addresses this question using a cybrid model approach in which mitochondria from donor platelets from 7 individuals that vary in TL and differ in mitoDNA variants are introduced into 143B cells that lack mitochondria. MitoDNA variants that exhibited reduced complex I activity showed telomere shortening in cybrids and increased telomere dysfunction. Interestingly, these phenotypes could be reduced with NAC antioxidant and NAD+ supplementation, suggesting that ROS and oxidative DNA damage at telomeres contributed to the telomere shortening. They further showed that cybrids with lower levels of ROS correlated with longer TL in the lymphocytes of the mitochondrial donors.
Strengths:
This study provides compelling evidence that mtDNA variants influence TL through a mechanism involving mitochondrial-derived ROS, potentially causing telomeric oxidative damage. The data are robust, and the manuscript is well written. However, the study could be strengthened by addressing the following questions and minor weaknesses below.
We thank the reviewer for this very positive evaluation of our work.
Weaknesses:
(1) Introduction. Line 81, the relationship between TL and the risk of lymphoid and myeloid leukemia is not straightforward. POT1 variants associated with long TL increase the risk for lymphoid and myeloproliferative neoplasms (see PMID: 41564438 for example).
We appreciate the reviewer raising the important link between pathogenic POT1 variants and lymphoid malignancies driven by elongated telomeres. We would like to clarify, however, that our introduction focused not on the pathological telomere attrition characteristic of telomere biology disorders, but rather on the natural, non-pathological variations observed in individuals with baseline telomere lengths on the shorter end of the spectrum.
Nevertheless, we will include this observation regarding patients with long telomeres in the introduction to underscore the complex relationship between telomere length and tumorigenesis.
The two following publications by the Armanios lab will be added:
DeBoy EA et al. Familial clonal hematopoiesis in a long telomere syndrome. 2023. N Engl J Med 388, 2422-2433.
Davidson-Swinton HR et al. Lymphoid malignancy and clonality in the POT1-mediated long telomere syndrome. 2026. Blood 147, 2226-2237.
(2) Figure 1. Since sex also influences TL, it would be good to know the sex of the selected individuals or explain why this is not necessary.
Because this study investigated the potential maternal inheritance of TL via the mitochondrial genome, our cohort consisted predominantly of female donors (6/7). Donor #5, the husband of Donor #7, was the only male included. We will update Figure 1D to include the sex of each donor and clarify this rationale in the text.
Notably, our analysis revealed minimal influence of sex on TL, which cannot account for the observed differences between the extreme groups.
(3) Please include a description of the 143B cells that were used for cybrid formation in the Results section when introducing the cybrids.
We will do so.
(4) Lines 155-156. The authors note that cybrids from donors 1 and 2 show "pronounced" telomere damage. This result indicates an increase in 53BP1-positive telomeres, which could be indicative of telomere dysfunction or damage. Quantification of the increased chromosome end fusions for cybrids 1 and 2 would strengthen the result.
We thank the reviewer for this suggestion. Following their advice, we used our metaphase spread FISH analyses to quantify chromosome end fusions in the parental 143B Rho0, Cybrid 1 and Cybrid 6 cells. However, because Cybrid 2 metaphase spreads were of insufficient quality for adequate chromosome analysis, we will restrict our telomere fusion comments exclusively to Cybrid 1 and include the quantifications in our revised manuscript.
Author response image 1.
The number of fusions and total chromosomes analyzed are indicated on each bar.
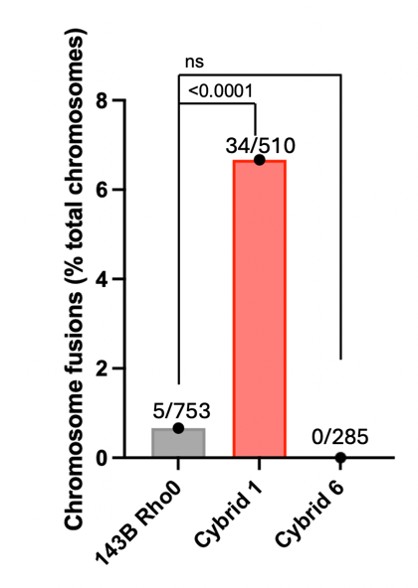
Do the increased fusions correlate with an increase in telomere signal-free ends? These should be apparent in the telomere FISH images of metaphase chromosomes.
