Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMichael LaubHoward Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, United States of America
- Senior EditorBavesh KanaUniversity of the Witwatersrand, Johannesburg, South Africa
Reviewer #1 (Public Review):
The paper from Hsu and co-workers describes a new automated method for analyzing the cell wall peptidoglycan composition of bacteria using liquid chromatography and mass spectrometry (LC/MS) combined with newly developed analysis software. The work has great potential for determining the composition of bacterial cell walls from diverse bacteria in high-throughput, allowing new connections between cell wall structure and other important biological functions like cell morphology or host-microbe interactions to be discovered. A downside to the method is that it does require some prior knowledge of an organisms peptidoglycan composition to generate the database for automated analysis. Nevertheless, the automation will allow rapid analysis of peptidoglycan composition under a variety of conditions and/or between closely related organisms once the general peptidoglycan structure is known. The methodology described will therefore be useful for the field.
The potential connection between the structure of different cell walls from bifidobacteria and cell stiffness proposed in the report is weak. The cells analyzed are from different strains such that there are many possible reasons for the change in physical measurements made by AFM. Conclusions relating cell wall composition to stiffness would be best drawn from a single strain of bacteria genetically modified to have an altered content of 3-3 crosslinks.
Reviewer #2 (Public Review):
The authors introduce "HAMA", a new automated pipeline for architectural analysis of the bacterial cell wall. Using MS/MS fragmentation and a computational pipeline, they validate the approach using well-characterized model organisms and then apply the platform to elucidate the PG architecture of several members of the human gut microbiota. They discover differences in the length of peptide crossbridges between two species of the genus Bifidobacterium and then show that these species also differ in cell envelope stiffness, resulting in the conclusion that PG "compactness" determines stiffness.
The pipeline is solid and revealing the poorly characterized PG architecture of the human gut microbiota is worthwhile and significant. However, it is unclear if or how their pipeline is superior to other existing techniques - PG architecture analysis is routinely done by many other labs; the only difference here seems to be that the authors chose gut microbes to interrogate.
I do not agree with their conclusions about the correlation between compactness and cell envelope stiffness. These experiments are done on two different species of bacteria and their experimental setup therefore does not allow them to isolate crossbridge length (which they propose indicates more or less compact PG) as the only differential property that can influence stiffness. These two species likely also differ in other ways that could modulate stiffness, e.g. turgor pressure, overall PG architecture (not just crossbridge length), membrane properties, teichoic acid composition etc.
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
The paper from Hsu and co-workers describes a new automated method for analyzing the cell wall peptidoglycan composition of bacteria using liquid chromatography and mass spectrometry (LC/MS) combined with newly developed analysis software. The work has great potential for determining the composition of bacterial cell walls from diverse bacteria in high-throughput, allowing new connections between cell wall structure and other important biological functions like cell morphology or host-microbe interactions to be discovered. In general, I find the paper to be well written and the methodology described to be useful for the field. However, there are areas where the details of the workflow could be clarified. I also think the claims connecting cell wall structure and stiffness of the cell surface are relatively weak. The text for this topic would benefit from a more thorough discussion of the weak points of the argument and a toning down of the conclusions drawn to make them more realistic.
Thank you for your thorough and insightful review of our manuscript. We greatly appreciate your positive and constructive feedbacks on our methodology. We have carefully reviewed your comments and have responded to each point as follows:
Specific points:
1) It was unclear to me from reading the paper whether or not prior knowledge of the peptidoglycan structure of an organism is required to build the "DBuilder" database for muropeptides. Based on the text as written, I was left wondering whether bacterial samples of unknown cell wall composition could be analyzed with the methods described, or whether some preliminary characterization of the composition is needed before the high-throughput analysis can be performed. The paper would be significantly improved if this point were explicitly addressed in the main text. We apologize for not making it clearer. The prior knowledge of the peptidoglycan structure of an organism is indeed required to build the “DBuilder” database to accurately identify muropeptides; otherwise, the false discovery rate might increase. While peptidoglycan structures of certain organisms might not have been extensively studied, users still remain the flexibility to adapt the muropeptide compositions based on their study, referencing closely related species for database construction. We have addressed this aspect in the main text to ensure a clearer understanding.
