Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorChris DoeHoward Hughes Medical Institute, Eugene, United States of America
- Senior EditorJohn HuguenardStanford University School of Medicine, Stanford, United States of America
Reviewer #1 (Public Review):
The overall tone of the rebuttal and lack of responses on several questions was surprising. Clearly, the authors took umbrage at the phrase 'no smoking gun' and provided a lengthy repetition of the fair argument about 'ticking boxes' on the classic list of criteria. They also make repeated historical references that descriptions of neurotransmitters include many papers, typically over decades, e.g. in the case of ACh and its discovery by Sir Henry Dale. While I empathize with the authors' apparent frustration (I quote: '...accept the reality that Rome was not built in a single day and that no transmitter was proven by a one single paper') I am a bit surprised at the complete brushing away of the argument, and in fact the discussion. In the original paper, the notion of a receptor was mentioned only in a single sentence and all three reviewers brought up this rather obvious question. The historical comparisons are difficult: Of course many papers contribute to the identification of a neurotransmitter, but there is a much higher burden of proof in 2023 compared to the work by Otto Loewi and Sir Henry Dale: most, if not all, currently accepted neurotransmitter have a clear biological function at the level of the brain and animal behavior or function - and were in fact first proposed to exist based on a functional biological experiment (e.g. Loewi's heart rate change). This, and the isolation of the chemical that does the job, were clear, unquestionable 'smoking guns' a hundred years ago. Fast forward 2023: Creatine has been carefully studied by the authors to tick many of the boxes for neurotransmitters, but there is no clear role for its function in an animal. The authors show convincing effects upon K+ stimulation and electrophysiological recordings that show altered neuronal activity using the slc6a8 and agat mutants as well as Cr application - but, as has been pointed out by other reviewers, these effects are not a clear-cut demonstration of a chemical transmitter function, however many boxes are ticked. The identification of a role of a neurotransmitter for brain function and animal behavior has reasonably more advanced possibilities in 2023 than a hundred years ago - and e.g. a discussion of approaches for possible receptor candidates should be possible.
Again, I reviewed this positively and agree that a lot of cumulative data are great to be put out there and allow the discovery to be more broadly discussed and tested. But I have to note, that the authors simply respond with the 'Rome was not built in a single day' statement to my suggestions on at least 'have some lead' how to approach the question of a receptor e.g. through agonists or antagonists (while clearly stating 'I do not think the publication of this manuscript should not be made dependent' on this). Similarly, in response to reviewer 2's concerns about a missing receptor, the authors' only (may I say snarky) response is ' We have deleted this sentence, though what could mediate postsynaptic responses other than receptors?' The bullet point by reviewer 3 ' • No candidate receptor for creatine has been identified postsynaptically.' is the one point by that reviewer that is simply ignored by the authors completely. Finally, I note that my reivew question on the K stimulation issues (e.g. 35 neurons that simply did not respond at all) was: ' Response: To avoid the disadvantage of K stimulation, we also performed optogenetic experiments recently and obtained encouraging preliminary results.' No details, not data - no response really.
In sum, I find this all a bit strange and the rebuttal surprising - all three reviewers were supportive and have carefully listed points of discussion that I found all valid and thoughtful. In response, the authors selectively responded scientifically to some experimental questions, but otherwise simply rather non-scientifically dismissed questions with 'Rome was not built in a day'-type answers, or less. I my view, the authors have disregarded the review process and the effort of three supportive reviewers, which should be part of the permanent record of this paper.
Reviewer #2 (Public Review):
Summary:
Bian et al studied creatine (Cr) in the context of central nervous system (CNS) function. They detected Cr in synaptic vesicles purified from mouse brains with anti-Synaptophysin using capillary electrophoresis-mass spectrometry. Cr levels in the synaptic vesicle fraction was reduced in mice lacking the Cr synthetase AGAT, or the Cr transporter SLC6A8. They provide evidence for Cr release within several minutes after treating brain slices with KCl. This KCl-induced Cr release was partially calcium dependent and was attenuated in slices obtained from AGAT and SLC6A8 mutant mice. Cr application also decreased the excitability of cortical pyramidal cells in one third of the cells tested. Finally, they provide evidence for SLC6A8-dependent Cr uptake into synaptosomes, and ATP-dependent Cr loading into synaptic vesicles. Based on these data, the authors propose that Cr may act as neurotransmitter in the CNS.
Strengths:
1. A major strength of the paper is the broad spectrum of tools used to investigate Cr.
2. The study provides evidence that Cr is present in/loaded into synaptic vesicles.
Weaknesses:
1. There is no significant decrease in Cr content pulled down by anti-Syp in AGAT-/- mice when normalized to IgG controls. Hence, blocking AGAT activity/Cr synthesis does not affect Cr levels in the synaptic vesicle fraction, arguing against a Cr enrichment.
