Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorXiaobing ShiVan Andel Institute, Grand Rapids, United States of America
- Senior EditorDetlef WeigelMax Planck Institute for Biology Tübingen, Tübingen, Germany
Reviewer #1 (Public Review):
In this work, Xie et al. developed SCA-seq, which is a multiOME mapping method that can obtain chromatin accessibility, methylation, and 3D genome information at the same time. SCA-seq first uses M.CviPI DNA methyltransferase to treat chromatin, then perform proximity ligation followed by long-read sequencing. This method is highly relevant to a few previously reported long read sequencing technologies. Specifically, NanoNome, SMAC-seq, and Fiber-seq have been reported to use m6A or GpC methyltransferase accessibility to map open chromatin, or open chromatin together with CpG methylation; Pore-C and MC-3C have been reported to use long read sequencing to map multiplex chromatin interactions, or together with CpG methylation. Therefore, as a combination of NanoNome/SMAC-seq/Fiber-seq and Pore-C/MC-3C, SCA-seq is one step forward. The authors tested SCA-seq in 293T cells and performed benchmark analyses testing the performance of SCA-seq in generating each data module (open chromatin and 3D genome). The QC metrics appear to be good and I am convinced that this is a valuable addition to the toolsets of multi-OMIC long-read sequencing mapping.
The revised manuscript addressed most of my questions except my concern about Fig. S9. This figure is about a theory that a chromatin region can become open due to interaction with other regions, and the author propose a mathematic model to compute such effects. I was concerned about the errors in the model of Fig. S9a, and I was also concerned about the lack of evidence or validation. In their responses, the authors admitted that they cannot provide biological evidence or validations but still chose to keep the figure and the text.
The revised Fig. S9a now uses a symmetric genome interaction matrix as I suggested. But Figure S9a still have a lot of problems. Firstly, the diagonal of the matrix in Fig. S9a still has many 0's, which I asked in my previous comments without an answer. The legend mentioned that the contacts were defined as 2, 0 or -2 but the revised Fig. S9a only shows 1,0, or -1 values. Furthermore, Fig. S9b,9c,9d all added a panel of CTCF+/- but there is no explanation in text or figure legend about these newly added panels. Given many unaddressed problems, I would still suggest deleting this figure.
In my opinion, this paper does not need Fig. S9 to support its major story. The model in this figure is independent of SCA-seq. I think it should be spinoff as an independent paper if the authors can provide more convincing analysis or experiments. I understand eLife lets authors to decide what to include in their paper. If the authors insist to include Fig. S9, I strongly suggest they should at least provide adequate explanation about all the figure panels. At this point, the Fig. S9 is not solid and clearly have many errors. The readers should ignore this part.
Reviewer #2 (Public Review):
In this manuscript, Xie et al presented a new method derived from PORE-C, SCA-seq, for simultaneously measuring chromatin accessibility, genome 3D and CpG DNA methylation. SCA-seq provides a useful tool to the scientific communities to interrogate the genome structure-function relationship.
The revised manuscript has clarified almost of the concerns raised in the previous round of review, though I still have two minor concerns,
1) In fig 2a, there is no number presented in the Venn diagram (although the left panel indeed showed the numbers of the different categories, including the numbers in the right panel would be more straightforward).
2) The authors clarified the discrepancy between sfig 7a and sfig 7g. However, the remaining question is, why is there a big difference in the percentage of the cardinality count of concatemers of the different groups between the chr7 and the whole genome?
Author Response
The following is the authors’ response to the original reviews.
In this manuscript, Xie et al report the development of SCA-seq, a multiOME mapping method that can obtain chromatin accessibility, methylation, and 3D genome information at the same time. This method is highly relevant to a few previously reported long read sequencing technologies. Specifically, NanoNome, SMAC-seq, and Fiber-seq have been reported to use m6A or GpC methyltransferase accessibility to map open chromatin, or open chromatin together with CpG methylation; Pore-C and MC-3C have been reported to use long read sequencing to map multiplex chromatin interactions, or together with CpG methylation. Therefore, as a combination of NanoNome/SMAC-seq/Fiber-seq and Pore-C/MC-3C, SCA-seq is one step forward. The authors tested SCA-seq in 293T cells and performed benchmark analyses testing the performance of SCA-seq in generating each data module (open chromatin and 3D genome). The QC metrics appear to be good and the methods, data and analyses broadly support the claims. However, there are some concerns regarding data analysis and conclusions, and some important information seems to be missing.
- The chromatin accessibility tracks from SCA-seq seem to be noisy, with higher background than DNase-seq and ATAC-seq (Fig. 2f, Fig. 4a and Fig. S5). Also, SCA-seq is much less sensitive than both DNase-seq and ATAC-seq (Figs. 2a and 2b). This and other limitations of SCA-seq (high background, high sequencing cost, requirement of specific equipment, etc) need to be carefully discussed.