“(Section HAMA platform: a High-throughput Automated Muropeptide Analysis for Identification of PGN Fragments) …(i) DBuilder... Based on their known (or putative) PGN structures, all possible combinations of GlcNAc, MurNAc and peptide were input into DBuilder to generate a comprehensive database that contains monomeric, dimeric, and trimeric muropeptides (Figure 1b)."
2) The potential connection between the structure of different cell walls from bifidobacteria and cell stiffness is pretty weak. The cells analyzed are from different strains such that there are many possible reasons for the change in physical measurements made by AFM. I think this point needs to be explicitly addressed in the main text. Given the many possible explanations for the observed measurement differences (lines 445-448, for example), the authors could remove this portion of the paper entirely. Conclusions relating cell wall composition to stiffness would be best drawn from a single strain of bacteria genetically modified to have an altered content of 3-3 crosslinks.
We understand your concern regarding the weak connection between cell wall structure and cell stiffness. We will make a clear and explicit statement in the main text to acknowledge that the cells analyzed are derived from different strains, introducing the possibility of various factors influencing the observed changes in physical measurements as determined by AFM. Furthermore, we greatly appreciate your suggestion to consider genetically modified strains to investigate the role of cross-bridge length in determining cell envelope stiffness. In this regard, we are in the process of developing a CRISPR/Cas genome editing toolbox for Bifidobacterium longum, and we plan on this avenue of investigation for future work.
Reviewer #2 (Public Review):
The authors introduce "HAMA", a new automated pipeline for architectural analysis of the bacterial cell wall. Using MS/MS fragmentation and a computational pipeline, they validate the approach using well-characterized model organisms and then apply the platform to elucidate the PG architecture of several members of the human gut microbiota. They discover differences in the length of peptide crossbridges between two species of the genus Bifidobacterium and then show that these species also differ in cell envelope stiffness, resulting in the conclusion that crossbridge length determines stiffness.
We appreciate your thoughtful review of our manuscript and your recognition of the potential significance of our work in elucidating the poorly characterized peptidoglycan (PGN) architecture of the human gut microbiota.
The pipeline is solid and revealing the poorly characterized PG architecture of the human gut microbiota is worthwhile and significant. However, it is unclear if or how their pipeline is superior to other existing techniques - PG architecture analysis is routinely done by many other labs; the only difference here seems to be that the authors chose gut microbes to interrogate.
We apologize if this could have been clearer. The HAMA platform stands apart from other pipelines by utilizing automatic analysis of LC-MS/MS data to identify muropeptides. In contrast, most of the routine PGN architecture analyses often use LC-UV/Vis or LC-MS platform, where only the automatic analyzing PGFinder software is supported. To our best knowledge, a comparable pipeline on automatically analyzing LC-MS/MS data was reported by Bern et al., which they used commercial Byonic software with an in-house FASTA database and specific glycan modifications. They achieved accurate and sensitive identification on monomer muropeptides, but struggled with cross-linked muropeptides due to the limitations of the Byonic software. We believe that our pipeline introducing the automatic and comprehensive analysis on muropeptide identification (particularly for Gram-positive bacterial peptidoglycans) would be a valuable addition to the field. To enhance clarity, we have adjusted the context as follows:
(Introduction) … Although they both demonstrated great success in identifying muropeptide monomers, the accurate identification of muropeptide multimers and other various bacterial PGN structures still remains unresolved. This is because deciphering the compositions requires MS/MS fragmentation, but it is still challenging to automatically annotate MS/MS spectra from these complex muropeptide structures."