2. There is no difference in KCl-induced Cr release between SLC6A8-/Y and SLC6A8+/Y when normalizing the data to the respective controls. Thus, the data are not consistent with the idea that depolarization-induced Cr release requires SLC6A8.
3. The rationale of grouping the excitability data into responders and non-responders is not convincing because the threshold of 10% decrease in AP rate is arbitrary. The data do therefore not support the conclusion that Cr reduces neuronal excitability.
Reviewer #3 (Public Review):
SUMMARY:
The manuscript by Bian et al. promotes the idea that creatine is a new neurotransmitter. The authors conduct an impressive combination of mass spectrometry (Fig. 1), genetics (Figs. 2, 3, 6), biochemistry (Figs. 2, 3, 8), immunostaining (Fig. 4), electrophysiology (Figs. 5, 6, 7), and EM (Fig. 8) in order to offer support for the hypothesis that creatine is a CNS neurotransmitter.
STRENGTHS:
There are many strengths to this study.
• The combinatorial approach is a strength. There is no shortage of data in this study.
• The careful consideration of specific criteria that creatine would need to meet in order to be considered a neurotransmitter is a strength.
• The comparison studies that the authors have done in parallel with classical neurotransmitters is helpful.
• Demonstration that creatine has inhibitory effects is another strength.
• The new genetic mutations for Slc6a8 and AGAT are strengths and potentially incredibly helpful for downstream work.
WEAKNESSES:
• Some data are indirect. Even though Slc6a8 and AGAT are helpful sentinels for the presence of creatine, they are not creatine themselves. Of note, these molecules themselves are not essential for making the case that creatine is a neurotransmitter.
• Regarding Slc6a8, it seems to work only as a reuptake transporter - not as a transporter into SVs. Therefore, we do not know what the transporter into the TVs is.
• Puzzlingly, Slc6a8 and AGAT are in different cells, setting up the complicated model that creatine is created in one cell type and then processed as a neurotransmitter in another. This matter will likely need to be resolved in future studies.
• No candidate receptor for creatine has been identified postsynaptically. This will likely need to be resolved in future studies.
• Because no candidate receptor has been identified, it is important to fully consider other possibilities for roles of creatine that would explain these observations other than it being a neurotransmitter? There is some attention to this in the Discussion.
There are several criteria that define a neurotransmitter. The authors nicely delineated many criteria in their discussion, but it is worth it for readers to do the same with their own understanding of the data.
By this reviewer's understanding (and combining some textbook definitions together) a neurotransmitter: 1) must be present within the presynaptic neuron and stored in vesicles; 2) must be released by depolarization of the presynaptic terminal; 3) must require Ca2+ influx upon depolarization prior to release; 4) must bind specific receptors present on the postsynaptic cell; 5) exogenous transmitter can mimic presynaptic release; 6) there exists a mechanism of removal of the neurotransmitter from the synaptic cleft.
For a paper to claim that the published work has identified a new neurotransmitter, several of these criteria would be met - and the paper would acknowledge in the discussion which ones have not been met. For this particular paper, this reviewer finds that condition 1 is clearly met.
Conditions 2 and 3 seem to be met by electrophysiology, but there are caveats here. High KCl stimulation is a blunt instrument that will depolarize absolutely everything in the prep all at once and could result in any number of non-specific biological reactions as a result of K+ rushing into all neurons in the prep. Moreover, the results in 0 Ca2+ are puzzling. For creatine (and for the other neurotransmitters), why is there such a massive uptick in release, even when the extracellular saline is devoid of calcium?
Condition 4 is not discussed in detail at all. In the discussion, the authors elide the criterion of receptors specified by Purves by inferring that the existence of postsynaptic responses implies the existence of receptors. True, but does it specifically imply the existence of creatinergic receptors? This reviewer does not think that is necessarily the case. The authors should be appropriately circumspect and consider other modes of inhibition that are induced by activation or potentiation of other receptors (e.g., GABAergic or glycinergic).
Condition 5 may be met, because authors applied exogenous creatine and observed inhibition. However, this is tough to know without understanding the effects of endogenous release of creatine. if they were to test if the absence of creatine caused excess excitation (at putative creatinergic synapses), then that would be supportive of the same. Nicely, Ghirardini et al., 2023 study cited by the reviewers does provide support for this exact notion in pyramidal neurons.
For condition 6, the authors made a great effort with Slc6a8. This is a very tough criterion to understand or prove for many synapses and neurotransmitters.
In terms of fundamental neuroscience, the story should be impactful. There are certainly more neurotransmitters out there than currently identified and by textbook criteria, creatine seems to be one of them taking all of the data in this study and others into account.
