Abstract
Dystrophin is a critical interacting protein of Nav1.5 that determines its membrane anchoring in cardiomyocytes. Long noncoding RNAs (lncRNAs) are involved in the regulation of cardiac ion channels, while their influence on sodium channel remains unexplored. Our preliminary data showed that lncRNA-Dachshund homolog 1 (lncDACH1) can bind to dystrophin, which drove us to investigate if lncDACH1 can regulate sodium channel by interfering with dystrophin. Western blot and immunofluorescent staining showed that cardiomyocyte-specific transgenic overexpression of lncDACH1(lncDACH1-TG) reduced the membrane distribution of dystrophin and Nav1.5 in cardiomyocytes. Meanwhile, peak INa were reduced in the hearts of lncDACH1-TG mice than wild-type (WT) controls. The opposite data of western blot, immunofluorescent staining and patch clamp were collected from lncDACH1 cardiomyocyte conditional knockout (lncDACH1-cKO) mice. Moreover, increased ventricular arrhythmia susceptibility was observed in lncDACH1-TG mice in vivo and ex vivo. The conservative fragment of lncDACH1 inhibited membrane distribution of dystrophin and Nav1.5, and promoted the inducibility of ventricular arrhythmia. Strikingly, activation of dystrophin transcription by dCas9-SAM system in lncDACH1-TG mice rescued the impaired membrane distribution of dystrophin and Nav1.5, and prevented the occurrence of ventricular arrhythmia. Furthermore, lncDACH1 was increased in transaortic constriction (TAC) induced failing hearts, which promoted the inducibility of ventricular arrhythmia. And the expression of lncDACH1 is regulated by hydroxyacyl-CoA dehydrogenase subunit beta (hadhb), which binds to lncDACH1 and decreases its stability. The human homologue of lncDACH1 inhibited the membrane distribution of Nav1.5 in human iPS-differentiated cardiomyocytes. The findings provide novel insights into the mechanism of Nav1.5 membrane targeting and the development of ventricular arrhythmias.
Introduction
The voltage gated sodium channel mediates the 0 phase depolarizing inward sodium currents of cardiomyocytes1. The expression and function of sodium channel is regulated at multiple levels encompassing gene mutation, post-transcriptional modification, post-translational modification and protein trafficking etc1,2. The disruption of either process is arrhythmogenic and occasionally causes sudden death2,3.
The membrane targeting and localization of pore-forming subunit of sodium channel Nav1.5 was regulated by several interacting proteins such as ankyrin-G, MOG1, syntrophin and dystrophin etc2,4. Dystrophin is a cytoplasmic protein that is encoded by duchenne muscular dystrophy (DMD) gene5. It distributes mainly on the cellular membrane of skeletal muscle cells and cardiomyocytes, and acts as a scaffold for Nav1.56. In cardiomyocytes, dystrophin controls the expression and membrane anchoring of Nav1.5. Gavillet et al. showed that knockout of dystrophin in cardiomyocytes reduced peak sodium current, Nav1.5 protein expression and conduction velocity in mice 6. Subsequently, they confirmed that knockout of dystrophin inhibits membrane distribution of Nav1.5 due to the disruption of dystrophin-syntrophin complex7.
Long noncoding RNAs (LncRNAs) are a new class of RNAs that are more than 200 nts long and possess little protein coding property.8 LncRNAs have been shown to regulate multiple biological processes and participate in the pathogenesis of various diseases including cardiac diseases9. LncRNAs were shown to regulate cardiac electrophysiological property by altering the function of different ion channels. For example, the increased expression of lncRNA-Kcna2as in heart failure reduced Iks and prolonged action potential duration (APD)10. LncRNA-MALAT1 enhanced arrhythmia susceptibility by suppressing Ito and prolonging APD11. In a previous study, we found that lncRNA-CCRR (cardiac conduction regulatory) interacts with connexin-43 interacting protein 85(CIP85) to promote connexin-43 membrane distribution and improve the impaired cardiac conduction of failing hearts12. However, to date, it remains unknown whether and how lncRNA regulates sodium channel.
LncDACH1 is an intronic lncRNA located on the first intron of DACH1 gene13. We previously showed that lncDACH1 impairs cardiac function by promoting the degradation of sarco-endoplasmic reticulum ATPase 2a (SERCA2a), and exacerbates cardiac ischemia injury by inhibiting Yes-associated protein 1(YAP1) mediated proliferation of neonatal cardiomyocytes13,14. During analyzing the interacting proteins of lncDACH1 identified by mass spectrometry, we found dystrophin that drove us to hypothesize that lncDACH1 may be a critical regulator of sodium channel Nav1.5.
Therefore, in this study we explored whether lncDACH1 regulates Nav1.5 by interacting with dystrophin. We found that lncDACH1 inhibited the membrane trafficking of Nav1.5 by binding to dystrophin, which led to reduced sodium current and increased ventricular arrhythmia susceptibility. The study highlights a novel mechanism for the regulation of sodium channel trafficking, and reveals a potential therapeutic target for sodium channel dysfunction related cardiac arrhythmias.
