Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorYamini DalalNational Cancer Institute, Bethesda, United States of America
- Senior EditorDetlef WeigelMax Planck Institute for Biology Tübingen, Tübingen, Germany
Reviewer #1 (Public Review):
This manuscript by Tan et al is using cryo-electron tomography to investigate the structure of yeast nucleosomes both ex vivo (nuclear lysates) and in situ (lamellae and cryosections). The sheer number of experiments and results are astounding and comparable with an entire PhD thesis. However, as is always the case, it is hard to prove that something is not there. In this case, canonical nucleosomes. In their path to find the nucleosomes, the authors also stumble over new insights into nucleosome arrangement that indicates that the positions of the histones is more flexible than previously believed.
Major strengths and weaknesses:
Personally, I am not ready to agree with their conclusion that heterogenous non-canonical nucleosomes predominate in yeast cells, but this reviewer is not an expert in the field of nucleosomes and can't judge how well these results fit into previous results in the field. As a technological expert though, I think the authors have done everything possible to test that hypothesis with today's available methods. One can debate whether it is necessary to have 35 supplementary figures, but after working through them all, I see that the nature of the argument needs all that support, precisely because it is so hard to show what is not there. The massive amount of work that has gone into this manuscript and the state-of-the art nature of the technology should be warmly commended. I also think the authors have done a really great job with including all their results to the benefit of the scientific community. Yet, I am left with some questions and comments:
Could the nucleosomes change into other shapes that were predetermined in situ? Could the authors expand on if there was a structure or two that was more common than the others of the classes they found? Or would this not have been found because of the template matching and later reference particle used?
Could it simply be that the yeast nucleoplasm is differently structured than that of HeLa cells and it was harder to find nucleosomes by template matching in these cells? The authors argue against crowding in the discussion, but maybe it is just a nucleoplasm texture that side-tracks the programs?
The title of the paper is not well reflected in the main figures. The title of Figure 2 says "Canonical nucleosomes are rare in wild-type cells", but that is not shown/quantified in that figure. Rare is comparison to what? I suggest adding a comparative view from the HeLa cells, like the text does in lines 195-199. A measure of nucleosomes detected per volume nucleoplasm would also facilitate a comparison.
If the cell contains mostly non-canonical nucleosomes, are they really non-canonical? Maybe a change of language is required once this is somewhat sure (say, after line 303).
The authors could explain more why they sometimes use conventional the 2D followed by 3D classification approach and sometimes "direct 3-D classification". Why, for example, do they do 2D followed by 3D in Figure S5A? This Figure could be considered a regular figure since it shows the main message of the paper.
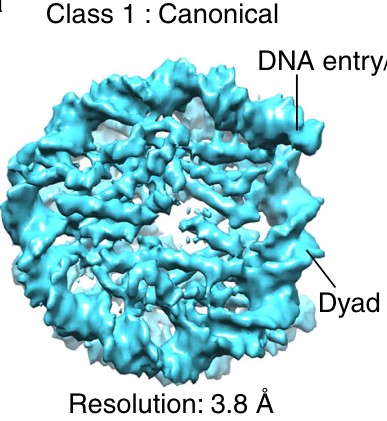
Figure 1: Why is there a gap in the middle of the nucleosome in panel B? The authors write that this is a higher resolution structure (18Å), but in the even higher resolution crystallography structure (3Å resolution), there is no gap in the middle.
Reviewer #2 (Public Review):
Nucleosome structures inside cells remain unclear. Tan et al. tackled this problem using cryo-ET and 3-D classification analysis of yeast cells. The authors found that the fraction of canonical nucleosomes in the cell could be less than 10% of total nucleosomes. The finding is consistent with the unstable property of yeast nucleosomes and the high proportion of the actively transcribed yeast genome. The authors made an important point in understanding chromatin structure in situ. Overall, the paper is well-written and informative to the chromatin/chromosome field.
Reviewer #3 (Public Review):
Several labs in the 1970s published fundamental work revealing that almost all eukaryotes organize their DNA into repeating units called nucleosomes, which form the chromatin fiber. Decades of elegant biochemical and structural work indicated a primarily octameric organization of the nucleosome with 2 copies of each histone H2A, H2B, H3 and H4, wrapping 147bp of DNA in a left handed toroid, to which linker histone would bind.
