Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorIshier RaoteInstitut Jacques Monod, Paris, France
- Senior EditorFelix CampeloUniversitat Pompeu Fabra, Barcelona, Spain
Reviewer #1 (Public review):
Summary:
The authors present a novel approach to subcellular spatial proteomics by combining laser microdissection with expansion microscopy and LC-MS/MS analysis (SPEx). They implement two different workflows for LMD and LC-MS/MS quantification:
(1)The standard approach, where an area of interest is cut out by LMD, subjected to proteomics analysis, and compared to the rest of the cell without the dissected ROI.
(2) The subtraction approach, where ROIs are removed, and the remaining cellular material is compared to samples containing both the surrounding material and the ROI.
The authors assess the technique by applying it to subcellular targets of various sizes, volumes, and protein compositions such as the nucleus, nucleoli, and Golgi. They demonstrate that SPEx can identify proteins enriched or reduced in ROIs.
Strengths:
The broad, relatively easy, and inexpensive applicability of this approach to potentially many cell types and subcellular areas of interest provides an exciting alternative to subcellular fractionation, native immunoprecipitation, or genetically encoded proximity labeling constructs. Moreover, by visually selecting ROIs for subsequent analysis, subcellular context or organelle morphology can be taken into account, as discussed by the authors in the discussion section.
Weaknesses:
While strongly supporting the sharing of this approach, we have a number of comments and questions that will improve the impact of the manuscript:
(1) General:
a) The manuscript would benefit from restructuring and language revision. In its current form, the writing is sometimes dense and verbose (in particular, the Results section). This makes it difficult to follow the authors' arguments.
b) The authors mention the possibility of selecting organelles based on morphology. This is left for the discussion, but it seems like a missed opportunity - the authors could compare individual organelles in different morphological states, e.g., connected vs. fragmented mitochondria.
(2) Technical:
a) Why do the authors strive and optimize for a 10x expansion factor? Is SPEx compatible with a more standard 4x expansion, as e.g., used in the classic U-ExM approach (https://www.nature.com/articles/s41592-018-0238-1)? This could be added to the discussion.
b) The U-ExM approach shows improved ultrastructural preservation when using 3%FA with 0.1% glutaraldehyde fixation (GA). Is SPEx compatible with the use of low amounts of GA for fixation?
c) Related to the above, was the anchoring efficiency reduced only to achieve a 10x expansion factor or does this additionally affect the proteome coverage?
d) Have the authors considered using alternative anchoring approaches, such as GMA (https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0291506#pone.0291506.s001), which potentially increase the amount of sample retained in the hydrogel, thus allowing for better proteome coverage? This could be added to the discussion.
e) The limitation of the approach to near-2D samples should be mentioned, and alternative approaches for more 3D samples could be discussed.
f) How are peptides that are directly anchored to the hydrogel dealt with during LC-MS/MS analysis? Are they excluded, or can they be identified during the spectral search? The latter would allow us to get a deeper structural understanding of how proteins are actually anchored into hydrogels, which so far has not been assessed.
An alternative approach to address this question would be to investigate if the peptide coverage of proteins detected by SPEx is enriched for peptides representing the folded core of proteins as opposed to the surface-exposed regions, which likely get more anchored into the hydrogel.
g) Same question regarding peptides with NHS labeling. Can they be identified, or do they just compete for ionization and thus negatively affect coverage and dynamic range of the LC-MS/MS approach?
h) How are the primary and secondary antibodies affecting the proteomics analysis identified as contaminants?
i) Have the authors observed differences in proteomics coverage of only antibody vs NHS-labeling? Depending on the questions above, could pure antibody-based labeling increase proteomic coverage?
Reviewer #2 (Public review):
Summary:
This study introduces a method that combines physical expansion of cells, imaging-guided isolation of defined regions, and protein identification to enable compartment-resolved analysis of protein composition at the subcellular scale. The authors aim to address a central limitation in existing approaches, namely the loss of spatial information during sample preparation or the indirect nature of proximity-based labeling methods. Using several cellular compartments as examples, they demonstrate that their approach can recover compartment-enriched protein sets and identify candidate proteins with previously unassigned localization.
Strengths:
A major strength of this work is the conceptual simplicity and accessibility of the approach. By combining established techniques in a modular way, the method avoids the need for genetic manipulation or specialized labeling strategies, making it broadly adaptable across experimental systems. The ability to directly select regions of interest based on imaging represents a clear advantage over indirect enrichment strategies and allows flexible targeting of both membrane-bound and non-membrane-bound compartments.