Results
LncDACH1 binds to dystrophin and reduces Nav1.5 membrane distribution in cardiomyocytes of lncDACH1 transgenic mice
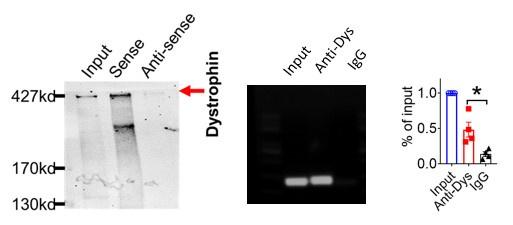
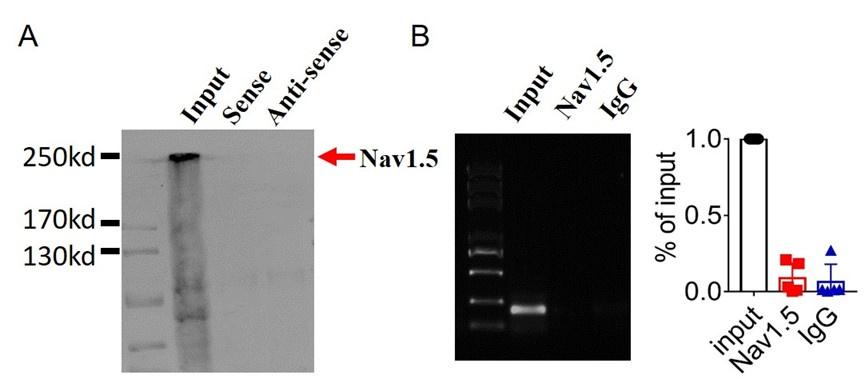
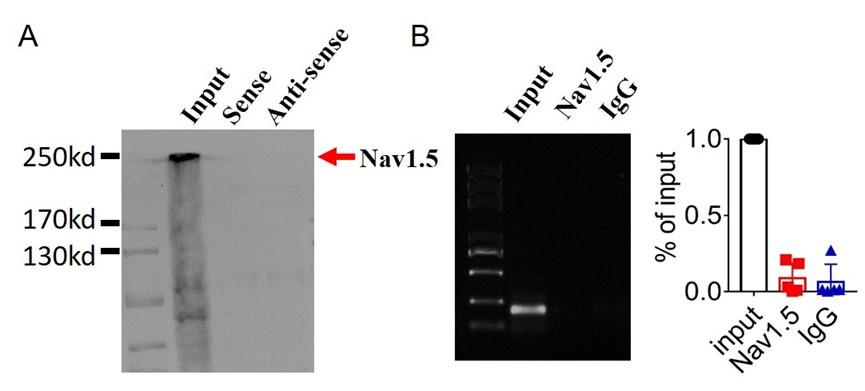
We first validated the binding between lncDACH1 and dystrophin. The RNA pulldown plus immunoblot assay confirmed that lncDACH1 can successfully pulldown dystrophin (Fig. 1a). These data indicated that lncDACH does not interact with Nav1.5 directly. Consistently, the RNA immunoprecipitation (RIP) study showed that the antibody for dystrophin precipitated lncDACH1, while the negative control IgG did not (Fig. 1b). Conversely, lncDACH1 failed to pulldown Nav1.5 and anti-Nav1.5 did not precipitate lncDACH1 (Supplementary Fig. 1).

Binding of lncDACH1 to dystrophin and the effects of cardiomyocyte-specific transgenic overexpression of lncDACH1 (lncDACH1-TG) on the expression and function of sodium channel.
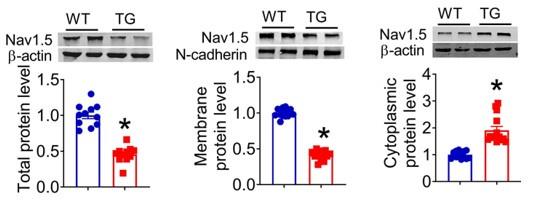
a, Blotting of dystrophin pulled-down by lncDACH1. b, LncDACH1 precipitated by the antibody of dystrophin. N=4. * P<0.05 vs IgG by one-way ANOVA, followed by Tukey’s post-hoc analysis. c,d, The total, membrane and cytoplasm levels of dystrophin and Nav1.5 by Western blot. N-cadherin is the loading control for membrane extracts. N=10-11 for total protein; N= 7-14 for membrane protein; N= 8-14 for cytoplasm protein. *P<0.05 vs WT group. P-values were determined by unpaired t test. e, Distribution of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes. f, The mRNA levels of dystrophin and SCN5A. N=8-11. g, Peak INa currents, I-V curve and kinetics of INa. N=9-15 cells from 3 mice. *P<0.05 vs WT group. h, Conduction velocity of perfused hearts by optical mapping recordings. N=7. *P<0.05 vs WT group. P-values were determined by unpaired t test.
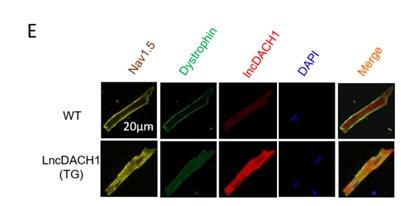
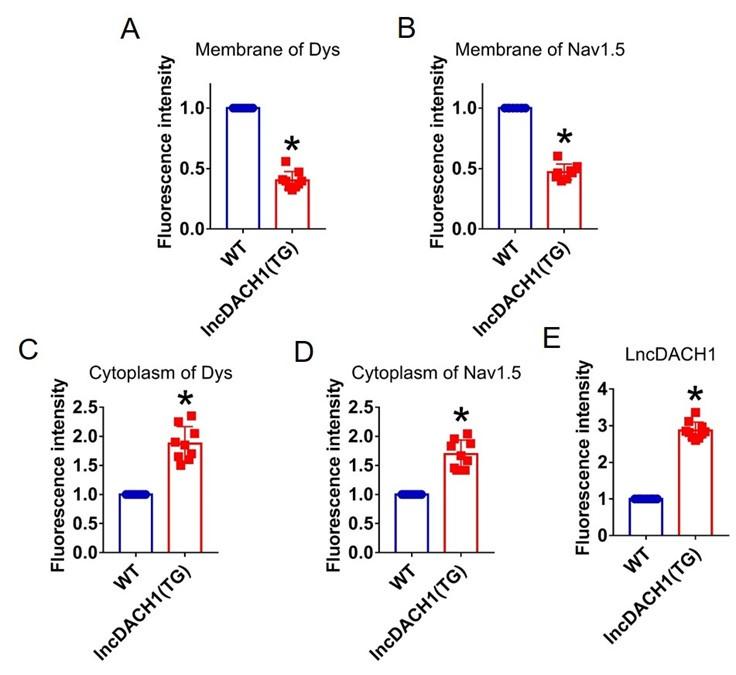
We next explored the influence of lncDACH1 on cellular distribution of dystrophin. The western blot data showed that the total protein of dystrophin did not change, while the membrane fraction was reduced, and the cytoplasmic fraction was increased in the hearts of lncDACH1-TG mice than wild-type (WT) controls (Fig. 1c). Accordingly, the membrane and total protein levels of Nav1.5 were reduced, while cytoplasmic Nav1.5 increased in the hearts of lncDACH1-TG mice than WT controls (Fig. 1d). The reduced membrane distribution of dystrophin and Nav1.5 in the cardiomyocytes of lncDACH1-Tg mice was further confirmed by immunofluorescent staining (Fig. 1e and Supplementary Fig. 2). The mRNA levels of dystrophin and SCN5A did not change (Fig. 1f). We then evaluated the functional change of sodium channel. Consistent with reduction of membrane Nav1.5, the peak INa was significantly decreased in the ventricular myocytes of lncDACH1-TG mice than WT controls, while the kinetics of INa (activation, inactivation, and recovery) did not change (Fig. 1g). Meanwhile, the conduction velocity was slower in the hearts of lncDACH1-TG than WT mice (Fig. 1h).