This was true for most species studied (except, yeast lack linker histone) and was recapitulated in stunning detail by in vitro reconstitutions by salt dialysis or chaperone-mediated assembly of nucleosomes. Thus, these landmark studies set the stage for an exploding number of papers on the topic of chromatin in the past 45 years.
An emerging counterpoint to the prevailing idea of static particles is that nucleosomes are much more dynamic and can undergo spontaneous transformation. Such dynamics could arise from intrinsic instability due to DNA structural deformation, specific histone variants or their mutations, post-translational histone modifications which weaken the main contacts, protein partners, and predominantly, from active processes like ATP-dependent chromatin remodeling, transcription, repair and replication.
This paper is important because it tests this idea whole-scale, applying novel cryo-EM tomography tools to examine the state of chromatin in yeast lysates or cryo-sections. The experimental work is meticulously performed, with vast amount of data collected. The main findings are interpreted by the authors to suggest that majority of yeast nucleosomes lack a stable octameric conformation. The findings are not surprising in that alternative conformations of nucleosomes might exist in vivo, but rather in the sheer scale of such particles reported, relative to the traditional form expected from decades of biochemical, biophysical and structural data. Thus, it is likely that this work will be perceived as controversial. Nonetheless, we believe these kinds of tools represent an important advance for in situ analysis of chromatin. We also think the field should have the opportunity to carefully evaluate the data and assess whether the claims are supported, or consider what additional experiments could be done to further test the conceptual claims made. It is our hope that such work will spark thought-provoking debate in a collegial fashion, and lead to the development of exciting new tools which can interrogate native chromatin shape in vivo. Most importantly, it will be critical to assess biological implications associated with more dynamic - or static forms- of nucleosomes, the associated chromatin fiber, and its three-dimensional organization, for nuclear or mitotic function.
Author Response
The following is the authors’ response to the original reviews.
eLife assessment
This important paper exploits new cryo-EM tomography tools to examine the state of chromatin in situ. The experimental work is meticulously performed and convincing, with a vast amount of data collected. The main findings are interpreted by the authors to suggest that the majority of yeast nucleosomes lack a stable octameric conformation. Despite the possibly controversial nature of this report, it is our hope that such work will spark thought-provoking debate, and further the development of exciting new tools that can interrogate native chromatin shape and associated function in vivo.
We thank the Editors and Reviewers for their thoughtful and helpful comments. We also appreciate the extraordinary amount of effort needed to assess both the lengthy manuscript and the previous reviews. Below, we provide our point-by-point response in bold blue font. Nearly all comments have been addressed in the revised manuscript. For a subset of comments that would require us to speculate, we have taken a conservative approach because we either lack key information or technical expertise: Instead of adding the speculative replies to the main text, we think it is better to leave them in the rebuttal for posterity. Readers will thereby have access to our speculation and know that we did not feel confident enough to include these thoughts in the Version of Record.
Reviewer #1 (Public Review):
This manuscript by Tan et al is using cryo-electron tomography to investigate the structure of yeast nucleosomes both ex vivo (nuclear lysates) and in situ (lamellae and cryosections). The sheer number of experiments and results are astounding and comparable with an entire PhD thesis. However, as is always the case, it is hard to prove that something is not there. In this case, canonical nucleosomes. In their path to find the nucleosomes, the authors also stumble over new insights into nucleosome arrangement that indicates that the positions of the histones is more flexible than previously believed.
Please note that canonical nucleosomes are there in wild-type cells in situ, albeit rarer than what’s expected based on our HeLa cell analysis and especially the total number of yeast nucleosomes (canonical plus non-canonical). The negative result (absence of any canonical nucleosome classes in situ) was found in the histone-GFP mutants.
Major strengths and weaknesses:
Personally, I am not ready to agree with their conclusion that heterogenous non-canonical nucleosomes predominate in yeast cells, but this reviewer is not an expert in the field of nucleosomes and can't judge how well these results fit into previous results in the field. As a technological expert though, I think the authors have done everything possible to test that hypothesis with today's available methods. One can debate whether it is necessary to have 35 supplementary figures, but after working through them all, I see that the nature of the argument needs all that support, precisely because it is so hard to show what is not there. The massive amount of work that has gone into this manuscript and the state-of-the art nature of the technology should be warmly commended. I also think the authors have done a really great job with including all their results to the benefit of the scientific community. Yet, I am left with some questions and comments:
Could the nucleosomes change into other shapes that were predetermined in situ? Could the authors expand on if there was a structure or two that was more common than the others of the classes they found? Or would this not have been found because of the template matching and later reference particle used?