The experimental design is also a strong aspect of the study. The use of complementary comparison strategies-analyzing isolated compartments alongside matched "subtracted" controls-provides an internal framework for assessing enrichment and depletion, increasing confidence in spatial assignment. The application of the method across multiple organelles of different sizes and properties demonstrates versatility, and the reported specificity for several compartments is encouraging. In particular, the ability to profile small and biochemically challenging structures highlights a potentially important niche for the approach.
Weaknesses:
Despite these strengths, several methodological limitations constrain the interpretation of the results. The most important relates to spatial accuracy in three dimensions. While lateral resolution is improved through physical expansion, the lack of depth resolution introduces uncertainty regarding contributions from structures above and below the selected region. Although the authors argue that this does not substantially affect specificity, the current evidence is largely indirect, and a more rigorous quantification of potential contamination would strengthen this conclusion.
Quantitative interpretation also remains challenging. Because the measurements reflect total protein abundance rather than local concentration, differences in compartment size and protein density can influence enrichment values, particularly for small structures embedded within larger volumes. This issue is evident in the analysis of smaller compartments and complicates direct comparison across conditions. Additional normalization or modeling would help clarify how to interpret these measurements.
Another limitation concerns variability in the expansion process and its downstream consequences. Differences in expansion factor across samples may affect the definition of regions of interest and introduce variability in sampling, yet the impact of this variability is not fully explored. Similarly, the use of a modified chemical treatment to preserve proteins for downstream analysis is central to the workflow but is not extensively validated with respect to preservation of spatial organization.
While the identification of previously unannotated proteins is an appealing aspect of the study, validation is limited to a small number of examples, and broader support from independent datasets or literature context is lacking. In addition, the study primarily focuses on steady-state measurements in a single cell type, and therefore does not yet demonstrate the ability of the method to capture dynamic or condition-dependent changes in protein localization.
Finally, the positioning of the method relative to existing approaches could be more clearly articulated. Although qualitative comparisons are provided, a more systematic and quantitative benchmarking against alternative strategies would help readers better understand the specific advantages and trade-offs.
Reviewer #3 (Public review):
Franziscus et al. describe an elegant approach for spatially specific proteome analysis. To achieve this, they expand fixed cells and subsequently use a laser to micro-dissect a region of interest, which is then analyzed by mass spectrometry.
They demonstrate the effectiveness of their approach by analyzing the nucleus, nucleolus, and the Golgi, and benchmark their hits against previous datasets for these organelles.
The manuscript is very well written and nicely guides the reader through the applied methods. The presented data is convincing, and I do not see the need for additional experimental verification of the protocol. The only minor concern is the novelty of the method and the presentation. A combination of expansion, laser microdissection, and proteomics has been applied in the past (PMID: 36450705, PMID: 39477916). In the manuscript, one of these studies is cited, though it does not become clear that this approach is already described. However, Franziscus et al. describe the approach better and make it more accessible to the reader, especially since the other studies described this methodology in combination with tissue expansion and not in combination with single cell expansion as it is done here. I would ask the authors to be clearer in the introduction about what others have already done and what their contribution is here. In general, I am convinced that the community will benefit from the presented protocol to analyze organelle proteomics in detail.
Author Response:
Reviewer #1 (Public review):
Summary:
The authors present a novel approach to subcellular spatial proteomics by combining laser microdissection with expansion microscopy and LC-MS/MS analysis (SPEx). They implement two different workflows for LMD and LC-MS/MS quantification:
(1)The standard approach, where an area of interest is cut out by LMD, subjected to proteomics analysis, and compared to the rest of the cell without the dissected ROI.
(2) The subtraction approach, where ROIs are removed, and the remaining cellular material is compared to samples containing both the surrounding material and the ROI.
The authors assess the technique by applying it to subcellular targets of various sizes, volumes, and protein compositions such as the nucleus, nucleoli, and Golgi. They demonstrate that SPEx can identify proteins enriched or reduced in ROIs.
Strengths:
The broad, relatively easy, and inexpensive applicability of this approach to potentially many cell types and subcellular areas of interest provides an exciting alternative to subcellular fractionation, native immunoprecipitation, or genetically encoded proximity labeling constructs. Moreover, by visually selecting ROIs for subsequent analysis, subcellular context or organelle morphology can be taken into account, as discussed by the authors in the discussion section.