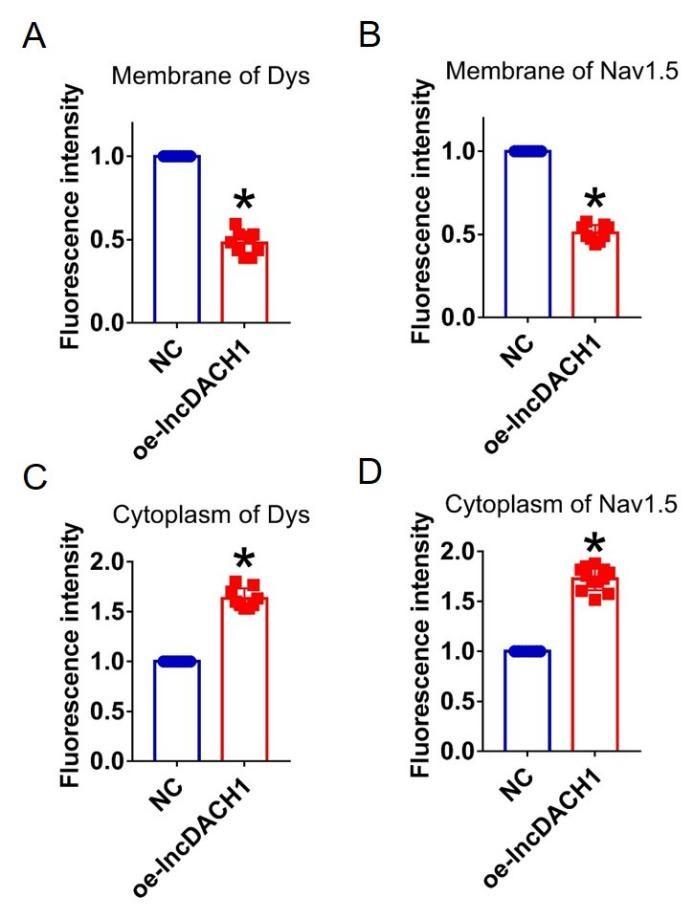
We then applied lncDACH1 adenovirus to cultured neonatal cardiomyocytes to confirm the regulation of lncDACH1 on sodium channel in vitro. Infection of adenovirus carrying lncDACH1 significantly upregulated the level of lncDACH1 (Fig. 2a) and remarkably inhibited peak INa with no change in kinetics (Fig. 2b, c). The western blot data showed that the total protein of dystrophin did not change, while the membrane fraction was reduced, and the cytoplasmic fraction increased in neonatal cardiomyocytes with lncDACH1 overexpression (Supplementary Fig. 3a). Consistently, the membrane protein level and total protein level of Nav1.5 were reduced, while cytoplasmic Nav1.5 increased in neonatal cardiomyocytes with lncDACH1 overexpression (Supplementary Fig. 3b). The membrane distribution of dystrophin and Nav1.5 was remarkably reduced by overexpression of lncDACH1 as indicated by immunofluorescent staining (Fig. 2d and Supplementary Fig. 4). The mRNA levels of dystrophin and SCN5A were not altered by lncDACH1 overexpression (Fig. 2e).

Effects of lncDACH1 overexpression on sodium channel expression and function in cultured neonatal cardiomyocytes.
a, Verification of the expression of lncDACH1 after transfection of adenovirus carrying lncDACH1. N=7-8 from 3 independent cultures. *P<0.05 vs NC (negative control, empty plasmid). P-values were determined by unpaired t test. b, c, Peak INa currents, I-V curves and kinetics of INa. N=12-16 cells from 3 independent cultures. *P<0.05 vs NC. d, Distribution of Nav1.5 and dystrophin by immunofluorescent staining. e, The mRNA levels of dystrophin and SCN5A. N=8-10 from 3 independent cultures.
Inhibition of lncDACH1 in cardiomyocytes increased membrane Nav1.5 distribution
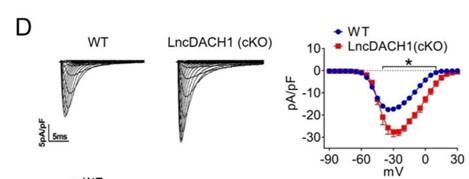
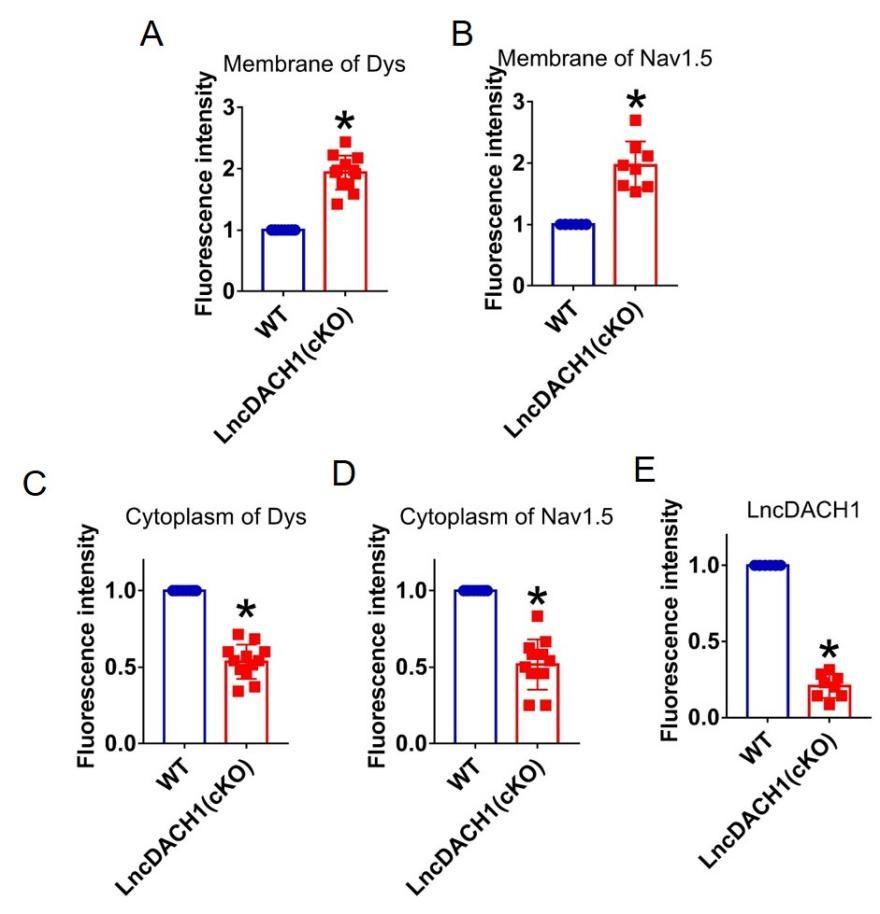
We then employed lncDACH1 cardiomyocyte conditional knockout(lncDACH1-cKO) mice to examine the regulation of lncDACH1 on dystrophin and Nav1.5. The western blot data showed that membrane distribution of dystrophin was increased in the hearts of lncDACH1-cKO mice than WT controls, while the total dystrophin protein and dystrophin mRNA did not change (Fig. 3a). Consistently, the membrane and total level of Nav1.5 was increased in the hearts of lncDACH1-cKO mice than WT controls, with no change on SCN5A mRNA (Fig. 3b). The change on dystrophin and Nav1.5 membrane distribution was further validated by immunofluorescent staining (Fig. 3c and Supplementary Fig. 5). Meanwhile, the peak INa was larger in cardiomyocytes of lncDACH1-cKO mice than WT controls, while the kinetics of INa (activation, inactivation, and recovery) did not change (Fig. 3d). Consistent with the increase of peak INa, the conduction velocity in the hearts of lncDACH1-cKO mice was faster than WT controls (Fig. 3e).