I do not agree with their conclusions about the correlation between crossbridge length and cell envelope stiffness. These experiments are done on two different species of bacteria and their experimental setup therefore does not allow them to isolate crossbridge length as the only differential property that can influence stiffness. These two species likely also differ in other ways that could modulate stiffness, e.g. turgor pressure, overall PG architecture (not just crossbridge length), membrane properties, teichoic acid composition etc.
Regarding the conclusions drawn about the correlation between cross-bridge length and cell envelope stiffness, we understand your point and appreciate your feedback. We revisit this section of our manuscript and tone down the conclusions drawn from this aspect of the study. We also recognize the importance of considering other potential factors that could influence stiffness, as you mentioned above. In light of this, we mentioned the need for further investigations, potentially involving genetically modified strains, in the main text to isolate and accurately determine the impact of bridge length on cell envelope stiffness.
Reviewer #1 (Recommendations For The Authors):
Minor points:
1) One thing to consider would be testing the robustness of the analysis pipeline with one the well-characterized bacteria studied, but genetically modifying them to change the cell wall composition in predictable ways. Does the analysis pipeline detect the expected changes?
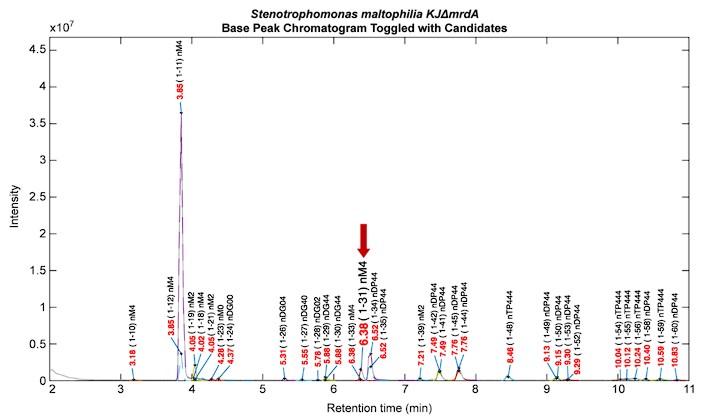
We appreciate the reviewer's suggestion and would like to provide a clear response. Regarding to testing the pipeline with genetically modified strains, our lab previously worked on genetically modified S. maltophilia (KJΔmrdA).1 Inactivation of mrdA turned out the increasing level of N-acetylglucosaminyl-1,6-anhydro-N-acetylmuramyl-L-alanyl-D-glutamyl-meso-diamnopimelic acid-D-alanine (GlcNAc-anhMurNAc tetrapeptide) in muropeptide profiles, which is the critical activator ligands for mutant strain ΔmrdA-mediated β-lactamase expression. In this case, our platform could provide rapid PGN analysis for verifying the expected change of muropeptide profiles (see Author response image 1). Besides, if the predictable changes involve genetically modifications on interpeptide bridges within the PGN structure, for example, the femA/B genes of S. aureus, which are encoded for the synthesis of interpeptide bridges,2 our current HAMA pipeline is capable of detecting these anticipated changes. However, if the genetically modifications involve the introduce of novel components to PGN structures, then it would need to create a dedicated database specific to the genetically modified strain.
Author response image 1.
2) Line 368: products catalyzed > products formed
The sentence has been revised.
“(Section Inferring PGN Cross-linking Types Based on Identified PGN Fragments) …Based on the muropeptide compositional analysis mentioned above, we found high abundances of M3/M3b monomer and D34 dimer in the PGNs of E. faecalis, E. faecium, L. acidophilus, B. breve, B. longum, and A. muciniphila, which may be the PGN products formed by Ldts.”
3) Lines 400-402: Is it possible the effect is related to porosity, not "hardness".