Weaknesses:
While strongly supporting the sharing of this approach, we have a number of comments and questions that will improve the impact of the manuscript:
We thank the reviewer for the careful evaluation of our manuscript and the generally positive assessment. We plan on improving our manuscript based on the reviewers’ comments.
(1) General:
a) The manuscript would benefit from restructuring and language revision. In its current form, the writing is sometimes dense and verbose (in particular, the Results section). This makes it difficult to follow the authors' arguments.
We will improve readability and clarity of the results section in the revised manuscript.
b) The authors mention the possibility of selecting organelles based on morphology. This is left for the discussion, but it seems like a missed opportunity - the authors could compare individual organelles in different morphological states, e.g., connected vs. fragmented mitochondria.
The authors agree with the reviewers’ assessment that investigating proteome of organelles based on morphology or cellular state is an exciting application of SPEx. While we plan experiments along this line in the future, we think that these experiments are beyond the scope of this manuscript, which is meant to describe the method and its general usefulness.
(2) Technical:
a) Why do the authors strive and optimize for a 10x expansion factor? Is SPEx compatible with a more standard 4x expansion, as e.g., used in the classic U-ExM approach (https://www.nature.com/articles/s41592-018-0238-1)? This could be added to the discussion.
We aimed for 10x expansion solely because our ultimate goal is to cut out very small structures. Isolating structures as small as nucleoli would not be as reliable with a lower expansion factor (i.e. 4x) expansion. We did not assess the compatibility with U-ExM. We would assume that SPEx would also work with U-ExM as expansion method; omitting protease treatment, however. Still, we performed pilots with just 4x expansion (using TREx) in the early stages of optimization. We were able to isolate single cells and obtain similar protein coverage as with 10x expansion. We will further clarify our motivation to use 10x expansion in the discussion.
We would also like to point out whether to U-ExM the standard method or not is rather subjective. Even though TREx was published three years later, it is also very widely used. The original expansion microscopy method was published three years prior to U-ExM.
b) The U-ExM approach shows improved ultrastructural preservation when using 3%FA with 0.1% glutaraldehyde fixation (GA). Is SPEx compatible with the use of low amounts of GA for fixation?
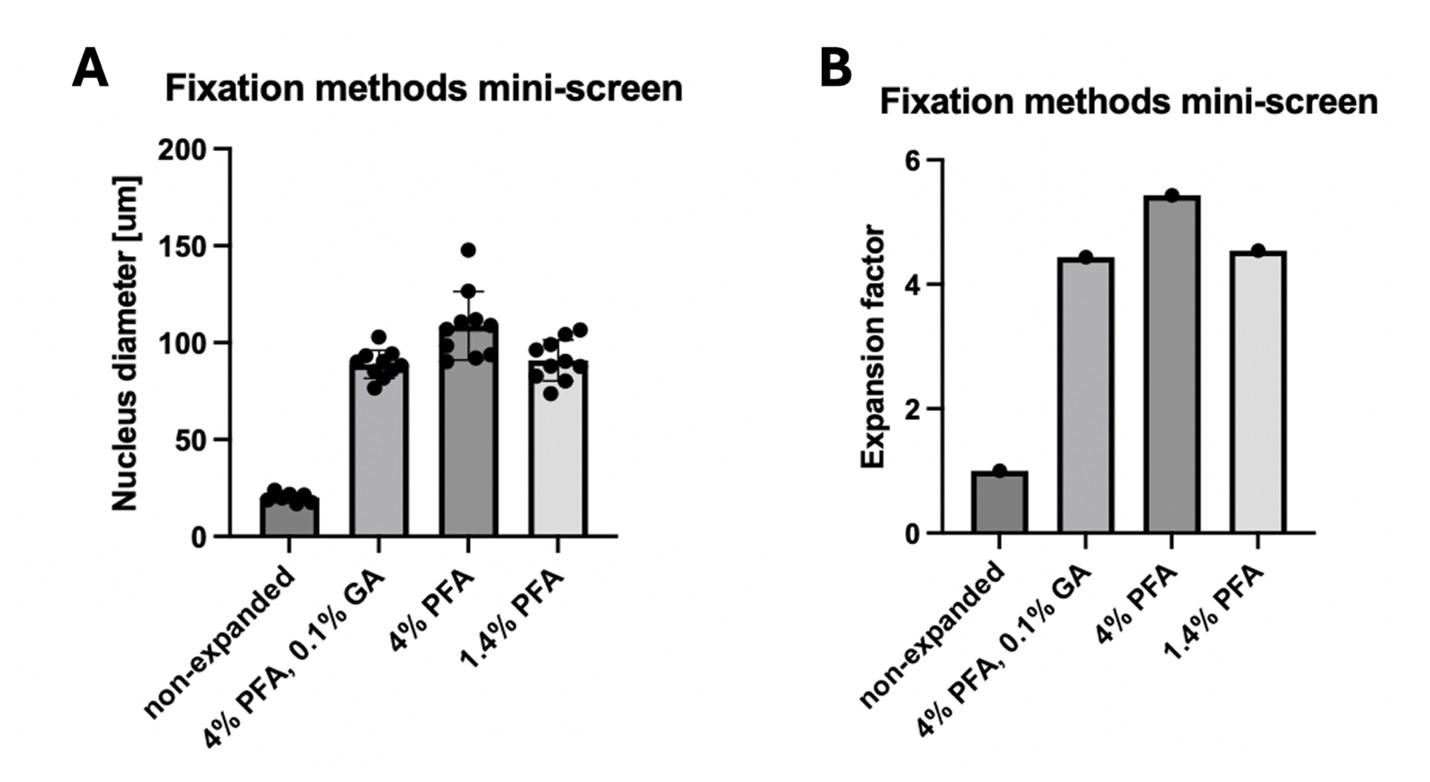