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
This is an interesting and somewhat unusual paper supporting the idea that creatine is a neurotransmitter in the central nervous system of vertebrates. The idea is not entirely new, and the authors carefully weigh the evidence, both past and newly acquired, to make their case. The strength of the paper lies in the importance of the potential discovery - as the authors point out, creatine ticks more boxes on criteria of neurotransmitters than some of the ones listed in textbooks - and the list of known transmitters (currently 16) certainly is textbook material. A further strength of the manuscript is the careful consideration of a list of criteria for transmitters and newly acquired evidence for four of these criteria: 1. evidence that creatine is stored in synaptic vesicles, 2. mutants for creatine synthesis and a vesicular transporter show reduced storage and release of creatine, 3. functional measurement that creatine release has an excitatory or inhibitory (here inhibitory) effect in vivo, and 4. ATP-dependence. The key weakness of the paper is that there is no single clear 'smoking gun', like a postsynaptic creatine receptor, that would really demonstrate the function as a transmitter. Instead, the evidence is of a cumulative nature, and not all bits of evidence are equally strong. On balance, I found the path to discovery and the evidence assembled in this manuscript to establish a clear possibility, positive evidence, and to provide a foundation for further work in this direction.
it is notable that, historically, no neurotransmitter has ever been established in a single paper. While creatine will not be an exception, data presented in this paper are more than any previous paper in demonstrating the possibility of a new neurotransmitter. However, we added an entire paragraph in the Discussion part about differences between Cr and classic neurotransmitters such as Glu, beginning with the absence of a molecularly defined receptor at this point and the Ca2+ independent component of Cr release induced by extracellular K+.
We appreciate the reviewer for noting that evidence obtained by us now support that creatine satisfies all 4 criteria of transmitters.
We respectively disagree the point about a smoking gun: any of these four is a smoking gun, while the satisfication of all 4 is quite strong, more than a smoking gun.
We find it disagreeable that a receptor “would really demonstrate the function of a transmitter”. Textbook criteria for a transmitter usually require postsynaptic responses, not a molecularly defined receptor. A molecularly defined receptor for many of the known transmitters required many years of work, while they were accepted as transmitters before their receptors were finally molecularly defined. As long as there is a postsynaptic response, there is of course a receptor, though its molecular properties should be further studied. For examples, responses to choline were discovered in 1900 (Hunt, Am J Physiol 3, xviii-xix, 1900), those to acetylcholine in 1906 (Hunt and Taveau, Br Med J 2:1788-1789, 1906), those to supradrenal glands before 1894 (Oliver and Schäfer, J Physiol 18:230-276 1895). Henry Dale was awarded a Nobel prize in 1936 partly for his work on acetylcholine. Receptors for acetylcholine and noradrenaline were not molecularly defined until the 1970s and 1980s. Before then, they were only known by mediating responses to natural transmitters and synthesized chemicals.
There were two previous reports that creatine could be taken into brain slices (Almeida et al., 2006) or synaptosomes (Peral, Vázquez-Carretero and Ilundain, 2010). These were used by the reviewer to argue that the idea of creatine as a neurotransmitter “is not entirely new”. However, no one has followed up these studies for 10 years, thus they would not be considered as good smoking guns. While we have reproduced the synaptosome uptake result (together with our new finding that this uptake was dependent on SLC6A8), it should be noted that uptake of molecules into synaptosomes is not absolutely required for a neurotransmitter because degradation of a transmitter is equally valid. Furthermore, molecules required synaptically but not as a transmitter can also be transported into the synaptic terminal.
Our detection of Cr in the synaptic vesicles provides much stronger evidence supporting its importance. If a smoking gun is important, the detection of creatine in the SVs is the best smoking gun, whose discovery in fact was the reason leading us to study its release, postsynaptic responses as well as repeating the uptake experiment with genetic mutants.
Reviewer #2 (Public Review):
Summary:
Bian et al studied creatine (Cr) in the context of central nervous system (CNS) function. They detected Cr in synaptic vesicles purified from mouse brains with anti-Synaptophysin using capillary electrophoresis-mass spectrometry. Cr levels in the synaptic vesicle fraction were reduced in mice lacking the Cr synthetase AGAT, or the Cr transporter SLC6A8. They provide evidence for Cr release within several minutes after treating brain slices with KCl. This KCl-induced Cr release was partially calcium-dependent and was attenuated in slices obtained from AGAT and SLC6A8 mutant mice. Cr application also decreased the excitability of cortical pyramidal cells in one third of the cells tested. Finally, they provide evidence for SLC6A8-dependent Cr uptake into synaptosomes, and ATP-dependent Cr loading into synaptic vesicles. Based on these data, the authors propose that Cr may act as a neurotransmitter in the CNS.
Strengths:
1) A major strength of the paper is the broad spectrum of tools used to investigate Cr.
2) The study provides strong evidence that Cr is present in/loaded into synaptic vesicles.
Weaknesses:
(in sequential order)
1) Are Cr levels indeed reduced in Agat-/-? The decrease in Cr IgG in Agat-/- (and Agat+/-) is similar to the corresponding decrease in Syp (Fig. 3B). What is the explanation for this? Is the decrease in Cr in Agat-/- significant when considering the drop in IgG? The data should be normalized to the respective IgG control.