Thank you for the suggestion. The possibility of the slower hydrolysis rate of purified PGN in B. breve being related to porosity is indeed noteworthy. While this could be a potential factor, we would like to acknowledge the limited existing literature that directly addresses the relation between PGN architecture and porosity. It is plausible that current methods available for assessing cell wall porosity may have certain limitations, contributing to the scarcity of relevant studies. In light of this, we would like to propose a speculative explanation for the observed effect. It is plausible that the tighter PGN architecture resulting from shorter interpeptide bridges in B. breve could contribute to its harder texture. This speculation is grounded in the concept that a more compact PGN structure might lead to increased stiffness, aligning with our observations of higher cell stiffness in B. breve.
4) Lines 403-408: See point #2 above.
Thank you for the suggestion. We have explicitly addressed this point in the main text:
“(Section Exploring the Bridge Length-dependent Cell Envelope Stiffness in B. longum and B. breve) … Taken all together, we speculate that a tight peptidoglycan network woven by shorter interpeptide bridges or 3-3 cross-linkages could give bacteria stiffer cell walls. However, it is important to note that cell stiffness is a mechanical property that also depends on PGN thickness, overall architecture, and turgor pressure. These parameters may vary among different bacterial strains. Hence, carefully controlled, genetically engineered strains with similar characteristics will be needed to dissect the role of cross-bridge length in cell envelope stiffness.”
5) Lines 428-429: It is not clear to me how mapping the cell wall architecture provides structural information about the synthetic system. It is also not clear how antibiotic resistance can be inferred. More detail is needed here to flesh out these points.
Thank you for the suggestion. To provide further clarity on these important aspects, the context in the manuscript has been revised.
“(Discussion) …Importantly, our HAMA platform provides a powerful tool for mapping peptidoglycan architecture, giving structural information on the PGN biosynthesis system. This involves the ability to infer possible PGN cross-linkages based on the type of PGN fragments obtained from hydrolysis. For instance, the identification of 3-3 cross-linkage formed by L,D-transpeptidases (Ldts) is of particular significance. Unlike 4-3 cross-linkages, the 3-3 cross-linkage is resistant to inhibition by β-Lactam antibiotics, a class of antibiotics that commonly targets bacterial cell wall synthesis through interference with 4-3 cross-linkages. Therefore, by elucidating the specific cross-linkage types within the peptidoglycan architecture, our approach offers insights into antibiotic resistance mechanisms.”
6) Line 478: "maneuvers are proposed for" > "work is needed to generate". Also, delete "innovative". Also "in silico" > "in silico-based".
The sentence has been revised.
“(Discussion) …To achieve a more comprehensive identification of muropeptides, future work is needed to generate an expanded database, in silico-based fragmentation patterns, and improved MS/MS spectra acquisition.”
7) Line 485: "Its" > "It has potential"
The sentence has been revised.
“(Discussion) …It has potential applications in identifying activation ligands for antimicrobial resistance studies, characterizing key motifs recognized by pattern recognition receptors for host-microbiota immuno-interaction research, and mapping peptidoglycan in cell wall architecture studies.”
8) Figure 1 legend: Define Gb and Pb.
Gb and Pb are the abbreviations of glycosidic bonds and peptide bonds. We have revised the Figure legend 1 as follow:
“(Figure legend 1) …(b) DBuilder constructs a muropeptide database containing monomers, dimers, and trimers with two types of linkage: glycosidic bonds (Gb) and peptide bonds (Pb).”
9) Figure 2: It is hard to see what is going on in panel a and c with all the labels. Consider removing them and showing a zoomed inset with labels in addition to ab unlabeled full chromatogram.
We apologize for not making this clearer. The panel a and c in Figure 2 were directly generated by the Analyzer as a software screenshot of the peak annotations on chromatogram. Our intention was to present a comprehensive PGN mapping (approximately 70% of the peak area was assigned to muropeptide signals) using this platform. We understand the label density might affect clarity, so we have added the output tables of the whole muropeptide identifications as source data (Table 1–Source Data 1&2). Additionally, we have uploaded the Analyzer output files (see Additional Files), which can be better visualized in the Viewer program, and it also allows users zoom in for detailed labeling information.