We thank the reviewer for the important comment about noisy GpC methylation signal in SCA-seq. We acknowledge that the SCA-seq signal presented in Fig. 2f, Fig. 4a, and Fig. S5 in our first draft was indeed noisy, as we present the raw 1D genomic signal. In this revision, we have taken steps to reduce the noise in GpC methylation signal by identifying the accessible regions on each segment of every single molecule. For each segment, we performed the sliding window analysis (50bp window sliding by a 10 bp step) with binomial test to identify accessible windows that significantly deviate from background GpC methylation ratio. The overlapping accessible windows (p < 0.05 for binomial test and contain at least two GpC sites) on the single fragments are merged as accessible region. Then we retain the GpC methylation signal inside the accessible region to reduce the background noise (Sfig 5ab). The details of the noise filtering steps are described in the Methods section (page 22 lines 13-23).
Visually, we can observe from the updated exemplary view of 1D signal track that the noise is dramatically reduced in filtered SCA-seq GpC methylation signal compared to the raw signal (Sfig5c). The clean SCA-seq GpC methylation 1D signals were also updated (Fig2f and Fig4a). We have observed an increase in the TSS enrichment score, which is a commonly used metric for assessing the signal-to-noise ratios in ATAC-seq data quality control. Specifically, the TSS enrichment score increased to 2.74 when using the filtered signal, compared to 1.93 when using the raw signal (Sfig5d). After noise filtering, 80% of SCA-seq 1D peaks overlaps with peaks called by ATAC-seq and/or DNase-seq (Fig2ab), compared to 74% from the raw signal in the first draft.
We thank the reviewer for raising up the concern about the sequencing cost and requirement of specific equipment. The sequencing cost is approximately 1300 USD per sample to sequence 30X depth human sample and obtain saturated GpC methylation signal (Sfig4d) as well as loop signal similar to the NGS-based Hi-C (Fig3gh). Considering that SCA-seq simultaneously provides higher-order chromatin structure and chromatin accessibility at single molecule resolution, we believe the cost is acceptable. However, it is worth noting that SCA-seq requires a regular Oxford nanopore sequencer with R9.4.1 chip, which is currently available but might be discontinued by Oxford Nanopore in the future. We have addressed all these concerns in the discussion section.
- In Fig. 2f, many smaller peaks are present besides the major peaks. Are they caused by baseline DNA methylation? How many of the small methylation signals are called peaks? In Fig. 4a, it seems that the authors define many more enhancers from SCA-seq data than what will be defined from ATAC-seq or DHS. Are those additional enhancers false positives? Also, it is difficult to distinguish the gray "inaccessible segments" from the light purple "accessible segments.
We thank the reviewer for bringing up these concerns.
Regarding the smaller peaks in the 1D genomic GpC methylation signal, we have addressed this issue by implementing the noise filtering in this revision, the small peaks on 1D tracks are greatly reduced (Fig2f, Sfig5c). It is important to note that SCA-seq generates accessibility signals specifically on ligation junctions, which differs from the one-dimensional (1D) signals obtained through ATAC-seq or DNase-seq. The presence of remaining small peaks in the SCA-seq data can be attributed to the varied sequencing depth, which is influenced by the enriched spatial interactions occurring in regions of the genome that are enriched with ligation junctions. In general, the SCA-seq 1D peaks are well correlated with the high confidence peaks from 1D track of ATAC-seq and DNase-seq (Fig2b).
We apologize for the lack of clarity in our enhancer annotation. The enhancer regions were obtained from The Ensembl Regulatory Build (PMID: 25887522). We have now included this information in the method section (page 24 line 16).
We thank the reviewer for pointing out this visualization problem. The color scheme has been revised, with purple now representing the inaccessible segments and yellow representing the accessible segments.
- For 3D genome analysis, it is important to provide information about data yield from SCA-seq. With 30X sequencing depth, how many contacts are obtained (with long-read sequencing, this should be the number of ligation junctions)? How is the number compared to Hi-C.
We thank the reviewer for raising up this crucial point about the sequencing yield that we missed. We have now included this information in the revised result section (page 11, lines 11-14).
We have checked the public data of a successful HEK293T Hi-C run (PMID: 34400762). The Hi-C experiment produced 699,464,541 reads (105G base), and we obtained 388,031,859 contacts.
From 100G bases of HEK293T SCA-seq data, we obtained 81,229,369 ligation junctions and 378,848,187 virtual pairwise contacts (3.8M pairwise contacts per Gb). The SCA-seq performance of virtual pairwise contact number per Gb is similar to that of PORE-C (PMID: 35637420).