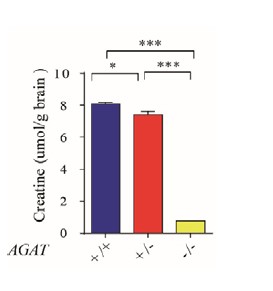
We measured the Cr concentration in the whole brain lysates using Creatine Assay Kit (Sigma, MAK079). Cr levels in the brain were reduced in Agat-/- mice. The Cr concentration in AGAT-/- mice was reduced to about 1/10 of AGAT+/+ and AGAT+/- mice (Author response image 1).
Author response image 1.
Cr concentration in brain from AGAT+/+, AGAT+/- and AGAT-/- mice (n=5 male mice for each group). , p<0.05, **, p<0.001, one-way ANOVA with Tukey’s correction.
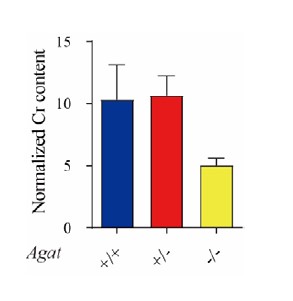
As pointed by the reviewer, the decrease in Cr IgG in Agat-/- seems similar to the corresponding decrease in Syp (Fig. 3B in the paper). Cr pulled down by IgG was 0.46 ± 0.04, 0.37 ± 0.06 and 0.17 ±0.03 pmol/μg anti-syp antibody for Agat+/+, Agat+/-, and Agat-/- mice respectively. There was a trend of reduction Cr IgG in Agat-/-, however, there were no statistically significant differences between Agat-/- and Agat+/+, or between Agat-/- and Agat+/-, as determined by one-way ANOVA (Fig. 3B in the paper). Due to the fact that Agat-/- reduced Cr concentration in the brain, we speculate that the apparent drop in Cr pulled down by IgG may have partially resulted from the overall reduction of Cr content in the brain.
The absolute content of Cr pulled down by Syp in Agat-/- mice was reduced to 21.6% of Agat+/+ mice and 23.6% of Agat+/- mice (Fig. 3B in the paper). As suggested by the reviewer, we normalized the Cr pulled down by Syp to the respective IgG control (Author response image 2). The normalized Cr content in AGAT-/- mice has a tendency to decrease, but not statistically significant, as compared to Agat+/+ and Agat+/- mice (n=10 for each group, one-way ANOVA).
Author response image 2.
Normalized Cr content in brain from AGAT+/+, AGAT+/- and AGAT-/- mice (n=10 for each group). Cr pulled down by anti-Syp antibody was normalized to that of IgG.
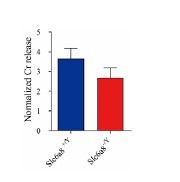
2) The data supporting that depolarization-induced Cr release is SLC6A8 dependent is not convincing because the relative increase in KCl-induced Cr release is similar between SLC6A8-/Y and SLC6A8+/Y (Fig. 5D). The data should be also normalized to the respective controls.
As suggested by the reviewer, we normalized the Cr release during KCl stimulation to the baseline (Author response image 3). The ratio of Cr release evoked by high KCl stimulation to the baseline was similar in WT and Slc6a8 knockouts. This suggests that Cr is not released through SLC6A8 transporter.
Author response image 3.
Normalized Cr release from slices from Slc6a8+/Y and Slc6a8-/Y mice (n=7 slices for each group). Cr released evoked by high KCl stimulation was normalized to baseline.
However, without Slc6a8, KCl-induced release of Cr was significantly reduced (Figure 5D in the paper). This is because Slc6a8 is a transporter to Cr uptake into synaptic terminals (Figure 5D and 8C in the paper). Therefore, Cr content in SVs (Figure 2C in the paper) indirectly reduced Cr release.
3) The majority (almost 3/4) of depolarization-induced Cr release is Ca2+ independent (Fig. 5G). Furthermore, KCl-induced, Ca2+-independent release persists in SLC6A8-/Y (Fig. 5G). What is the model for Ca2+-independent Cr release? Why is there Ca2+-independent Cr release from SLC6A8 KO neurons? How does this relate to the prominent decrease in Ca2+-dependent Cr release in SLC6A8-/Y (Fig. 5G)? They show a prominent decrease in Cr control levels in SLC6A8-/Y in Fig. 5D. Were the data shown in Fig. 5D obtained in the presence or absence of Ca2+? Could the decrease in Ca2+-dependent Cr release in SLC6A8-/Y (Fig. 5G) be due to decreased Cr baseline levels in the presence of Ca2+ (Fig. 5D)?