10) Figure 3: It is worth pointing out what features of the MS/MS fingerprints are helping to discriminate between species.
Thank you for the suggestion. We have revised Figure 3 and the legend as follow:
“(Figure legend 3) …The sequence of each isomer was determined using in silico MS/MS fragmentation matching, with the identified sequence having the highest matching score. The key MS/MS fragments that discriminate between two isomers are labeled in bold brown.”
Author response image 2.
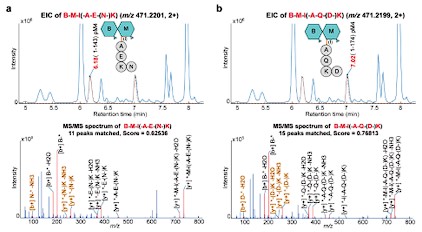
11) Figure 4 and 5 legend: Can you condense the long descriptions of the abbreviations - or at least only refer to them once?
Certainly, to enhance clarity and conciseness in the figure legends, we have revised Figure legend 5 as follow:
“(Figure legend 5) …(b) Heatmap displaying …. Symbols: M, monomer; D, dimer; T, trimer (numbers indicate amino acids in stem peptides). Description of symbol abbreviations as in Figure legend 4, with the addition of "Glycan-T" representing trimers linked by glycosidic bonds.”
Reviewer #2 (Recommendations For The Authors):
- Please read the manuscript carefully for spelling errors.
We appreciate your careful review of our manuscript. We have thoroughly rechecked the entire manuscript for spelling errors and have made the necessary corrections to ensure the accuracy and quality of the text.
- Line 46 - "multilayered" is likely only true for Gram-positive bacteria.
We thank reviewer #2 for bringing up this concern. Indeed, Gram-negative bacteria mostly possess single layer of peptidoglycan, but could be up to three layers in some part of the cell surface.3, 4 In order to reduce the confusion, we have rewritten the context as follow: “(Introduction) …PGN is a net-like polymeric structure composed of various muropeptide molecules, with their glycans linearly conjugated and short peptide chains cross-linked through transpeptidation.”
- Methods section: It seems like pellets from a 10 mL bacterial culture were ultimately suspended in 1.5 L (750 mL water + 750 mL tris) - why such a large volume? And how were PG fragments subsequently washed (centrifugation? There is no information on this in the Methods).
We apologize for the mislabeling on the units. The accurate volume should be “1.5 mL (750 µL water + 750 µL tris)”. We have updated the correct volume in the Methods section (lines 99-100). For the washing process of purified PGN, we added 1 mL water, centrifuged at 10,000 rpm for 5 minutes, and removed supernatant. This information has added to the Methods section (lines 95-98).
- Line 183 - why were 6 modifications chose as the cutoff? Please make rationale more clear.
We thank reviewer #2 for the comments. We set the maximum modification number of 6 in the assumption of one modification on each sugar of a trimeric muropeptide. A lower cutoff could effectively limit the identification of muropeptides with unlikely numbers of modifications, whereas a higher cutoff could allow for having multiple modifications on a muropeptide. In our hand, muropeptide modifications of E. coli are mostly N-deacetyl-MurNAc and anhydro-MurNAc, and modifications of gut microbes used here are mostly N-deacetyl-GlcNAc, anhydro-MurNAc, O-acetyl-MurNAc, loss of GlcNAc, and amidated iso-Glu. While we recommend starting data analysis with the cutoff of 6 modifications, users are free to adjust this based on their studies.