We tried different fixation methods in the early stages of this study (where expansion was not yet close to 10x). We saw a mild negative effect of GA on the expansion factor, so we avoided it in the later experiments since it also did not seem necessary to preserve the structure of our organelles of interest. However, the use of GA would generally be compatible with SPEx, potentially at the cost of a mild negative effect on expansion factor (see Author response image 1) and proteome coverage. We can add this information to the discussion.
Author response image 1.
Fixation methods mini-screen. Cells were fixed with the indicated reagents for 10 minutes at 37°C. After TREx expansion, the diameter of the nucleus was measured (A) and the resulting expansion factor compared to the non-expanded control was determined (B).
Related to the above, was the anchoring efficiency reduced only to achieve a 10x expansion factor or does this additionally affect the proteome coverage?
We solely lowered the anchoring in order to allow for higher expansion factors. In earlier pilots we performed proteomic analysis on samples that were just expanded 4x using standard TREx expansion (also using the original anchoring strategy from the TREx publication, consisting of 0.2 mg/ml AcX for overnight at RT). We presented the results of this pilot in Fig S1A. We still detected over 2,000 proteins from 10 cells, a coverage, which is highly similar to what we found in the final experiments (Figure 2F), in which the anchoring was lower yielding 10x expansion. Based on these data, we hypothesize that anchoring (and expansion factor!) has a negligible impact on protein coverage. We will clarify this in the manuscript.
d) Have the authors considered using alternative anchoring approaches, such as GMA (https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0291506#pone.0291506.s001), which potentially increase the amount of sample retained in the hydrogel, thus allowing for better proteome coverage? This could be added to the discussion.
We did not use alternative anchoring approaches. We modified the TREx protocol to fit our purposes and since this was sufficient, we did not explore alternatives. However, using anchoring approaches, in which higher amounts of sample could be retained in the gel might be beneficial for the proteomics coverage. We will keep this suggestion in mind for future experiments. Thank you for the suggestion!
e) The limitation of the approach to near-2D samples should be mentioned, and alternative approaches for more 3D samples could be discussed.
The authors agree that SPEx is limited to near-2D samples at this point. We suggest that SPEx is applicable for 3D samples (e.g. in tissues) by performing cryosectioning. TREx has been shown to be compatible with sectioned tissue (Damstra et al., 2022). We will elaborate this in the discussion.
f) How are peptides that are directly anchored to the hydrogel dealt with during LC-MS/MS analysis? Are they excluded, or can they be identified during the spectral search? The latter would allow us to get a deeper structural understanding of how proteins are actually anchored into hydrogels, which so far has not been assessed.