Our best guess (speculation) is that one of the class averages that is smaller than the canonical nucleosome contains one or more non-canonical nucleosome classes. However, we do not feel confident enough to single out any of these classes precisely because we do not yet know if they arise from one non-canonical nucleosome structure or from multiple – and therefore mis-classified – non-canonical nucleosome structures (potentially with other non-nucleosome complexes mixed in). We feel it is better to leave this discussion out of the manuscript, or risk sending the community on wild goose chases.
Our template-matching workflow uses a low-enough cross-correlation threshold that any nucleosome-sized particle (plus minus a few nanometers) would be picked, which is why the number of hits is so large. So unless the noncanonical nucleosomes quadrupled in size or lost most of their histones, they should be grouped with one or more of the other 99 class averages (WT cells) or any of the 100 class averages (cells with GFP-tagged histones). As to whether the later reference particle could have prevented us from detecting one of the non-canonical nucleosome structures, we are unable to tell because we’d really have to know what an in situ non-canonical nucleosome looks like first.
Could it simply be that the yeast nucleoplasm is differently structured than that of HeLa cells and it was harder to find nucleosomes by template matching in these cells? The authors argue against crowding in the discussion, but maybe it is just a nucleoplasm texture that side-tracks the programs?
Presumably, the nucleoplasmic “side-tracking” texture would come from some molecules in the yeast nucleus. These molecules would be too small to visualize as discrete particles in the tomographic slices, but they would contribute textures that can be “seen” by the programs – in particular RELION, which does the discrimination between structural states. We are not sure what types of density textures would side-track RELION’s classification routines.
The title of the paper is not well reflected in the main figures. The title of Figure 2 says "Canonical nucleosomes are rare in wild-type cells", but that is not shown/quantified in that figure. Rare is comparison to what? I suggest adding a comparative view from the HeLa cells, like the text does in lines 195-199. A measure of nucleosomes detected per volume nucleoplasm would also facilitate a comparison.
Figure 2’s title is indeed unclear and does not align with the paper’s title and key conclusion. The rarity here is relative to the expected number of nucleosomes (canonical plus non-canonical). We have changed the title to:
“Canonical nucleosomes are a minority of the expected total in wild-type cells”.
We would prefer to leave the reference to HeLa cells to the main text instead of as a figure panel because the comparison is not straightforward for a graphical presentation. Instead, we now report the total number of nucleosomes estimated for this particular yeast tomogram (~7,600) versus the number of canonical nucleosomes classified (297; 594 if we assume we missed half of them). This information is in the revised figure legend:
“In this tomogram, we estimate there are ~7,600 nucleosomes (see Methods on how the calculation is done), of which 297 are canonical structures. Accounting for the missing disc views, we estimate there are ~594 canonical nucleosomes in this cryolamella (< 8% the expected number of nucleosomes).”
If the cell contains mostly non-canonical nucleosomes, are they really non-canonical? Maybe a change of language is required once this is somewhat sure (say, after line 303).
This is an interesting semantic and philosophical point. From the yeast cell’s “perspective”, the canonical nucleosome structure would be the form that is in the majority. That being said, we do not know if there is one structure that is the majority. From the chromatin field’s point of view, the canonical nucleosome is the form that is most commonly seen in all the historical – and most contemporary – literature, namely something that resembles the crystal structure of Luger et al, 1997. Given these two lines of thinking, we added the following clarification as lines 312 – 316:
“At present, we do not know what the non-canonical nucleosome structures are, meaning that we cannot even determine if one non-canonical structure is the majority. Until we know the non-canonical nucleosomes’ structures, we will use the term non-canonical to describe all the nucleosomes that do not have the canonical (crystal) structure.”
The authors could explain more why they sometimes use conventional the 2D followed by 3D classification approach and sometimes "direct 3-D classification". Why, for example, do they do 2D followed by 3D in Figure S5A? This Figure could be considered a regular figure since it shows the main message of the paper.
Since the classification of subtomograms in situ is still a work in progress, we felt it would be better to show one instance of 2-D classification for lysates and one for lamellae. While it is true that we could have presented direct 3-D classification for the entire paper, we anticipate that readers will be interested to see what the in situ 2-D class averages look like.