While this is a strong argument, the telomeres within these cybrid models are critically short. Consequently, the FISH signal intensity falls below the threshold required for reliable quantification of telomere-free ends.
(5) Lines 168-169. What is the evidence that the "in vitro metabolic shift" causes acute oxidative stress?
The reviewer is correct that we have not formally demonstrated this in our experimental system. Instead, our assumption was based on the fact that the initial phase of Rho0 cell repopulation involves a temporary ROS burst, partly driven by incompletely assembled ETC supercomplexes that are known to elevate ROS levels (Maranzana et al, 2013). This is supported by our experimental observation that the NAC antioxidant, combined with NR, potently inhibits telomere shortening in cybrids with low CI activity. We propose to add this explanation in the revised manuscript.
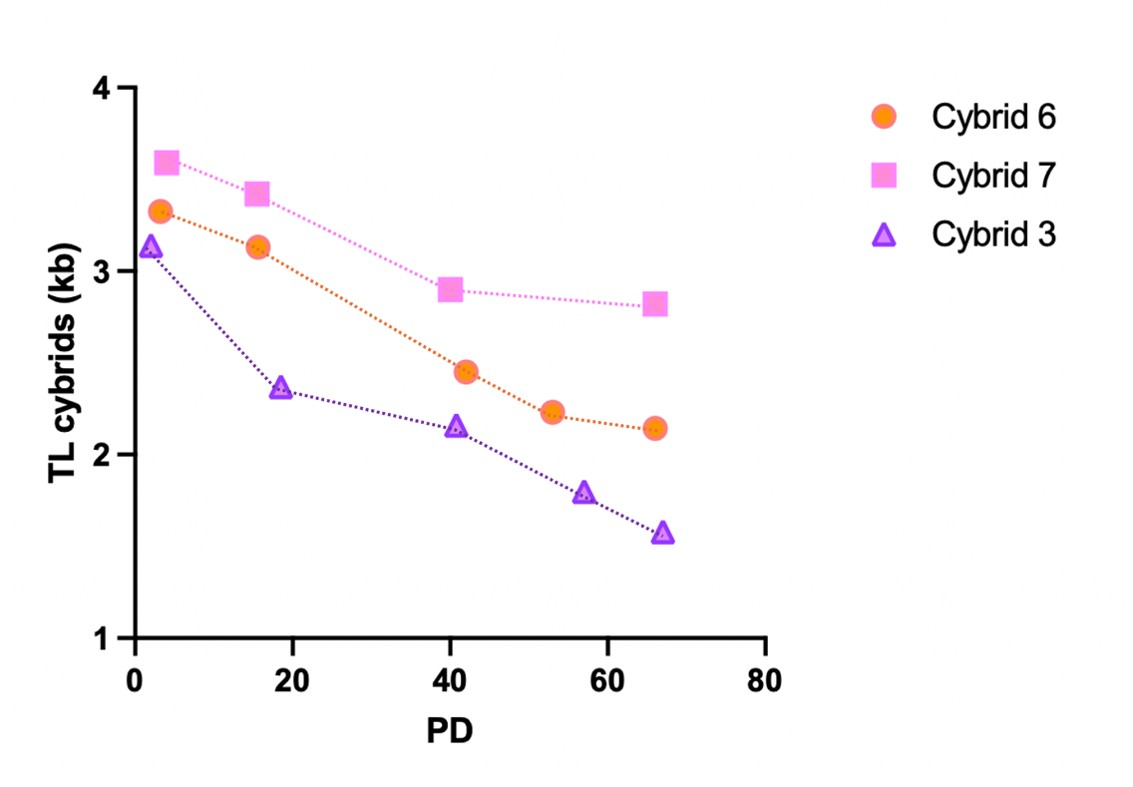
(6) Why did the elevated ROS in cybrid #3 (Figure 4C) not translate to shorter telomeres in the cybrid (Figure 2A)? Perhaps there is a difference between factors that determine TL in the cybrid vs the donor's lymphocytes?