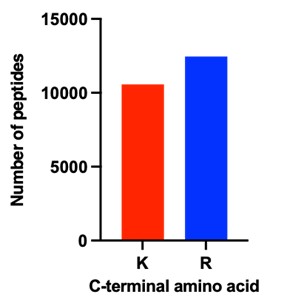
The reviewer raises an interesting point. In general, peptides carrying the anchoring modification are analysed by LC-MS, but we did not include these specific modifications in the database search. Overall, we assumed that the labeling would be low and stochastic and hence should, if at all, only minimally affect the detection of peptides. Nevertheless, in response to the reviewers’ comment, we searched the MS data again for the crosslinking reagent linked to lysine residues. However, we could not get any confident hit for any peptide containing this modification. Since we cannot exclude that the modification precludes the identification of the corresponding peptides, we compared the number peptides generated by trypsin cleavage after arginine and lysine. As the human genome contains similar proportions of both amino acids, one would expect similar numbers of both peptide types being identified. Any modifications of lysine by the anchoring reagent used, would prevent tryptic cleavage and thus reduce the number of lysine peptides. As shown in Author response image 2, the number of lysine terminating is only slightly lower compared to arginine terminating peptides. Notably, the proteomics results of a different fixed human tissue sample directly extracted by laser capture micro dissection without expansion showed a very similar lysine to arginine peptide ratio. This indicates that the large majority of lysine residues is not modified and affected by the hydrogel anchoring.
Author response image 2.
Number of peptides identified either terminating with lysine (K) or arginine (R) across all samples shown in Figure 5F.
An alternative approach to address this question would be to investigate if the peptide coverage of proteins detected by SPEx is enriched for peptides representing the folded core of proteins as opposed to the surface-exposed regions, which likely get more anchored into the hydrogel.
Because of the negligible amounts of modified peptides, we did not investigate this potential bias of surface-exposed versus folded-core peptides.
g) Same question regarding peptides with NHS labeling. Can they be identified, or do they just compete for ionization and thus negatively affect coverage and dynamic range of the LC-MS/MS approach?
The reviewer raises a similar point as above for another lysine labeling used during the SPEx protocol. Again, we specifically looked for this modification by re-searching the raw MS data, but still could not identify any peptides, carrying this modification on a lysine residue. Even though we cannot exclude that this rather large modification prevents detection, considering the high number of lysine terminating peptides in our dataset (see Figure 2), we would expect that also this labeling step is stochastic and affects only a minor proportion of the proteins.
h) How are the primary and secondary antibodies affecting the proteomics analysis identified as contaminants?
We thank the reviewer for this comment. Since antibodies bind to proteins in a non-covalent manner, they will be released during the denaturing steps of the protocol. Of course, the antibodies will stay in the sample, be digested and analyzed and could, if very abundant, affect the analysis of the proteins from the samples. To check this possibility, we re-searched the MS data including the sequences of the antibodies used. To our surprise, we could not detect any peptides of these antibodies. This suggests that the concentrations of the antibodies used are much lower than those of the sample proteins and thus should not have any impact on the proteomics results. We interpret this result also as a benefit of our method compared to organellar-IP.
i) Have the authors observed differences in proteomics coverage of only antibody vs NHS-labeling? Depending on the questions above, could pure antibody-based labeling increase proteomic coverage?
We did not perform this comparative analysis, since we always used NHS dyes. In the experiments presented in this manuscript, NHS dyes allowed easy visualization of the whole cell without the use of antibodies. This NHS staining was essential for this particular setup for sample acquisition. We cut out entire cells, cells lacking the nucleus and cells lacking the Golgi apparatus, which served as critical controls. However, other ways of detecting cell boundaries could be used to avoid NHS staining. As shown above, both, the anchor and NHS labeling are likewise sparse and stochastic. Moreover, we could not detect any impact of the antibody labeling to our results. Thus, we assume that both labeling procedures could be used.
Reviewer #2 (Public review):
Summary:
This study introduces a method that combines physical expansion of cells, imaging-guided isolation of defined regions, and protein identification to enable compartment-resolved analysis of protein composition at the subcellular scale. The authors aim to address a central limitation in existing approaches, namely the loss of spatial information during sample preparation or the indirect nature of proximity-based labeling methods. Using several cellular compartments as examples, they demonstrate that their approach can recover compartment-enriched protein sets and identify candidate proteins with previously unassigned localization.
Strengths:
A major strength of this work is the conceptual simplicity and accessibility of the approach. By combining established techniques in a modular way, the method avoids the need for genetic manipulation or specialized labeling strategies, making it broadly adaptable across experimental systems. The ability to directly select regions of interest based on imaging represents a clear advantage over indirect enrichment strategies and allows flexible targeting of both membrane-bound and non-membrane-bound compartments.