These are interesting questions that, at this point, could only be answered by references to literature. For example, one possibility was that Ca2+-independent Cr release might occurs in glia, since as pointed by the reviewer in Point 6, high GAMT levels were reported for astrocytes and oligodendrites (Schmidt et al. 2004; Rosko et al. 2023). As reported, other neuromodulators such as taurine can be released from astrocytes (Philibert, Rogers, and Dutton 1989) or slices (Saransaari and Oja 2006) in Ca2+ independent manner. In addition, in the absence of potassium stimulation, Ca2+ depletion lead to increased release of taurine in cultured astrocytes (Takuma et al. 1996) or in striatum in vivo (Molchanova, Oja, and Saransaari 2005). Similarly, in SLC6A8 KO slices, Ca2+ depletion (Figure 5G) also increased creatine baseline levels as compared to that in normal ACSF (Figure 5D). Another possibility was that Ca2+-independent Cr release might occurs in neurons lacking SLC6a8 expression.
As mentioned in the paper, data shown in Figure 5D was obtained in the presence Ca2+. Reduction of Ca2+-dependent Cr release evoked by potassium in SLC6A8-/Y (Figure 5G) may be due to decreased Cr baseline levels in the presence of Ca2+ and reduced Cr in synaptic vesicles (Figure 5D).
4) Cr levels are strongly reduced in Agat-/- (Figure 6B). However, KCl-induced Cr release persists after loss of AGAT (Figure 6B). These data do not support that Cr release is Agat dependent.
Although KCl-induced Cr release persisted in AGAT-/- mutants, it was dropped to 11.6% of WT mice (Figure 6B). AGAT is not directly involved in the release, but required for providing sufficient Cr.
5) The authors show that Cr application decreases excitability in ~1/3 of the tested neurons (Figure 7). How were responders and non-responders defined? What justifies this classification? The data for all Cr-treated cells should be pooled. Are there indeed two distributions (responders/non-responders)? Running statistics on pre-selected groups (Figure 7H-J) is meaningless. Given that the effects could be seen 2-8 minutes after Cr application - at what time points were the data shown in Figure 7E-J collected? Is the Cr group shown in Figure 7F significantly different from the control group/wash?
The responders were defined by three criteria: (1) When Cr was applied, the rheobase was increased as compared to both control and wash conditions. (2) The number of total evoked spikes was decreased during Cr application than both control and wash. (3) The number of total evoked spikes was decreased at least by 10% than control or wash.
For all the individual responders, when Cr was applied, the rheobase was increased (Figure 7E and 7F). While in individual non-responders, the rheobase was either identical to both control and wash (n=19/35), identical to either control or wash (n=11/35), between control and wash (n=2/35) or smaller than both control and wash (n=3/35) following Cr application. Thus, the responders and non-responders were separatable. When the rheobase data were pulled together, many points were overlapped, so we did not pull the data here.
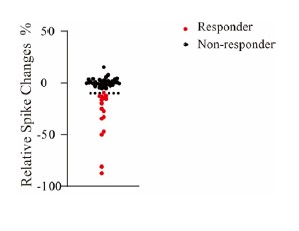
As suggested, we pulled the data of the ratio of spike changes in response to 100 μM Cr application for all neurons together (Author response image 4). Evoked spikes of non-responders were typically (34/35) changed in the range of -10% to 10%.
Author response image 4.
Relative changes of total evoked spikes in response to 100 μM Cr. Responders are represented by red dots and non-responders by black dots. Dashed black line indicates 10%. Relative change = (Cr-(Control +wash)/2)/((Control +wash)/2)*100%.
In Figure 7E-J, we collected data at time points when the maximal response was reached. The Cr group shown in Figure 7F was indeed significantly different from the control group/wash (p<0.05, paired t test, for data points collected under 75-500 pA current injection).
6) Indirect effects: The phenotypes could be partially caused by indirect effects of perturbing the Cr/PCr/CK system, which is known to play essential roles in ATP regeneration, Ca2+ homeostasis, neurotransmission, intracellular signaling systems, axonal and dendritic transport... Similarly, high GAMT levels were reported for astrocytes (e.g., Schmidt et al. 2004; doi: 10.1093/hmg/ddh112), and changes in astrocytic Cr may underlie the phenotypes. Cr has been also reported to be an osmolyte: a hyperosmotic shock of astrocytes induced an increase in Cr uptake, suggesting that Cr can work as a compensatory osmolyte (Alfieri et al. 2006; doi: 10.1113/jphysiol.2006.115006). Potential indirect effects are also consistent with a trend towards decreased KCl-induced GABA (and Glutamate) release in SLC6A8-/Y (Figure 5C). These indirect effects may in part explain the phenotypes seen after perturbing Agat, SLC6A8, and should be thoroughly discussed.
We discussed the possibility of creatine/phosphocreatine as non-transmitters in discussion part. We added the possibility of astrocytic Cr in discussion part. KCl-induced GABA (and Glutamate) release in SLC6A8-/Y (Figure 5C) was not significant.
7) As stated by the authors, there is some evidence that Cr may act as a co-transmitter for GABAA receptors (although only at high concentrations). Would a GABAA blocker decrease the fraction of cells with decreased excitability after Cr exposure?