Conditional knockout of lncDACH1(lncDACH1-cKO) in cardiomyocytes increased peak sodium current, membrane Nav1.5 expression.
a, The total, membrane and cytoplasm levels of dystrophin by Western blot and dystrophin mRNA by qRT-PCR. N-cadherin is the loading control for membrane extracts. N=10 for total protein; N= 10 for membrane protein; N= 15 for cytoplasm protein. *P<0.05 vs WT group. P-values were determined by unpaired t test. b, The total, membrane and cytoplasm levels of Nav1.5 by Western blot and SCN5A mRNA by qRT-PCR. N-cadherin is the loading control for membrane extracts. N=10 for total protein; N= 8 for membrane protein; N= 8 for cytoplasm protein. *P<0.05 vs WT group. P-values were determined by unpaired t test. c, distribution of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes. d, Peak INa currents, I-V curve and kinetics of INa. N=10-15 cells; N=3 mice of WT; N=4 mice of LncDACH1(cKO). *P<0.05 vs WT group. e, Conduction velocity of perfused hearts by optical mapping recordings. N=7 and 5. * P<0.05 vs WT group. P-values were determined by unpaired t test.
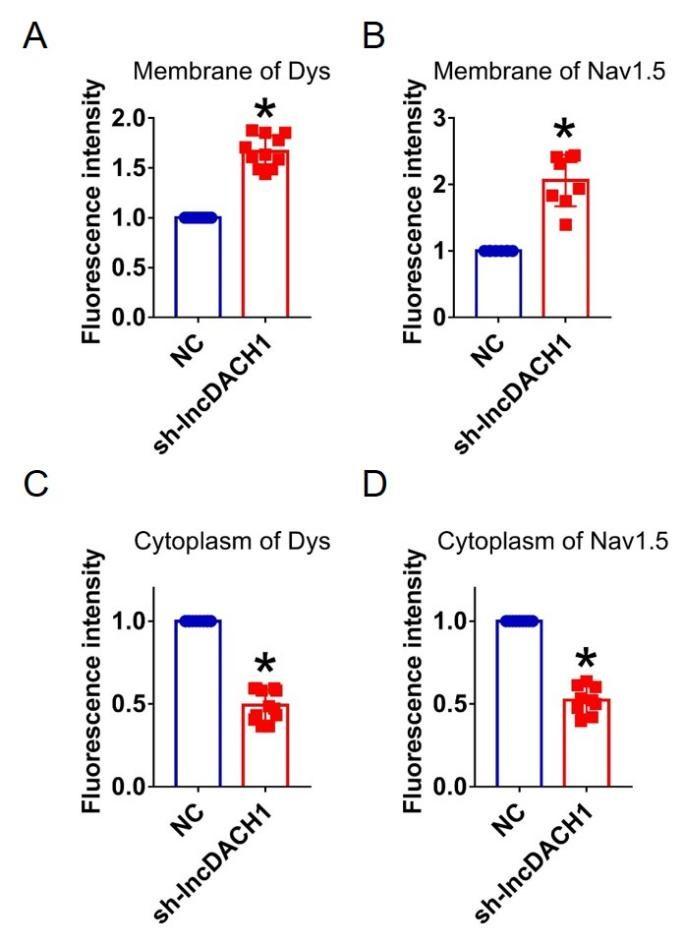
We further confirmed effects of lncDACH1 knockdown with its shRNA on sodium channel in cultured neonatal cardiomyocytes in vitro. Infection of adenovirus carrying shRNA for lncDACH1 significantly reduced the level of lncDACH1 (Fig. 4a). The patch-clamp recordings showed that knockdown of lncDACH1 significantly increased the current density of peak INa with no change on channel kinetics (Fig. 4b, c). The western blot data showed that the total protein of dystrophin did not change, while the membrane fraction was increased, and the cytoplasmic fraction reduced after lncDACH1 knockdown (Supplementary Fig. 6a). Consistently, the membrane and total protein levels of Nav1.5 were increased, while cytoplasmic Nav1.5 reduced after lncDACH1 knockdown (Supplementary Fig. 6b). Membrane distribution of dystrophin and Nav1.5 were both increased after knockdown of lncDACH1 as indicated by immunofluorescent staining (Fig. 4d and Supplementary Fig. 7). The mRNA levels of dystrophin and SCN5A were not altered by lncDACH1 knockdown (Fig. 4e).

Effects of lncDACH1 knockdown on sodium channel expression and function in cultured neonatal cardiomyocytes.
a, Verification of the expression of lncDACH1 after infection of adenovirus carrying lncDACH1 shRNA. N=15 from 3 independent cultures. *P<0.05 vs NC (negative control, empty plasmids). P-values were determined by unpaired t test. b,c, Peak INa currents, I-V curve and kinetics of INa. N=9-15 from 3 independent cultures. *P<0.05 vs NC. d, Distribution of Nav1.5 and dystrophin by immunofluorescent staining. e, The mRNA levels of dystrophin and SCN5A. N=11-15 from 3 independent cultures.
In addition, neither knockout nor transgenic overexpression of lncDACH1 changed the expression of DACH1 and its neighbor genes (KLHL1, BORA, MZt1, DIS3) (Supplementary Fig. 8).