The experimental design is also a strong aspect of the study. The use of complementary comparison strategies-analyzing isolated compartments alongside matched "subtracted" controls-provides an internal framework for assessing enrichment and depletion, increasing confidence in spatial assignment. The application of the method across multiple organelles of different sizes and properties demonstrates versatility, and the reported specificity for several compartments is encouraging. In particular, the ability to profile small and biochemically challenging structures highlights a potentially important niche for the approach.
Weaknesses:
Despite these strengths, several methodological limitations constrain the interpretation of the results. The most important relates to spatial accuracy in three dimensions. While lateral resolution is improved through physical expansion, the lack of depth resolution introduces uncertainty regarding contributions from structures above and below the selected region. Although the authors argue that this does not substantially affect specificity, the current evidence is largely indirect, and a more rigorous quantification of potential contamination would strengthen this conclusion.
Quantitative interpretation also remains challenging. Because the measurements reflect total protein abundance rather than local concentration, differences in compartment size and protein density can influence enrichment values, particularly for small structures embedded within larger volumes. This issue is evident in the analysis of smaller compartments and complicates direct comparison across conditions. Additional normalization or modeling would help clarify how to interpret these measurements.
Another limitation concerns variability in the expansion process and its downstream consequences. Differences in expansion factor across samples may affect the definition of regions of interest and introduce variability in sampling, yet the impact of this variability is not fully explored. Similarly, the use of a modified chemical treatment to preserve proteins for downstream analysis is central to the workflow but is not extensively validated with respect to preservation of spatial organization.
While the identification of previously unannotated proteins is an appealing aspect of the study, validation is limited to a small number of examples, and broader support from independent datasets or literature context is lacking. In addition, the study primarily focuses on steady-state measurements in a single cell type, and therefore does not yet demonstrate the ability of the method to capture dynamic or condition-dependent changes in protein localization.
Finally, the positioning of the method relative to existing approaches could be more clearly articulated. Although qualitative comparisons are provided, a more systematic and quantitative benchmarking against alternative strategies would help readers better understand the specific advantages and trade-offs.
We thank the reviewer for the careful evaluation of the manuscript and for the constructive feedback. We think the reviewer raises valid points and will address them in the revised manuscript.
Reviewer #3 (Public review):
Franziscus et al. describe an elegant approach for spatially specific proteome analysis. To achieve this, they expand fixed cells and subsequently use a laser to micro-dissect a region of interest, which is then analyzed by mass spectrometry.
They demonstrate the effectiveness of their approach by analyzing the nucleus, nucleolus, and the Golgi, and benchmark their hits against previous datasets for these organelles.
The manuscript is very well written and nicely guides the reader through the applied methods. The presented data is convincing, and I do not see the need for additional experimental verification of the protocol. The only minor concern is the novelty of the method and the presentation. A combination of expansion, laser microdissection, and proteomics has been applied in the past (PMID: 36450705, PMID: 39477916). In the manuscript, one of these studies is cited, though it does not become clear that this approach is already described. However, Franziscus et al. describe the approach better and make it more accessible to the reader, especially since the other studies described this methodology in combination with tissue expansion and not in combination with single cell expansion as it is done here. I would ask the authors to be clearer in the introduction about what others have already done and what their contribution is here. In general, I am convinced that the community will benefit from the presented protocol to analyze organelle proteomics in detail.
We thank the reviewer for the careful evaluation of our manuscript and overwhelmingly positive assessment. We apologize for the omission of the mentioned citations, and will adjust the introduction to make it clearer what has already been done and what the advance our method provides.
References
Damstra HG, Mohar B, Eddison M, Akhmanova A, Kapitein LC, Tillberg PW. 2022. Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). eLife 11:e73775. DOI: https://doi.org/10.7554/eLife.73775
Gambarotto D, Hamel V, Guichard P. 2021. Ultrastructure expansion microscopy (U-ExM). Methods in Cell Biology 161:57–81. DOI: https://doi.org/10.1016/bs.mcb.2020.05.006, PMID: 33478697
Liffner B, Silva TLA e., Vega-Rodriguez J, Absalon S. 2024. Mosquito Tissue Ultrastructure-Expansion Microscopy (MoTissU-ExM) enables ultrastructural and anatomical analysis of malaria parasites and their mosquito. BMC Methods 1:13. DOI: https://doi.org/10.1186/s44330-024-00013-4