We performed another experiment in CA1 pyramidal neurons in hippocampus showing that Cr at 100 μM did not change GABAergic neurotransmission (n=8, Author response image 5). Inhibitory postsynaptic currents (IPSCs) recorded in the presence of glutamate receptor blockers (10 μM APV and 10 μM CNQX) were not changed by 100 μM creatine in hippocampal CA1 pyramidal neurons (Bgroup data of IPSC frequency (B) and amplitude (C) averaged in 1 min duration). These did not support Cr activation of GABAA receptors.
Author response image 5.
IPSCs recorded in in hippocampal CA1 pyramidal neurons. (A) representative raw traces before (Control), during (Creatine) and after (Wash) the application of 100 μM creatine. (B&C) group data of IPSC frequency (B) and amplitude (C) averaged in 1 min duration.
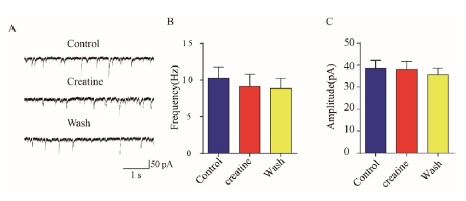
8) The statement "Our results have also satisfied the criteria of Purves et al. 67,68, because the presence of postsynaptic receptors can be inferred by postsynaptic responses." (l.568) is not supported by the data and should be removed.
We have deleted this sentence, though what could mediate postsynaptic responses other than receptors?
Reviewer #3 (Public Review):
SUMMARY:
The manuscript by Bian et al. promotes the idea that creatine is a new neurotransmitter. The authors conduct an impressive combination of mass spectrometry (Fig. 1), genetics (Figs. 2, 3, 6), biochemistry (Figs. 2, 3, 8), immunostaining (Fig. 4), electrophysiology (Figs. 5, 6, 7), and EM (Fig. 8) in order to offer support for the hypothesis that creatine is a CNS neurotransmitter.
We thank the reviewer for the summary.
STRENGTHS:
There are many strengths to this study.
• The combinatorial approach is a strength. There is no shortage of data in this study.
• The careful consideration of specific criteria that creatine would need to meet in order to be considered a neurotransmitter is a strength.
• The comparison studies that the authors have done in parallel with classical neurotransmitters are helpful.
• Demonstration that creatine has inhibitory effects is another strength.
• The new genetic mutations for Slc6a8 and AGAT are strengths and potentially incredibly helpful for downstream work.
WEAKNESSES:
• Some data are indirect. Even though Slc6a8 and AGAT are helpful sentinels for the presence of creatine, they are not creatine themselves. Therefore, the conclusions that are drawn should be circumspect.
SLC6A8 and AGAT mutants are not essential for Cr’s role as a neurotransmitter.
• Regarding Slc6a8, it seems to work only as a reuptake transporter - not as a transporter into SVs. Therefore, we do not know what the transporter is.
Indeed, SLC6A8 is only a transporter on the cytoplasmic membrane, not a transporter on synaptic vesicles. We have shown biochemistry here, and we have unpublished data that showed other SLCs on SVs, which did not include SLC6A8.
• Puzzlingly, Slc6a8 and AGAT are in different cells, setting up the complicated model that creatine is created in one cell type and then processed as a neurotransmitter in another.
• No candidate receptor for creatine has been identified postsynaptically.
• Because no candidate receptor has been identified, is it possible that creatine is exerting its effects indirectly through other inhibitory receptors (e.g., GABAergic Rs)?
As shown in our response to Question 7 of Reviewer 2, Cr did not exert its effects through inhibitory GABAA receptors.
• More broadly, what are the other possibilities for roles of creatine that would explain these observations other than it being a neurotransmitter? Could it simply be a modifier that exists in the SVs (lots of molecules exist in SVs)?
We discussed the possibility of a non-transmitter role for creatine/phosphocreatine in discussion part.
• The biochemical studies are helpful in terms of comparing relevant molecules (e.g., Figs. 8 and S1), but the images of the westerns are all so fuzzy that there are questions about processing and the accuracy of the quantification.
Multiple members (>4) have carried out SV purifications repeatedly over the last decade in our group, we are highly confident of SV purifications presented in Figs. 8 and S1.
There are several criteria that define a neurotransmitter. The authors nicely delineated many criteria in their discussion, but it is worth it for readers to do the same with their own understanding of the data.
By this reviewer's understanding (and the Purves' textbook definition) a neurotransmitter: 1) must be present within the presynaptic neuron and stored in vesicles; 2) must be released by depolarization of the presynaptic terminal; 3) must require Ca2+ influx upon depolarization prior to release; 4) must bind specific receptors present on the postsynaptic cell; 5) exogenous transmitter can mimic presynaptic release; 6) there exists a mechanism of removal of the neurotransmitter from the synaptic cleft.