Transgenic overexpression of lncDACH1 is arrhythmogenic in mice
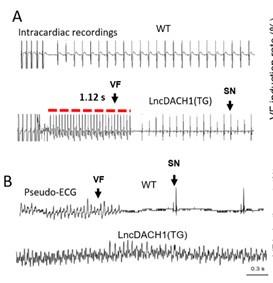
We next evaluated whether the inhibition of Nav1.5 by lncDACH1 is arrhythmogenic in lncDACH1-TG mice. Electrical pacing technique was employed to evaluate the arrhythmia susceptibility of intact hearts in vivo and isolated hearts ex vivo. The in vivo study showed that programmed pacing induced more ventricular arrhythmia in lncDACH1-TG mice that WT controls. Both induction rate and episodes of ventricular arrhythmia were higher in lncDACH1-TG mice (Fig. 5a). In consistent with the results of in vivo., the ex vivo electrical pacing study demonstrated that ventricular arrhythmia was more frequently occurred in lncDACH1-TG mice (Fig. 5b). The optimal mapping study revealed that there are more breaking points in the perfused heart of lncDACH1-TG mice than WT controls (Fig. 5c, d). Conversely, no ventricular arrhythmia was induced in the hearts of lncDACH1-cKO mice (Fig. 5e).

Increased arrhythmia susceptibility in lncDACH1-TG mice.
a, Ventricular fibrillation (VF) induced by S1S2 pacing in intact mice. The red lines in the ECG traces indicate VF duration. N=10-16. SN: sinus rhythm b, Ventricular fibrillation (VF) induced byS1S1 pacing of perfused hearts. c, Break points during VT of WT and lncDACH1-TG mice by optical mapping. Consecutive phase maps sampled at 10-ms interval during VF from WT and TG mice. Phase singularities (wavebreaks) are indicated by phase maps. Upper panels showed corresponding optical recording of VF at asterisk site. N=8. d, The number of phase singularities and dominant frequency of WT and TG mice. e, Ventricular fibrillation (VF) induced by S1S1 pacing in perfused hearts. N=5-7 mice.
The conservative fragment of lncDACH1 reduced peak sodium current and promoted ventricular arrhythmia
The sequence blasting data showed that the fragment of lncDACH1 from 835 to 2085 nts is conservative between human and mouse. We then cut lncDACH1 into different fragments (Fig. 6a) to evaluate the sequence that is responsible for the binding with dystrophin. The data showed that only fragments containing the conserved sequence, fragments a and b, can pulldown dystrophin, indicating that the conserved sequence may be the functional region of lncDACH1 (Fig. 6a).

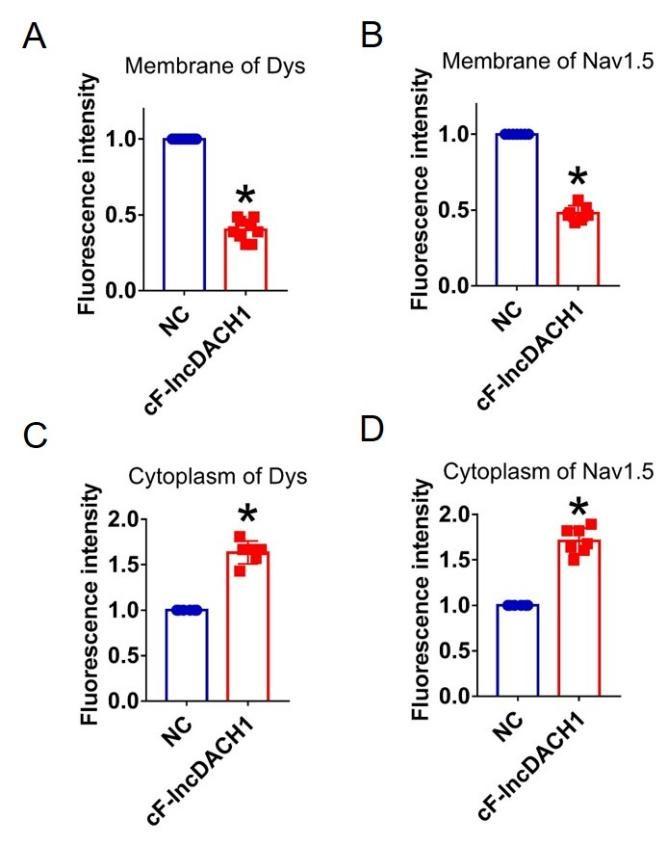
The conserved fragment of lncDACH1(cF-lncDACH1) inhibited sodium channel function in mice.
a, Pulldown of dystrophin by fragments of lncDACH1 as indicated. b, Verification of the expression of cF-lncDACH1 after injection of adeno-virus carrying cF-lncDACH1. N=5. *P<0.05 vs NC (negative control, Adeno-virus carrying empty plasmid). P-values were determined by unpaired t test. c, The total, membrane and cytoplasm levels of dystrophin and Nav1.5 by Western blot. N-cadherin is the loading control for membrane extracts. N=11-12 for total protein; N= 10 for membrane protein; N= 5-11 for cytoplasm protein. *P<0.05 vs NC group. P-values were determined by unpaired t test. d, Distribution of dystrophin and Nav1.5 in isolated cardiomyocytes. e, Representative traces and I-V curve of peak INa currents. N=12-15 cells; N=3 mice of NC; N=4 mice of cF-lncDACH1. *P<0.05 vs NC group. f, Conduction velocity of perfused hearts by optical mapping recordings. N=9-10. *P<0.05 vs NC group. P-values were determined by unpaired t test. g, Break points during VT by optical mapping. h, Ventricular (VF) induced by S1S2 pacing in intact mice. The induction rate, average episodes and duration of ventricular (VF) were determined from ECG recordings. The red lines in the ECG traces indicate VF duration. N=11 for NC; N= 18 for cF-lncDACH1. *P<0.05 vs NC group.
We then examined the influence of the conserved sequence from 835 to 2085 nts (conserved fragment of lncDACH1, cF-lncDACH1) on cardiac sodium channel. The adenovirus carrying cF-lncDACH1 was constructed and administered to mice. The successful overexpression of cF-lncDACH1 was validated by qRT-PCR (Fig. 6b). Administration of cF-lncDACH1 reduced the membrane distribution, and increased cytoplasmic expression of both dystrophin and Nav1.5 as indicated by western blot and immunofluorescent data (Fig. 6c, d and Supplementary Fig. 9). The mRNA levels of dystrophin and Nav1.5 were not affected by cF-lncDACH1 (Supplementary Fig. 10a). Overexpression of cF-lncDACH1 reduced peak INa currents (Fig. 6e), and produced no influence on channel kinetics (Supplementary Fig. 10b). The optical mapping data showed that administration of cF-lncDACH1 reduced conduction velocity and increased break points of ventricular arrhythmias (Fig. 6f, g). The induction rate and episodes of ventricular tachycardia (VT) were higher in cF-lncDACH1 group than controls (Fig. 6h).