The main message is that there are canonical nucleosomes in situ (at least in wild-type cells), but they are a minority. Therefore, the conventional classification for Figure S5A should not be a main figure because it does not show any canonical nucleosome class averages in situ.
Figure 1: Why is there a gap in the middle of the nucleosome in panel B? The authors write that this is a higher resolution structure (18Å), but in the even higher resolution crystallography structure (3Å resolution), there is no gap in the middle.
There is a lower concentration of amino acids at the middle in the disc view; unfortunately, the space-filling model in Figure 1A hides this feature. The gap exists in experimental cryo-EM density maps. See Author response image 1 for an example (pubmed.ncbi.nlm.nih.gov/29626188). The size of the gap depends on the contour level and probably the contrast mechanism, as the gap is less visible in the VPP subtomogram averages. To clarify this confusing phenomenon, we added the following lines to the figure legend:
“The gap in the disc view of the nuclear-lysate-based average is due to the lower concentration of amino acids there, which is not visible in panel A due to space-filling rendering. This gap’s visibility may also depend on the contrast mechanism because it is not visible in the VPP averages.”
Author response image 1.
Reviewer #2 (Public Review):
Nucleosome structures inside cells remain unclear. Tan et al. tackled this problem using cryo-ET and 3-D classification analysis of yeast cells. The authors found that the fraction of canonical nucleosomes in the cell could be less than 10% of total nucleosomes. The finding is consistent with the unstable property of yeast nucleosomes and the high proportion of the actively transcribed yeast genome. The authors made an important point in understanding chromatin structure in situ. Overall, the paper is well-written and informative to the chromatin/chromosome field.
We thank Reviewer 2 for their positive assessment.
Reviewer #3 (Public Review):
Several labs in the 1970s published fundamental work revealing that almost all eukaryotes organize their DNA into repeating units called nucleosomes, which form the chromatin fiber. Decades of elegant biochemical and structural work indicated a primarily octameric organization of the nucleosome with 2 copies of each histone H2A, H2B, H3 and H4, wrapping 147bp of DNA in a left handed toroid, to which linker histone would bind.
This was true for most species studied (except, yeast lack linker histone) and was recapitulated in stunning detail by in vitro reconstitutions by salt dialysis or chaperone-mediated assembly of nucleosomes. Thus, these landmark studies set the stage for an exploding number of papers on the topic of chromatin in the past 45 years.
An emerging counterpoint to the prevailing idea of static particles is that nucleosomes are much more dynamic and can undergo spontaneous transformation. Such dynamics could arise from intrinsic instability due to DNA structural deformation, specific histone variants or their mutations, post-translational histone modifications which weaken the main contacts, protein partners, and predominantly, from active processes like ATP-dependent chromatin remodeling, transcription, repair and replication.
This paper is important because it tests this idea whole-scale, applying novel cryo-EM tomography tools to examine the state of chromatin in yeast lysates or cryo-sections. The experimental work is meticulously performed, with vast amount of data collected. The main findings are interpreted by the authors to suggest that majority of yeast nucleosomes lack a stable octameric conformation. The findings are not surprising in that alternative conformations of nucleosomes might exist in vivo, but rather in the sheer scale of such particles reported, relative to the traditional form expected from decades of biochemical, biophysical and structural data. Thus, it is likely that this work will be perceived as controversial. Nonetheless, we believe these kinds of tools represent an important advance for in situ analysis of chromatin. We also think the field should have the opportunity to carefully evaluate the data and assess whether the claims are supported, or consider what additional experiments could be done to further test the conceptual claims made. It is our hope that such work will spark thought-provoking debate in a collegial fashion, and lead to the development of exciting new tools which can interrogate native chromatin shape in vivo. Most importantly, it will be critical to assess biological implications associated with more dynamic - or static forms- of nucleosomes, the associated chromatin fiber, and its three-dimensional organization, for nuclear or mitotic function.
Thank you for putting our work in the context of the field’s trajectory. We hope our EMPIAR entry, which includes all the raw data used in this paper, will be useful for the community. As more labs (hopefully) upload their raw data and as image-processing continues to advance, the field will be able to revisit the question of non-canonical nucleosomes in budding yeast and other organisms.