6 criteria seem to be only required by the reviewer. As discussed in our Discussion part, Purves’ textbook did not list 6 criteria but only three criteria, “the substance must be present within the presynaptic neuron; the substance must be released in response to presynaptic depolarization, and the release must be Ca2+ dependent; specific receptors for the substance be present on the postsynaptic cell” (Purves et al., 2001, 2016).
Kandel et al. (2013, 2021) listed 4 criteria for a neurotransmitter: “it is synthesized in the presynaptic neuron; it is present within vesicles and is released in amounts sufficient to exert a defined action on the postsynaptic neuron or effector organ; when administered exogenously in reasonable concentrations it mimics the action of the endogenous transmitter; a specific mechanism usually exists for removing the substance from the synaptic cleft”.
While we agree that any neuroscientist can have his/her own criteria, it is more reasonable to accept the textbooks that have been widely read for decades.
For a paper to claim that the work has identified a new neurotransmitter, several of these criteria would be met - and the paper would acknowledge in the discussion which ones have not been met. For this particular paper, this reviewer finds that condition 1 is clearly met.
Conditions 2 and 3 seem to be met by electrophysiology, but there are caveats here. High KCl stimulation is a blunt instrument that will depolarize absolutely everything in the prep all at once and could result in any number of non-specific biological reactions as a result of K+ rushing into all neurons in the prep. Moreover, the results in 0 Ca2+ are puzzling. For creatine (and for the other neurotransmitters), why is there such a massive uptick in release, even when the extracellular saline is devoid of calcium?
To avoid the disadvantage of high KCl stimulation, we performed optogenetic experiments recently, with encouraging preliminary data. We do not know the source of Ca2+-independent release of Cr and neurotransmitters, though astrocytes are a possibility.
Condition 4 is not discussed in detail at all. In the discussion, the authors elide the criterion of receptors specified by Purves by inferring that the existence of postsynaptic responses implies the existence of receptors. True, but does it specifically imply the existence of creatinergic receptors? This reviewer does not think that is necessarily the case. The authors should be appropriately circumspect and consider other modes of inhibition that are induced by activation or potentiation of other receptors (e.g., GABAergic or glycinergic).
Our results did not support Cr stimulation of inhibitory GABAA receptors (see our answer to Point 7 in of Reviewer 2).
Condition 5 may be met, because the authors applied exogenous creatine and observed inhibition (Fig. 7). However, this is tough to know without understanding the effects of endogenous release of creatine. if they were to test if the absence of creatine caused excess excitation (at putative creatinergic synapses), then that would be supportive of the same.
After the submission of our manuscript, we found a recent paper showing that slc6a8 knockout led to increased excitation in pyramidal neurons in the prefrontal cortex (PFC), with increased firing frequency (Ghirardini et al., 2023). Because we have shown that slc6a8 knockout would cause decrease of Cr in SVs (Figure 2 in our paper), this result provide the evidence described as Condition 5 of this reviewer: that decrease of Cr in SVs led to excess excitation.
For condition 6, the authors made a great effort with Slc6a8. This is a very tough criterion to understand for many synapses and neurotransmitters.
In terms of fundamental neuroscience, the story would be impactful if proven correct. There are certainly more neurotransmitters out there than currently identified.
The impact as framed by the authors in the abstract and introduction for intellectual disability is uncertain (forming a "new basis for ID pathogenesis") and it seems quite speculative beyond the data in this paper.
We deleted this sentence.
Reviewer #1 (Recommendations For The Authors):
To strengthen the manuscript, I suggest the following considerations:
1) The key missing evidence to my mind is a receptor - but this is clearly outside the scope of this paper. Yet, I am surprised that in the list of criteria for neurotransmitters in general there is no mention of a receptor. Furthermore, many receptors have been identified through receptor agonists or antagonists, like neurotoxins or drugs. The authors do not talk about putative receptors except for a sentence in the discussion where they speculate on a GPCR. There are numerous GPCR agonists and antagonists, which may be a long-shot, or something even a bit more designed based on knowledge about creatine? I do not think the publication of this manuscript should have been made dependent on finding an agonist or antagonist of this specific unknown receptor (if it exists), but it would be good to have at least some leads on this from the authors what has been tried or what could be done? How about a manipulation of G-protein-coupled signal transduction to support the idea that there IS such a GPCR? There may be a real opportunity here to test existing compounds in wild type, the slc6a8 and agat mutants.
We will keep trying, but accept the reality that Rome was not built in a single day and that no transmitter was proven by one single paper.
A key new puzzle piece of evidence is the identification of creatine in synaptic vesicles. The experiment relies heavily on the purity of the SV fraction using the anti-synaptophysin antibody. I am quite sure that these preparations contain many other compartments - and of course a big mix of synaptic (and other) vesicles. Would it be possible to purify with an anti slc6a8 antibody?
Sl6a8 is expressed in on the plasma membrane of neurons7-9, instead of synaptic vesicles. Consistent with this, we could not detect obvious Slc6a8-HA signal in our starting material (Lane S in Author response image 6) that was used for SV purification. We have tried to purify SVs by HA antibody in Slc6a8 mice and SV markers could not be detected.