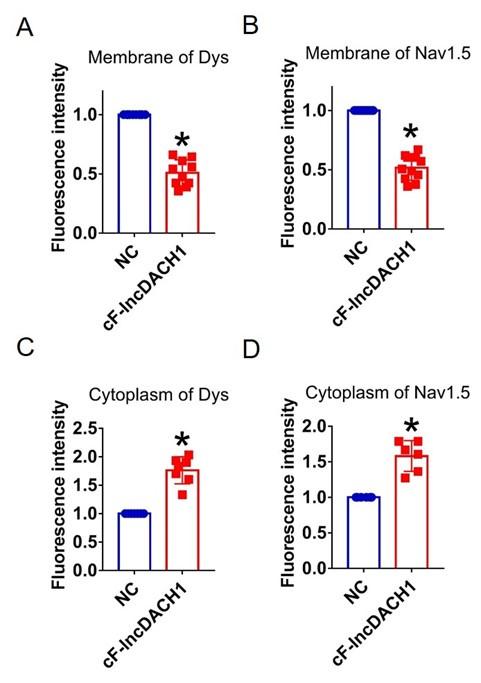
In cultured neonatal cardiomyocytes, overexpression of cF-lncDACH1 reduced peak INa with no change in kinetics, inhibited membrane distribution of dystrophin and Nav1.5, and produced no influence on the mRNA levels of dystrophin and SCN5A (Supplementary Fig.11, 12). The western blot data showed that the total protein of dystrophin did not change, while the membrane fraction was reduced, and the cytoplasmic fraction increased after overexpression of cF-lncDACH1 (Supplementary Fig. 13a). Consistently, the membrane and total protein levels of Nav1.5 were reduced, while cytoplasmic Nav1.5 increased after overexpression of cF-lncDACH1 (Supplementary Fig. 13b).
We also blasted the lncDACH1 in different species and found that lncDACH1 is conserved among sheep, pig, dog, rat, human and mouse (Supplementary Fig. 14).
Activation of dystrophin transcription by dCas9-SAM system prevented the reduction of sodium current in lncDACH1 transgenic mice
As lncDACH1 reduced Nav1.5 membrane targeting by interacting with dystrophin, we reasoned that overexpression of dystrophin may rescue the inhibition of Nav1.5 by lncDACH1. To test this notion, we constructed the AAV9 virus carrying dCas9-SAM system that can activate dystrophin transcription (AAV9-Dys-Act) to perform rescuing experiments on lncDACH1-TG mice (Supplementary Fig. 15). Tail vein injection of AAV9-Dys-Act virus significantly increased the mRNA level of dystrophin in the hearts of both WT and lncDACH1-TG mice (Fig. 7a). The western blot data showed that overexpression of dystrophin with AAV9-Dys-Act virus increased both total and membrane protein expression of dystrophin, and rescued the reduction of dystrophin expression in lnDACH1-TG mice (Fig. 7a). The mRNA level of SCN5A was not influenced by AAV9-Dys-Act virus (Fig. 7b). Strikingly, AAV9-Dys-Act virus administration restored total and membrane expression of Nav1.5 in lnDACH1-TG mice (Fig. 7b). In addition, AAV9-Dys-Act virus injection rescued the reduction of peak INa current in lnDACH1-TG mice (Fig. 7c). The kinetics of INa current did not change among groups (Fig. 7c). Activation of dystrophin transcription with AAV9-Dys-Act virus restored the conduction velocity in lncDACH1-TG mice (Fig. 7d). Both in vivo and ex vivo data indicated that activation of dystrophin transcription reduced the susceptibility to ventricular arrhythmia of lncDACH1-TG mice (Fig. 7e, f).

Activation of dystrophin transcription by AAV9 virus carrying dCas9-SAM system (AAV9-Dys-Act) rescued the remodeling of sodium channel in lncDACH1-TG mice.
a, The mRNA level of dystrophin by real-time PCR (N=12-17) and the total, membrane and cytoplasm protein levels of dystrophin by western blot. N-cadherin is the loading control for membrane extracts. N=7 for total protein; N= 8 for membrane protein; N= 8 for cytoplasm protein. *P<0.05 vs NC group. #P<0.05 vs TG group. NC, AAV9 virus carrying dCas9-SAM system with control sgRNA; Dys, AAV9 virus carrying dCas9-SAM system with sgRNA targeting dystrophin promoter. b, The mRNA level of Nav1.5 by real-time PCR (N=10-12) and the total, membrane and cytoplasm protein levels of Nav1.5 by western blot. N-cadherin is the loading control for membrane extracts. N=10 for total protein; N= 5 for membrane protein; N= 6 for cytoplasm protein. *P<0.05 vs NC group. #P<0.05 vs TG group. c, Representative traces, I-V curves and kinetics of peak INa currents. N=11-20 cells; N=3 mice of WT+NC; N=3 mice of WT+Dys; N=3 mice of TG+NC; N=4 mice of TG+Dys. *P<0.05 vs NC group. #P<0.05 vs TG group. d, Conduction velocity of perfused hearts by optical mapping recordings. N=6-9. * P<0.05 vs NC group. #P<0.05 vs TG group. P-values were determined by unpaired t test. e, Ventricular fibrillation (VF) induced by S1S1 pacing in perfused hearts. N= 7-10. f, Ventricular fibrillation (VF) induced by S1S2 pacing in intact mice. N= 10-13. The data are analyzed by one-way ANOVA followed by Tukey’s post-hoc analysis.
Hadhb binds to lncDACH1 and promotes its decay
Reduced Nav1.5 expression and reduction of peak INa in heart failure have been reported by multiple studies16–19. We therefore evaluated the contribution of lncDACH1 on sodium channel remodeling in transaortic constriction (TAC) induced heart failure model in mice. We found that lncDACH1 was increased in failing hearts than sham controls (Fig. 8a). Although lncDACH1 was upregulated in failing hearts, the mRNA of its host gene DACH1 did not change (Fig. 8b). This finding excluded the transcription related mechanism on lncDACH1 upregulation during heart failure. By analyzing the RNA Pulldown plus Mass Spectrometry data, we identified three potential interacting proteins of lncDACH1 that have been shown to regulate RNA stability. They are ANP32a (acidic leucine-rich nuclear phosphoprotein 32A), eIF4A1(eukaryotic initiation factor 4A1) and hydroxyacyl-CoA dehydrogenase subunit beta (hadhb). We therefore speculated that it may be the change of RNA stability that renders to the expression change of lncDACH1. We then tested whether these proteins can affect lncDACH1 level by knocking down their expression with siRNA. The data showed that knockdown of hadhb increased the expression of lncDACH1, while knockdown of ANP32a and eIF4A1 produced no influence (Fig. 8c). The influence of hadhb on lncDACH1 stability was further validated by the fact that knockdown of hadhb increased the decaying half-life of lncDACH1(Fig. 8d). Furthermore, the sense sequence of lncDACH1 successfully pulled down hadhb, and the antibody of hadhb precipitated lncDACH1(Fig. 8e). Additionally, the protein level of hadhb was reduced in mouse failing hearts (Fig. 8f), which is inversely correlated to the upregulation of lncDACH1. The siRNA for hadhb reduced the expression of Nav1.5 (Fig. 8g). These data indicated that hadhb is an upstream regulator of lncDACH1 which determines the stability of lncDACH1.