Reviewer #1 (Recommendations For The Authors):
The manuscript sometimes reads like a part of a series rather than a stand-alone paper. Be sure to spell out what needs to be known from previous work to read this article. The introduction is very EM-technique focused but could do with more nucleosome information.
We have added a new paragraph that discusses the sources of structural variability to better prepare readers, as lines 50 – 59:
“In the context of chromatin, nucleosomes are not discrete particles because sequential nucleosomes are connected by short stretches of linker DNA. Variation in linker DNA structure is a source of chromatin conformational heterogeneity (Collepardo-Guevara and Schlick, 2014). Recent cryo-EM studies show that nucleosomes can deviate from the canonical form in vitro, primarily in the structure of DNA near the entry/exit site (Bilokapic et al., 2018; Fukushima et al., 2022; Sato et al., 2021; Zhou et al., 2021). In addition to DNA structural variability, nucleosomes in vitro have small changes in histone conformations (Bilokapic et al., 2018). Larger-scale variations of DNA and histone structure are not compatible with high-resolution analysis and may have been missed in single-particle cryo-EM studies.”
Line 165-6 "did not reveal a nucleosome class average in..". Add "canonical", since it otherwise suggests there were no nucleosomes.
Thank you for catching this error. Corrected.
Lines 177-182: Why are the disc views missed by the classification analysis? They should be there in the sample, as you say.
We suspect that RELION 3 is misclassifying the disc-view canonical nucleosomes into the other classes. The RELION developers suspect that view-dependent misclassification arises from RELION 3’s 3-D CTF model. RELION 4 is reported to be less biased by the particles’ views. We have started testing RELION 4 but do not have anything concrete to report yet.
Line 222: a GFP tag.
Fixed.
Line 382: "Note that the percentage .." I can't follow this sentence. Why would you need to know how many chromosome's worth of nucleosomes you are looking at to say the percentage of non-canonical nucleosomes?
Thank you for noticing this confusing wording. The sentence has been both simplified and clarified as follows in lines 396 – 398:
“Note that the percentage of canonical nucleosomes in lysates cannot be accurately estimated because we cannot determine how many nucleosomes in total are in each field of view.”
Line 397: "We're not implying that..." Please add a sentence clearly stating what you DO mean with mobility for H2A/H2B.
We have added the following clarifying sentence in lines 412 – 413:
“We mean that H2A-H2B is attached to the rest of the nucleosome and can have small differences in orientation.”
Line 428: repeated message from line 424. "in this figure, the blurring implies.."
Redundant phrase removed.
Line 439: "on a HeLa cell" - a single cell in the whole study?
Yes, that study was done on a single cell.
A general comment is that the authors could help the reader more by developing the figures and making them more pedagogical, a list of suggestions can be found below.
Thank you for the suggestions. We have applied all of them to the specific figure callouts and to the other figures that could use similar clarification.
Figure 2: Help the reader by avoiding abbreviations in the figure legend. VPP tomographic slice - spell out "Volta Phase Plate". Same with the term "remapped" (panel B) what does that mean?
We spelled out Volta phase plate in full and explained “remapped” the additional figure legend text:
“the class averages were oriented and positioned in the locations of their contributing subtomograms”.
Supplementary figures:
Figure S3: It is unclear what you mean with "two types of BY4741 nucleosomes". You then say that the canonical nucleosomes are shaded blue. So what color is then the non-canonical? All the greys? Some of them look just like random stuff, not nucleosomes.
“Two types” is a typo and has been removed and “nucleosomes” has been replaced with “candidate nucleosome template-matching hits” to accurately reflect the particles used in classification.
Figure S6: Top left says "3 tomograms (defocus)". I wonder if you meant to add the defocus range here. I have understood it like this is the same data as shown in Figure S5, which makes me wonder if this top cartoon should not be on top of that figure too (or exclusively there).
To make Figures S6 (and S5) clearer, we have copied the top cartoon from Figure S6 to S5.
Note that we corrected a typo for these figures (and the Table S7): the number of template-matched candidate nucleosomes should be 93,204, not 62,428.
The description in the parentheses (defocus) is shorthand for defocus phase contrast and was not intended to also display a defocus range. All of the revised figure legends now report the meaning of both this shorthand and of the Volta phase plate (VPP).