- Line 339 - define donor vs. acceptor here (can be added in parentheses after explaining the relevant chemical reactions further above in the text)
Thank you for the suggestion. To provide greater clarity regarding the roles of the donor and acceptor substrates in the transpeptidation process, we have revised the content in the manuscript as follows:
“(Section Inferring PGN Cross-linking Types Based on Identified PGN Fragments) …In general, there are two types of PGN cross-linkage…. Transpeptidation involves two stem peptides which function as acyl donor and acceptor substrates, respectively. As the enzyme names imply, the donor substrates that Ddts and Ldts bind to are terminated as D,D-stereocenters and L,D-stereocenters, which structurally means pentapeptides and tetrapeptides. During D,D-transpeptidation, Ddts recognize D-Ala4-D-Ala5 of the donor stem (pentapeptide) and remove the terminal D-Ala5 residue, forming an intermediate. The intermediate then cross-links the NH2 group in the third position of the neighboring acceptor stem, forming a 4-3 cross-link.”
- Line 366 following - can you calculate % crosslinks based on these numbers? What does "high abundance" of 3,3 crosslinks mean in this context? Is this the majority of PG?
Thank you for your questions. Calculating the percentage of crosslinks based on the muropeptide compositional numbers is a valid consideration. However, it's important to note that the muropeptides we analyzed were hydrolyzed by mutanolysin, and as such, deriving an accurate % crosslink value from these data might not provide a true representation of the crosslinking percentage within the PGN network. For a more precise determination of % crosslinks, methods such as solid-phase NMR on purified peptidoglycan would be required. Our research provides insights into the characterization of PGN fragments and allows us to infer potential PGN cross-linkage types and the enzymes involved based on the dominant muropeptide fragments. Regarding the phrase "high abundance" in the context, it indicates that the M3b/M4b monomer and D34 dimer muropeptides represent a significant portion of the hydrolysis products. These muropeptides are major constituents within the PGN fragments obtained from the enzymatic hydrolysis.
- Line 375 - I am not sure PG is a meaningful diffusion barrier for drugs and signaling molecules, give that even larger proteins can apparently diffuse through the pores.
Thank you for raising this point. Peptidoglycan indeed possesses relatively wide pores that allow for the diffusion of larger molecules, including proteins.5 Research has provided a rough estimate of the porosity of the PGN meshwork, suggesting that it allows for the diffusion of proteins with a maximum molecular mass of around 50 kDa.6 Considering this, we acknowledge that PGN may not serve as a significant diffusion barrier for drugs and signaling molecules. The porosity of the PGN scaffold, which is defined by the degree of cross-linking, plays a role in influencing the transport of molecules to the cell membrane. Thus, while PGN may not serve as a strict diffusion barrier, its structural characteristics still impact bacterial cell mechanics and interactions. We have revised the manuscript to reflect this understanding:
“(Section Exploring the Bridge Length-dependent Cell Envelope Stiffness in B. longum and B. breve) …The porosity of the PGN scaffold, defined by the degree of cross-linking, influences the transport of larger molecules such as proteins. Therefore, modifications to PGN structure are anticipated to significantly affect bacterial cell mechanics and interactions.”
- Line 400 - what does "slower hydrolysis rate" refer to, is this chemical hydrolysis or enzymatic (autolysins?). also, I am not sure hydrolysis rate of either modality allows for solid conclusions about how hard (line 402) the PG is.
Thank you for your comments. The hydrolysis rate here refers to the enzymatic hydrolysis, specifically the mutanolysin cleaving the β-N-acetylmuramyl-(1,4)-N-acetylglucosamine linkage. Indeed, there is no direct correlation between the hydrolysis rate and the hardness of PGN architecture, although the structure rigidity is a key determinant in protein digestion.7 Considering the enzymatic hydrolysis rate depending on the accessibility of the substrate to the enzyme, we proposed that the tighter PGN architecture could also lead to a slower hydrolysis rate. This speculation aligns with our observations of higher cell stiffness or more compact PGN structure of B. breve and its slower hydrolysis rate. We understand this is indirect proof, so the revised sentence now reads:
“(Section Exploring the Bridge Length-dependent Cell Envelope Stiffness in B. longum and B. breve) …Furthermore, B. breve also showed a slower enzymatic hydrolysis rate in purified PGNs, implying that the cell wall structure of B. breve is characterized by a compact PGN architecture.”