- Fig 3j. Because SCA-seq only do GpC methylation, the capability to detect the footprint at individual CTCF peaks depends on the density of GpC nearby. Have the authors taken GpC density into account when defining CTCF sites with or without footprint?
We appreciate the reviewer for bringing up the concern about the GpC site density at CTCF site. We would like to highlight that Battaglia et al. have demonstrated the feasibility of identifying transcription factor binding events using GpC labeling (PMID: 36195755). In our study, we have implemented a high-resolution sliding window approach to enhance the sensitivity of CTCF binding detection. We have taken GpC density into account by performing a sliding window (50 bp window, 10 bp step) binomial test on every single molecule overlapping with CTCF site to call accessible region. The detailed steps to call accessible region has been described in the answer of the first question. Based on the pattern in Fig3j, we identify CTCF footprints if the accessible regions are called nearby the CTCF sites (at least 20 bp away from the center of CTCF sites) but not on the CTCF sites.
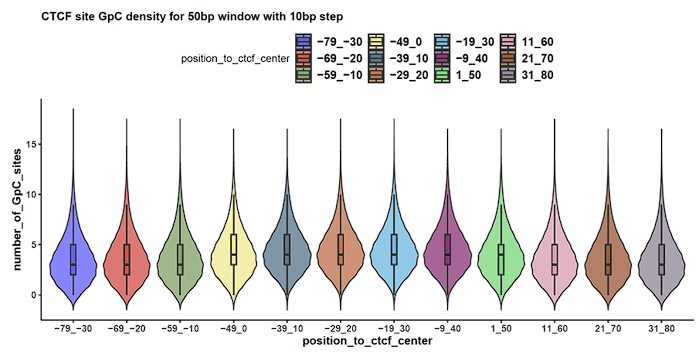
To ensure that the GpC site density is sufficient for binomial test of each sliding window of the regions around CTCF site genome-wide, we examined the number of GpC sites in each window. Our analysis revealed that GpC sites are evenly distributed, and over 87% of the windows contain at least 2 GpC sites, which qualifies them for a binomial test (Author response image 1). This indicates that we are able to detect the CTCF footprint at most of the CTCF sites, taking into consideration the GpC density.
Author response image 1.
Genome wide GpC site density at CTCF site centered region. Distribution of the number of GpC sites (y-axis) at each 50 bp sliding window region (x-axis) was presented in violin plots.
- This study only performs higher resolution chromatin interaction analysis based on individual read concatenates. It is unclear to me if the data have enough depth to perform loop analysis with Hi-C pipelines.
We thank the reviewer for highlighting this important concern about the depth of data for performing loop analysis. We have performed Aggregate peak analysis for SCA-seq and Hi-C side-by-side using hiccups function in Juicer (v1.9.9) (PMID: 27467249). We acknowledge that the level of loop signal enrichment is relatively weaker (one-fold less) in SCA-seq compared to Hi-C (Fig3h). This difference can be attributed to the lower sequencing yield per Gb in SCA-seq, which resulted in 4.93M pairwise contacts per Gb, compared to the 7M contacts per Gb in Hi-C. Despite this discrepancy, we were still able to observe the clear genome-wide loop enrichment pattern in SCA-seq (Fig3gh).
- It appears that SCA-seq is of low efficiency in detecting chromatin interactions. As shown in Fig. S7a, 65.4% of sequenced reads contained only one restriction enzyme (RE) fragment/segment (with no genomic contact), which is much higher than that reported in published PORE-C methods. In addition, Fig. S7g is very confusing and in conflict with Fig. S7a. For example, in Fig. S7g, 21.4% and 22.2% of CSA-seq concatemers contain one and two segments, whereas the numbers are 65.4% and 14.7% in Fig. S7a, respectively. Please explain.
We apologize for the confusion in sfig7a and sfig7g.
Sfig7a was intended to illustrate the cardinality count of concatemers with only chr7 segments included, representing the intra-chromosome cardinality instead of the genome-wide cardinality. We have revised sfig7a and its corresponding figure legend to clarify that the figure describes segments of intra-chromosome interactions.
On the other hand, sfig7g shows the concatemers including both intra-chromosome and inter-chromosome segments, which explains the differences in the percentages of different cardinality ranges compared to Figure S7a. Moreover, the percentages reported in Figure S7g are similar to what is typically reported in PORE-C methods when considering both intra- and inter-chromosome interactions.
To provide a comprehensive view of the genome-wide concatemer cardinality distribution, we have also included a histogram in Fig3k, which demonstrates the detailed distribution of cardinality for genome-wide concatemers.