We cannot fully explain the discrepancy, but we indeed suspect in vivo oxidative stress differs significantly from cell cultures (21% O2). Additionally, early cybrid replenishment involves an unknown telomere elongation step that may offset mitochondrial ROS-induced shortening.
To further investigate this question, we measured telomere length across varying population doublings (PDs) and observed the following shortening after 66-67 PDs:
- Cybrid 3: about 1.6 kb reduction
- Cybrid 6: about 1.2 kb reduction
- Cybrid 7: about 0.8 kb reduction
These results suggest that the rate of telomere shortening in culture may be higher in Cybrid 3 cells, possibly due to increased ROS levels.
We propose to include (as Supplementary figure) and discuss these data in the revised manuscript.
Author response image 2.
In Figure 4B, it appears that the statistical comparisons for mitochondrial superoxide are all relative to Cyb3. If so, why are the comparisons not with the parental 143B rho0 cell line? Please clarify.
We excluded Rho0 cells from our superoxide measurements because these cells were grown in a different culture medium. Instead, we focused on comparing mitochondrial ROS across cybrids to accurately correlate these values with the telomere length of the corresponding donors’ lymphocytes. Consequently, Rho0 cell measurements would not have contributed to this correlation analysis.
We propose to discuss this in the revised manuscript.
(7) Given the heterogeneity in TL and mtDNA variants in the human population, the conclusions could be further strengthened by increasing the number of donors and cybrids analyzed. However, there are admittedly practical factors. Overall, these findings are compelling and provide a solid foundation for expanding this analysis in the future. This is more of a comment than a weakness.
We thank the reviewer for this constructive feedback and entirely agree that including more donors would have added depth to our findings. While we acknowledge this limitation, pursuing this further is currently impossible without acquiring new ethical approvals and establishing fresh collaborations with clinicians.
Reviewer #2 (Public review):
Summary:
The authors aim to determine whether mitochondrial genotype influences telomere length. By generating cybrids harboring different mitochondrial backgrounds, the authors seek to establish a mechanistic link between mitochondrial status and telomere biology.
Strengths:
A major strength of the study is the use of cybrid technology, which provides a great approach to investigate the role of mitochondrial DNA independently of the nuclear genome. The authors also employ multiple complementary assays to assess telomere-related phenotypes associated with mitochondrial dysfunction. Together, these experiments generate an interesting dataset that will be of value to researchers interested in the intersection between mitochondrial biology, genome stability, aging, and development. These results also build on previous work supporting roles for ROS/mitochondria in driving telomere shortening.
We thank the reviewer for this very positive evaluation of our work.
Weaknesses:
The data support the conclusion that mitochondrial background is associated with differences in telomere length and telomere-related phenotypes. However, some of the mechanistic interpretations would benefit from additional evidence. In particular, the manuscript discusses mitochondrial influences on telomere shortening, yet telomere length in some experiments is assessed at a single time point. Consequently, the current data do not directly address the rate of telomere attrition. Differences observed between cybrid lines could potentially arise from events occurring during cybrid formation, clonal selection, or subsequent cell expansion. Longitudinal analyses across multiple passages, ideally beginning immediately after cybrid generation and controlling for population doublings, would help establish whether mitochondrial function directly affects telomere shortening dynamics. Some experimental results would also benefit from additional quantification, clarification, and some biological replicates are missing.