- Line 424 - I am not convinced this pipeline can detect PG architectures that other pipelines cannot; likely, the difference between previous analyses and theirs is due to different growth conditions (3,3 crosslink formation is often modulated by environmental factors/growth stage). In the next sentence, it sounds like mutanolysin treatment is a novelty in PG analysis (which it is not).
We apologize if this could have been clearer and we have revised the paragraph to describe our study more accurately. We agree that different growth conditions could influence PGN architecture and other pipelines could manually identify the PGN architectures or automatically identify them if they are not too complex. Our original intention was to highlight the ability of the HAMA program to automatically identify unreported PGN structure. Here are the revised sentences:
“(Discussion) …We speculate that this finding may be influenced by the comprehensive mass spectrometric approaches we employed or by variations in growth conditions. Moreover, we utilized the well-established enzymatic method involving mutanolysin to cleave the β-N-acetylmuramyl-(1,4)-N-acetylglucosamine linkage, which preserves the original peptide linkage in intact PGN subunits.”
- Line 440- 442: As outlined in more detail above: I don't think you can conclude something about the relationship between bridge length and envelope stiffness based on these data. Thank you for your valuable feedback. We agree that our data may not definitively support the direct conclusion about the relationship between bridge length and envelope stiffness in Bifidobacterium species. Instead, we will rephrase this section to accurately present the observed correlations without overgeneralizing:
“(Discussion) … Notably, our study suggested a potential correlation between the cell stiffness and the compactness of bacterial cell walls in Bifidobacterium species (Figure 5). B. longum, which predominantly harbors tetrapeptide bridges (Ser-Ala-Thr-Ala), exhibits a trend towards lower stiffness, whereas B. breve, characterized by PGN cross-linked with monopeptide bridges (Gly), demonstrates a trend towards higher stiffness. These findings suggested that it may be correlated between the increased rigidity and the more compact PGN architecture built by shorter cross-linked bridges.”
References: 1. Huang, Y.-W.; Wang, Y.; Lin, Y.; Lin, C.; Lin, Y.-T.; Hsu, C.-C.; Yang, T.-C., Impacts of Penicillin Binding Protein 2 Inactivation on β-Lactamase Expression and Muropeptide Profile in Stenotrophomonas maltophilia. mSystems 2017, 2 (4), 00077-00017.
-
Jarick, M.; Bertsche, U.; Stahl, M.; Schultz, D.; Methling, K.; Lalk, M.; Stigloher, C.; Steger, M.; Schlosser, A.; Ohlsen, K., The serine/threonine kinase Stk and the phosphatase Stp regulate cell wall synthesis in Staphylococcus aureus. Sci. Rep. 2018, 8 (1), 13693.
-
Labischinski, H.; Goodell, E. W.; Goodell, A.; Hochberg, M. L., Direct proof of a "more-than-single-layered" peptidoglycan architecture of Escherichia coli W7: a neutron small-angle scattering study. J. Bacteriol. 1991, 173 (2), 751-756.
-
Rohde, M., The Gram-Positive Bacterial Cell Wall. Microbiol. Spectr. 2019, 7 (3), gpp3-0044-2018.
-
Vollmer, W.; Höltje, J. V., The architecture of the murein (peptidoglycan) in gram-negative bacteria: vertical scaffold or horizontal layer(s)? J. Bacteriol. 2004, 186 (18), 5978-5987.
-
Vollmer, W.; Blanot, D.; De Pedro, M. A., Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32 (2), 149-167.
-
Li, Q.; Zhao, D.; Liu, H.; Zhang, M.; Jiang, S.; Xu, X.; Zhou, G.; Li, C., "Rigid" structure is a key determinant for the low digestibility of myoglobin. Food Chem.: X 2020, 7, 100094.