To help readers see the relationship between these two figures, we added the following clarifying text to the Figure S5 and S6 legends, respectively:
“This workflow uses the same template-matched candidate nucleosomes as in Figure S6; see below.”
“This workflow uses the same template-matched candidate nucleosomes as in Figure S5.”
Figure S7: In the first panel, it is unclear why the featureless cylinder is shown as it is not used as a reference here. Rather, it could be put throughout where it was used and then put the simulated EM-map alone here. If left in, it should be stated in the legend that it was not used here.
It would indeed be much clearer to show the featureless cylinder in all the other figures and leave the simulated nucleosome in this control figure. All figures are now updated. The figure legend was also updated as follows:
“(A) A simulated EM map from a crystal structure of the nucleosome was used as the template-matching and 3-D classification reference.”
Figure S18: Why are there classes where the GFP density is missing? Mention something about this in the figure legend.
We have appended the following speculations to explain the “missing” GFP densities:
“Some of the class averages are “missing” one or both expected GFP densities. The possible explanations include mobility of a subpopulation of GFPs or H2A-GFPs, incorrectly folded GFPs, or substitution of H2A for the variant histone H2A.Z.”
Reviewer #2 (Recommendations For The Authors):
My specific (rather minor) comments are the following:
1) Abstract:
yeast -> budding yeast.
All three instances in the abstract have been replaced with “budding yeast”.
It would be better to clarify what ex vivo means here.
We have appended “(in nuclear lysates)” to explain the meaning of ex vivo.
2) Some subtitles are unclear.
e.g., "in wild-type lysates" -> "wild-type yeast lysates"
Thank you for this suggestion. All unclear instances of subtitles and sample descriptions throughout the text have been corrected.
3) Page 6, Line 113. "...which detects more canonical nucleosomes." A similar thing was already mentioned in the same paragraph and seems redundant.
Thank you for noticing this redundant statement, which is now deleted.
4) Page 25, Line 525. "However, crowding is an unlikely explanation..." Please note that many macromolecules (proteins, RNAs, polysaccharides, etc.) were lost during the nuclei isolation process.
This is a good point. We have rewritten this paragraph to separate the discussion on technical versus biological effects of crowding, in lines 538 – 546:
“Another hypothesis for the low numbers of detected canonical nucleosomes is that the nucleoplasm is too crowded, making the image processing infeasible. However, crowding is an unlikely technical limitation because we were able to detect canonical nucleosome class averages in our most-crowded nuclear lysates, which are so crowded that most nucleosomes are butted against others (Figures S15 and S16). Crowding may instead have biological contributions to the different subtomogram-analysis outcomes in cell nuclei and nuclear lysates. For example, the crowding from other nuclear constituents (proteins, RNAs, polysaccharides, etc.) may contribute to in situ nucleosome structure, but is lost during nucleus isolation.”
5) Page 7, Line 126. "The subtomogram average..." Is there any explanation for this?
Presumably, the longer linker DNA length corresponds to the ordered portion of the ~22 bp linker between consecutive nucleosomes, given the ~168 bp nucleosome repeat length. We have appended the following explanation as the concluding sentence, lines 137 – 140:
“Because the nucleosome-repeat length of budding yeast chromatin is ~168 bp (Brogaard et al., 2012), this extra length of DNA may come from an ordered portion of the ~22 bp linker between adjacent nucleosomes.”
6) "Histone GFP-tagging strategy" subsection:
Since this subsection is a bit off the mainstream of the paper, it can be shortened and merged into the next one.
We have merged the “Histone GFP-tagging strategy” and “GFP is detectable on nucleosome subtomogram averages ex vivo” subsections and shortened the text as much as possible. The new subsection is entitled “Histone GFP-tagging and visualization ex vivo”
7) Page 16, Line 329. "Because all attempts to make H3- or H4-GFP "sole source" strains failed..." Is there a possible explanation here? Cytotoxic effect because of steric hindrance of nucleosomes?
Yes, it is possible that the GFP tag is interfering with the nucleosomes interactions with its numerous partners. It is also possible that the histone-GFP fusions do not import and/or assemble efficiently enough to support a bare-minimum number of functional nucleosomes. Given that the phenotypic consequences of fusion tags is an underexplored topic and that we don’t have any data on the (dead) transformants, we would prefer to leave out the speculation about the cause of death in the attempted creation of “sole source” strains.