Hadhb binds to lncDACH1 and promotes its decay.
a, The expression level of lncDACH1 in the hearts of TAC mice. N=5-8. *P<0.05 by unpaired t test. b, The mRNA level of DACH1 in the hearts of TAC mice. N=12-14. c, Effects of siRNAs for anp32a, eif4a1 and hadhb on the expression of lncDACH1. N=5-12 from 3 independent cultures. * P<0.05 by unpaired t test. d, The effects of hadhb siRNA on the decay of lncDACH1. N=6-15 from 3 independent cultures. e, Blotting of hadhb pulled-down by lncDACH1, and precipitation of lncDACH1 by anti-hadhb antibody. N=4. *P<0.05 vs IgG by one-way ANOVA followed by Tukey’s post-hoc analysis. f, The effects of heart failure on the protein expression of Hadhb. N=9. * P<0.05 vs sham group. P-values were determined by unpaired t test. g, The effects of hadhb siRNA on the protein expression of Nav1.5, N=6. * P<0.05 vs NC group. P-values were determined by unpaired t test. h, LncDACH1 inhibits Nav1.5 in human iPS differentiated cardiomyocytes. i, Schematic summary of the signaling pathway of lncDACH1 and arrhythmia.
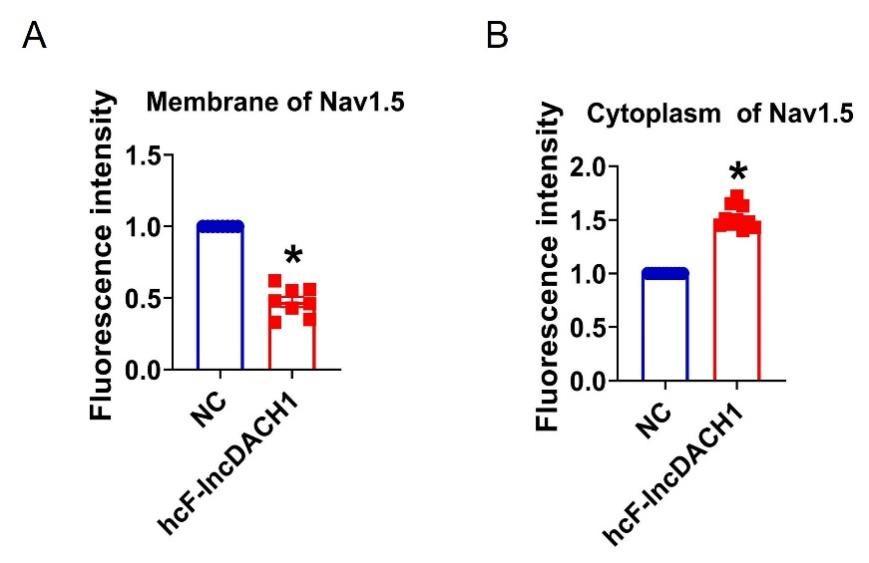
We lastly evaluated the human conserved sequence of lncDACH1(hcF-lncDACH1) on Nav1.5 distribution of human iPS induced cardiomyocytes. We found that overexpression of hcF-lncDACH1 reduced the membrane distribution of Nav1.5 (Fig. 8h and Supplemental Fig. 16). To confirm the arrhythmogenic effects of hcF-lncDACH1 in hiPSC-CM model, we overexpressed hcF-lncDACH1. We found that overexpression of hcF-lncDACH1 significantly inhibited sodium current and the Vmax of APD upstroke (Supplementary Fig. 17). These data indicate the arrhythmogenic effects of hcF-lncDACH1, and imply that knockdown of hcF-lncDACH1 may reduce the susceptibility of arrhythmia.
Discussion
In this study, we discovered that lncDACH1 is critical regulator of sodium channel in the heart. LncDACH1 binds to dystrophin and thus inhibits membrane trafficking of Nav1.5, which leads to the reduction of peak sodium current and impairment of cardiac conduction. Therefore, upregulation of lncDACH1 increased the susceptibility to ventricular arrhythmia (Fig. 8i).
LncRNAs have been established to be critical regulators of various biological processes20. The action modes of lncRNAs are complex. One major mechanism for them to exert their biological function in the cytoplasm is to interact with such as proteins, miRNAs, mRNAs to alter protein translation, enzyme activity, protein degradation, etc21. For instance, lncRNA- CCRR was shown to inhibit the endocytic trafficking of connexin-43 by binding to CIP8512. LncDACH1 mainly distributes in the cytoplasm, and can bind to SERCA2a to promote its ubiquitination and degradation13. LncDACH1 can also bind to protein phosphatase 1 catalytic subunit alpha (PP1A) to inhibit its dephosphorylation activity on yes-associated protein 1 (YAP1), leading to the cytoplasmic sequestration of YAP114. The unraveling of the molecular mechanism of Nav1.5 is critical for the insightful understanding of sodium channel function under physiological and pathological conditions. Several interacting proteins have been demonstrated to determine membrane distribution of Nav1.5 and sodium channel function4. Dystrophin is a well-characterized Nav1.5 partner protein. It indirectly interacts with Nav1.5 via syntrophin, which binds with the C-terminus of dystrophin and with the SIV motif on the C-terminus of Nav1.56,22. In this study, we found that lncDACH1 binds to dystrophin and leads to the impairment of Nav1.5 trafficking and reduced membrane distribution.
Although the membrane distribution of both dystrophin and Nav1.5 was inhibited by lncDACH1, the total protein level of dystrophin was not affected, while Nav1.5 was reduced. The mechanism for the differential influence of lncDACH1 on total protein levels of dystrophin and Nav1.5 is unclear. One explanation may be that Nav1.5 is a membrane channel protein. If they failed to target on the plasma membrane, they may be regarded as unnecessary protein and undergo the process of protein degradation. The E1-E3 enzymes in the ubiquitination systems have been shown to regulate the degradation of Nav1.5, which includes E1 enzyme UBE1(Ubiquitin-activating Enzyme1), UBA6(Ubiquitin-like modifier-activating enzyme 6), E2 enzyme, UBC9 (Ubiquitin-Conjugating Enzyme 9), and E3 ligase Nedd4-2 (neuronal precursor cell expressed developmentally downregulated 4-2) 23–25. LITAF (lipopolysaccharide-induced tumor necrosis factor-alpha factor), a protein encoding a regulator of endosomal trafficking, was shown to reduce surface Nav1.5 by promoting degradation of NEDD4-226. Therefore, cytoplasmic Nav1.5 that failed to target on plasma membrane may be quickly distinguished and then degraded by these ubiquitination enzymes(Supplementary Fig. 18).