We limited our TL measurements to the earliest viable time point after cybrid formation to avoid the confounding effects of cellular adaptation in culture. For instance, the early telomere shortening observed in Cybrid 1 and Cybrid 2 was later alleviated—likely due to the upregulation of the NAD+ salvage pathway genes NAMPT and NAPRT1.
To accurately capture the effects specific to cybrid formation, we isolated 4 independent clones per donor, all of which showed highly consistent TL values, as shown in Figure 2A and S3D.
While we acknowledge the reviewer's point about multi-passage longitudinal analyses, we did, in fact, measure telomere length across varying population doublings (PDs) in Cybrid 3 (high mito ROS levels), 6 and 7 (low mito ROS levels) and observed the following shortening after 66-67 PDs:
- Cybrid 3: about 1.6 kb reduction
- Cybrid 6: about 1.2 kb reduction
- Cybrid 7: about 0.8 kb reduction
These results suggest that the rate of telomere shortening in culture may be higher in Cybrid 3 cells, possibly due to increased ROS levels.
We propose to include a Supplementary figure and discuss these data in the revised manuscript.
We further propose to carefully edit the manuscript so as to clarify the text and, whenever possible, add quantifications. Among others, as suggested by Reviewer #1, we will add the quantification of chromosome end fusions in the parental 143B Rho0, Cybrid 1 and Cybrid 6 cells in our revised manuscript (See Author response image 1).
Overall, this study provides interesting evidence linking mitochondrial background to telomere biology. The cybrid models represent a useful resource for the field, and the work raises important questions regarding mitochondria-telomere communication.
Reviewer #3 (Public review):
Strengths:
Mahieu and colleagues address an interesting and underexplored question: whether non-pathogenic variation in the mitochondrial genome contributes to the inter-individual variability of human telomere length (TL). Using a Belgian Flow-FISH reference cohort (n=491) to identify donors at TL extremes, they generate transmitochondrial cybrids from platelets of seven donors of distinct mtDNA subhaplogroups and characterize the resulting cells with a broad and well-executed toolkit (TRF, TeSLA, ddTRAP, EPR-based mitoROS, Seahorse with permeabilized-cell ETC dissection, LC-MS metabolomics, telomeric PAR-FISH). The most compelling finding is that cybrids derived from donors with low complex I (CI) activity undergo rapid telomere shortening during the glycolysis-to-OXPHOS transition of cybrid formation, and that this is largely prevented by co-treatment with NAC and the NAD⁺ precursor nicotinamide riboside, supporting a model in which CI sustains the NAD⁺ pool required for PARP1-mediated repair of oxidative damage at telomeres. The authors further report an inverse correlation between donor lymphocyte TL and mitoROS in the corresponding cybrids, and provide preliminary evidence that the K1a-defining ATP6 A177T variant (m.G9055>A) may be enriched in long-telomere individuals.
We thank the reviewer for this very positive evaluation of our work.
Weaknesses:
(1) Statistical support and donor sampling for the central in vivo correlation (Figure 4C).
The inverse correlation between donor lymphocyte TL and cybrid mitoROS (R2=0.794, p=0.007) is the principal in vivo claim of the paper, but it is built on seven donors deliberately selected from the extremes of the Flow-FISH distribution. Sampling at the tails of the outcome variable can substantially inflate apparent correlation strength and significance. I would encourage the authors to (i) explicitly state this sampling structure where the correlation is introduced, (ii) report a leave-one-out sensitivity analysis to confirm the relationship is not driven by one or two donors (Cyb3 and Cyb6 appear to anchor the line), and (iii) where feasible, extend the analysis to additional donors with intermediate TL to test whether the relationship holds across the full distribution. Even a modest expansion (e.g., 4 to 5 additional donors at P25 to P75) would substantially strengthen this central claim.