- I disagree with the rationale of the entire Fig. S9. Biologically there is no evidence that chromatin accessibility will change due to genome interactions (the opposite is more likely), therefore the definition of "expected chromatin accessibility" is hard to believe. If the authors truly believe this is possible, they will need to test their hypothesis by deleting cohesin and check if the chromatin accessibility driven by "power center" are truly abolished. The math in Fig. S9 is also confusing. Firstly, the dimension of the contact matrix in Fig. S9 appears to be wrong, it should have 8 rows. Secondly, I don't understand why the interaction matrix is not symmetric. Third, if I understand correctly the diagonal of the matrix should be all 1, it is also hard to understand why the matrix only has 1, 0 or -1. It appears that the authors assume that the observed accessibility is a simple sum of the expected accessibility of all its interacting regions; this is wrong. In my opinion, the whole Fig. S9 should be deleted unless the authors can make sense of it and ideally also provide more evidence.
I apologize for any confusion caused by the rationale and figures in Fig. S9. The purpose of the hypothesis presented in the figure is to explore the potential relationship between chromatin accessibility and genome interactions. While there is currently no direct biological evidence supporting this hypothesis, it is a possibility that warrants further investigation.
Regarding the suggestion to delete Fig. S9 unless more evidence is provided, it is important to note that this paper primarily focuses on the methodology and theoretical framework. Experimental validation of the hypothesis falls outside the scope of this particular study.
We have made corrections to the schematic matrix in Fig. S9 to accurately represent the dimensions and symmetry. The numbers in the matrix represent mean accessible values of the contacts. Specifically, accessible-accessible contacts are represented by 2, accessible-inaccessible contacts are represented by 0, and inaccessible-inaccessible contacts are represented by -2.
Minor concerns:
- The authors may want to clearly demonstrate the specificity and sensitivity of the ATAC part and the efficiency of the Hi-C part of SCA-seq.
We appreciate the reviewer’s suggestion to demonstrate the specificity and sensitivity of the ATAC-seq part and the efficiency of the Hi-C part in SCA-seq.
We considered the non-peak region genomic bins shared by ATAC-seq and DNase-seq as true negatives and the overlapping peaks of ATAC-seq and DNase-seq as true positives. Based on these criteria, the specificity of SCA-seq 1D peaks is calculated as TN / N, where TN represents the number of true negatives (89107) and N represents the sum of true negatives and false positives (89107 + 9345). The resulting specificity is 0.91. The sensitivity of SCA-seq 1D peaks is calculated as TP / P, where TP represents the number of true positives (33190) and P represents the sum of true positives and false negatives (33190 + 11758). The resulting sensitivity is 0.73.
We evaluate the efficiency of spatial interaction by the restriction enzyme digested fragments recovered in the pairwise contacts that contain ligation junctions. In SCA-seq, the efficiency is calculated as the number of dpnII digested fragments recovered by pairwise contacts (5625908) divided by the total number of in silico dpnII digested fragments (7127633). The resulting efficiency is 0.79.
We have now included this information in the revised result section (page 8 lines 15-18)
- Fig 4g, colors with apparent differences might be used to clearly discriminate the three types of interactions (I-I, I-A and A-A).
We appreciate the reviewer for bringing up the issue regarding the visualization in Fig 4g. The color scheme has been revised, with purple now representing I-I interactions, orange representing I-A interactions, and red representing A-A interactions. We believe that these modifications have significantly improved the clarity.
- Fig. 4c, when fitting an unknown curve, R-square becomes meaningless.
We appreciate the reviewer for pointing out the issue regarding the interpretation of R-square. We have removed the R-square value from Fig. 4c.
- Fig 5a, "oCGIs comprised 65% CGIs that did not directly contact enhancers or promoters". Should it be "oCGIs comprised 65% of all CGIs"?
We appreciate the reviewer for pointing out the clarification needed in Fig 5a. We have revised the phrase in the figure legend to accurately state that “oCGIs comprised 65% of all CGIs”. Thank you for bringing this to our attention.
- Page 15 lines 5-8, "By examining the methylation status on reads, as expected, these read segments demonstrated lower CpG methylation and higher chromatin accessibility (GpC methylation), which further supports their roles in gene activation (Fig 5b)". This statement seems to be inconsistent with the figure legend.
We appreciate the reviewer for pointing out the inconsistency in the legend of Fig 5b. We have revised the legend of Fig 5b to accurately highlight the low CpG methylation on oCGI regions. Thank you for bringing this to our attention.
- Language editing and proof reading are needed.
I apologize for any errors or mistakes in the language. We have carefully reviewed the manuscript and made the necessary language editing and proofreading revisions to ensure its quality for publication.