Author response image 6.
Lack of Slc6a8-HA in our starting material. In Slc6a8-HA knock-in mice, the HA signal was present in whole brain homogenate (H), but not obvious in supernatants (S) following 35000 × centrifugation. In contrast, SV marker Syp was present in supernatants.

The K stimulation protocol in slices is relatively crude, as all neurons in the slice get simultaneously overactivated - and some of the effects on Ca-dependent release are not very strong (e.g. the 35 neurons that were not responsive to creatine at all). A primary neuronal culture of neurons that respond to creatine would strengthen this section.
To avoid the disadvantage of K stimulation, we also performed optogenetic experiments recently and obtained encouraging preliminary results.
Reviewer #2 (Recommendations For The Authors):
1) The different sections of the manuscript are not separated by headers.
2) The beginning of the results section either does not reference the underlying literature or refers to unpublished data.
We have kept a bit background in the beginning of the Results section.
3) The text contains many opinions and historical information that are not required (e.g., "It has never been easy to discover a new neurotransmitter, especially one in the central nervous system (CNS). We have been searching for new neurotransmitters for 12 years."; l. 17).
This is a field that has been dormant for decades and such background introductions are helpful for at least some readers.
4) Almeida et al. (2008; doi: 10.1002/syn.20280) provided evidence for electrical activity-, and Ca2+-dependent Cr release from rat brain slices. This paper should be introduced in the introduction.
Those were stand-alone papers which have not been reproduced or paid attention to. Our introduction part did not mention them because our research did not begin with those papers. We had no idea that those papers existed when we began. We started with SV purification and only read those papers afterwards. Thus, they were not necessary background to our paper but can be discussed after we discovered Cr in SVs.
5) Fig. 7: A Y-scale for the stimulation protocol is missing.
Revised.
Reviewer #3 (Recommendations For The Authors):
The main suggestion by this reviewer (beyond the details in the public review) is to consider the full spectrum of biology that is consistent with these results. By my reading, creatine could be a neurotransmitter, but other possibilities also exist, and the authors need to highlight those too.
We have discussed non-transmitter role in the discussion.
References
Ghirardini, E., G. Sagona, A. Marquez-Galera, F. Calugi, C. M. Navarron, F. Cacciante, S. Chen, F. Di Vetta, L. Dada, R. Mazziotti, L. Lupori, E. Putignano, P. Baldi, J. P. Lopez-Atalaya, T. Pizzorusso, and L. Baroncelli. 2023. Cell-specific vulnerability to metabolic failure: the crucial role of parvalbumin expressing neurons in creatine transporter deficiency. Acta Neuropathol Commun, 11: 34. doi: 10.1186/s40478-023-01533-w.
Lowe, M. T., Faull, R. L., Christie, D. L. & Waldvogel, H. J. Distribution of the creatine transporter throughout the human brain reveals a spectrum of creatine transporter immunoreactivity. J Comp Neurol 523, 699-725 (2015). https://doi.org:10.1002/cne.23667
Mak, C. S. et al. Immunohistochemical localisation of the creatine transporter in the rat brain. Neuroscience 163, 571-585 (2009). https://doi.org:10.1016/j.neuroscience.2009.06.065.
Molchanova, S. M., Oja, S. S. & Saransaari, P. Mechanisms of enhanced taurine release under Ca2+ depletion. Neurochem Int 47, 343-349 (2005). https://doi.org:10.1016/j.neuint.2005.04.027
Philibert, R. A., Rogers, K. L. & Dutton, G. R. K+-evoked taurine efflux from cerebellar astrocytes: on the roles of Ca2+ and Na+. Neurochem Res 14, 43-48 (1989). https://doi.org:10.1007/BF00969756
Rosko, L. M. et al. Cerebral Creatine Deficiency Affects the Timing of Oligodendrocyte Myelination. J Neurosci 43, 1143-1153 (2023). https://doi.org:10.1523/JNEUROSCI.2120-21.2022
Saransaari, P. & Oja, S. S. Characteristics of taurine release in slices from adult and developing mouse brain stem. Amino Acids 31, 35-43 (2006). https://doi.org:10.1007/s00726-006-0290-5
Schmidt, A. et al. Severely altered guanidino compound levels, disturbed body weight homeostasis and impaired fertility in a mouse model of guanidinoacetate N-methyltransferase (GAMT) deficiency. Hum Mol Genet 13, 905-921 (2004). https://doi.org:10.1093/hmg/ddh112
Speer, O. et al. Creatine transporters: a reappraisal. Mol Cell Biochem 256-257, 407-424 (2004). https://doi.org:10.1023/b:mcbi.0000009886.98508.e7
Takuma, K. et al. Ca2+ depletion facilitates taurine release in cultured rat astrocytes. Jpn J Pharmacol 72, 75-78 (1996). https://doi.org:10.1254/jjp.72.75