We thank the reviewer for this insightful comment. As suggested, we will explicitly describe the sampling structure upon introducing the correlation analysis. Furthermore, we have conducted the requested leave-one-out analysis:
- removing Cyb3: R2=0.730; p=0.0302
- removing Cyb6: R2=0.782; p=0.0194
- removing Cyb1: R2=0.740; p=0.0278
While we agree that additional donors would enhance the study, further experiments are however currently impossible without new ethical clearances and additional clinical partnerships.
(2) Reconciling the cybrid CI / TL relationship (Fig 3B) with the absence of a CI / TL relationship in donor lymphocytes (Figure 4A).
Figure 3B shows a strong correlation between CI activity and TL in cybrids (R2=0.87), while Figure 4A shows no correlation between donor CI activity (measured in the same cybrids) and donor lymphocyte TL. The authors acknowledge this, but the manuscript subsequently builds toward a CI-centric model of in vivo TL regulation, which seems to outrun the data. The most internally consistent interpretation is that the cybrid CI phenotype reports a sensitized in vitro response to the acute oxidative stress of the metabolic shift, rather than a steady-state determinant of leukocyte TL. I would suggest reframing the abstract, significance statement, and Discussion to make this distinction clearer. The in vitro CI / NAD⁺ / PARP1 axis is a strong finding on its own, while the in vivo role of CI activity (as opposed to ROS more broadly) is not yet established here. Donor #1's profile (very long lymphocyte TL, low CI activity, severe shortening in cybrids, no telomere inheritance in offspring) is informative in this regard and could be discussed more directly as a case that helps delineate where the cybrid model does and does not recapitulate in vivo biology.
We acknowledge that our study does not establish the in vivo role of CI activity in TL regulation. Our abstract specifically highlights an in vitro phenomenon: “Under the specific conditions of cybrid formation, which involve a metabolic shift from glycolysis to oxidative phosphorylation, mtDNA variants associated with reduced CI activity induced rapid telomere shortening, …”.
We are nevertheless happy to revise the text to clearly separate our in vitro results from in vivo biology as requested.
(3) The K1a / ATP6 A177T inheritance claim.
The proposal that K1a (and specifically ATP6 A177T) contributes to maternal inheritance of long telomeres is intriguing but currently rests on three pedigrees (one of which, donor #1, does not support the hypothesis) and a chi-square test that does not reach significance (p=0.153, Figure 4F). The supporting evidence is also limited by the fact that platelet-mediated mitochondrial transfer delivers donor mitochondrial proteins, lipids, and residual mtRNA in addition to mtDNA, making it difficult to attribute the cybrid phenotype of donor #6 specifically to the ATP6 A177T variant. I would recommend either: (a) extending the genotyping screen to additional unrelated donors and, if feasible, confirming the effect of ATP6 A177T through an isogenic approach (e.g., mtDNA base editing in a clean background), or (b) softening the relevant statements to "suggestive trend warranting larger studies," and presenting the K1a observation as hypothesis-generating rather than supportive. The Ashkenazi-centenarian connection raised in the Discussion is an excellent direction for follow-up and could be framed accordingly.
We agree that the evidence for the AT6 A177T inheritance claim remains inconclusive. To clarify, we do not argue that this mitochondrial variant is solely responsible for longer telomeres; indeed, the mtDNA genome of donor #1 suggests otherwise. Furthermore, the phenotypic impact of such mtDNA variants likely depends on nuclear variants in other telomere-related genes (e.g., hTERT or hTR), meaning AT6 A177T may not consistently result in elongated telomeres. Unfortunately, our ethical protocol precludes screening additional unrelated donors. We will revise the text to soften our statements accordingly.
New references:
DeBoy EA et al. Familial clonal hematopoiesis in a long telomere syndrome. 2023. N Engl J Med 388, 2422-2433.
Davidson-Swinton HR et al. Lymphoid malignancy and clonality in the POT1-mediated long telomere syndrome. 2026. Blood 147, 2226-2237.
Maranzana E et al. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. 2013. Antioxid Redox Signal 19, 1469-1480.