The dysfunction of sodium channel is associated with various arrhythmias. Reduced peak INa due to SCN5A loss-of-function mutation can cause a series of arrhythmias such as atrial fibrillation, Brugada syndrome, long QT syndrome, sudden cardiac death, and ventricular tachycardia etc27,28. Consistently, we found that accompanied with the reduction of peak INa, overexpression of lncDACH1 reduced ventricular conduction velocity and increased the susceptibility to ventricular arrhythmia in mice. The increased peak INa due to SCN5A gain-of-function mutation is associated arrhythmias such as atrial fibrillation, long QT syndrome; polymorphic ventricular complexes and ventricular arrhythmia27,28. In this study, although the peak INa increased in lncDACH1-cKO mice, the susceptibility to arrhythmia did not increase. One difference of our data with SCN5A gain-of-function mutation is that the kinetics of peak INa often change during mutation, while it is not the case of lncDACH1 knockout. One possible explanation may be that lncDACH1 does not alter Nav1.5 gating, the late Na current may not be enhanced to the same effect as observed with LQT gain-of-function Nav1.5 mutations, in which APD prolongation is attributed to gating defects that increase late Na current.
Sodium channel remodeling is occurs commonly in cardiac diseases especially heart failure. Despite of some discrepancy, the main observations are that the peak INa, SCN5A mRNA and Nav1.5 protein are reduced during heart failure of both human patients and animal models 16–19. In this study, we found that lncDACH1 was increased during heart failure, indicating that it may contribute to sodium channel remodeling and arrhythmogenesis during heart failure by interfering the action of dystrophin. Interestingly, we found that activation of dystrophin with dCas9-SAM system restored the membrane distribution of Nav1.5 in cardiomyocytes of lncDACH1-TG mice, which implies its potential in counteracting sodium channel remodeling of patients with heart failure.
The low sequence conservation of lncRNAs among species is a critical issue that limits the extrapolation of data from animal studies to human beings29,30. In this study, we found that the conserved fragment of lncDACH1 exhibits the same effect as lncDACH1 on Nav1.5 trafficking and arrhythmogenesis. Moreover, the human conservative homologous fragment of lncDACH1 can inhibit the membrane distribution of Nav1.5 in cardiomyocytes derived from induced pluripotent stem cells (iPS-CMs). These findings hint the clinical relevance of lncDACH1, which holds the potential to become a therapeutic target for treating sodium channel remodeling in clinic.
In conclusion, lncDACH1 is a novel regulator of sodium channel, which suppresses the membrane trafficking of Nav1.5 by disturbing the function of dystrophin. The current work enriched our understanding of the biology of sodium channel trafficking and function, and indicated that lncRNAs possess the potential to become therapeutic targets for ventricular arrhythmias.
Methods
Animals
Neonatal (within 3-day post born) and adult C57BL/6 mice (8 to 10 weeks old) were provided by the animal center at the Second Affiliated Hospital of Harbin Medical University. Use of animals was approved by the Ethic Committees of Harbin Medical University and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Neonatal cardiomyocytes preparation
Neonatal cardiomyocytes were isolated from 3-day-old mice in accordance with the following procedures. Briefly, after dissection, hearts were washed and minced in 0.25% trypsin. Pooled cell suspensions were centrifuged and resuspended in Dulbecco's modified Eagle's medium (DMEM Hyclone, USA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. The suspension was incubated in culture flasks for 90 min, which makes fibroblasts preferentially adhere to the bottom of the culture flasks. Neonatal cardiomyocytes were removed from the culture flasks and the medium was changed. Cell cultures were incubated for 48 h at 37 °C in a humidified atmosphere of 95% oxygen and 5% carbon dioxide before any experimentation.
Generation of cardiac myocyte-specific lncDACH1 overexpressing mice
Cardiomyocyte-specific lncDACH1 overexpressing mice driven by murine αMHC promoter on a C57BL/6 background was generated by Biocytogen Co., Ltd (Beijing, China) as demonstrated in previous study13.
Generation of cardiomyocyte-specific lncDACH1 knockout mice
LncDACH1 conditional KO mice (lncDACH1 Flox/Flox) was generated by using CRISPR/Cas9 technique on C57BL/6 background mice by Biocytogen Co., Ltd (Beijing, China) as demonstrated in previous study13.
Construction of adeno-associated virus 9 (AAV9) carrying deactivated clustered regularly interspaced short palindromic repeats associated protein 9 nuclease- synergistic activation mediator(dCas9-SAM) system to activate the transcription of dystrophin
Adeno-associated virus 9(AAV9) carrying dCas9-SAM system to activate the transcription of dystrophin was constructed as reported previously with brief modifications15. The sgRNA targeting on the promoter region of dystrophin was designed and cloned into the multiple cloning site of plasmid GV639 (EFS-NLS-dSaCas9-NLS-VP64-bGHpA-U6). The constructed plasmid was packaged into AAV9 virus. The sequence of sgRNA is: 5′- CGCTTCCGCGGCCCGTTCAA -3′; The mock-sgRNA target sequence (5′- CGCTTCCGCGGCCCGTTCAA -3′) was used as negative control. The obtained AAV9 virus volume was administered into C57BL/6 mice via tail vein injection at 1×1011 genome containing particles (GC)/animal in 100μl.
Construction of adenovirus carrying cF-lncDACH1 and in vivo gene delivery
Adenovirus vectors carrying cF-lncDACH1(OE- cF-lncDACH1) and a negative control (NC) and a CAG promoter conjugated with green fluorescent protein (GFP) were constructed by Genechem Co., Ltd. (Shanghai, China). OE- cF-lncDACH1, control constructs at 1×109 genome containing particles (GC)/animal in 100μl volume was administered into C57BL/6 mice with body weights ranging from 18~22g via tail vein injection. Seventy-two hours after injection, the mice were subjected to further study.
Construction of adenovirus carrying lncDACH1, lncDACH1 siRNA, conserved fragment of human lncDACH1 and infection
Adenovirus vectors carrying lncDACH1(OE-lncDACH1), a short RNA fragment for silencing lncDACH1 (Si-lncDACH1) or conserved fragment of human lncDACH1(hcF-lncDACH1) and a CAG promoter conjugated with green fluorescent protein (GFP) were constructed by Genechem Co., Ltd. (Shanghai, China). Neonatal cardiomyocytes were infected with adenovirus for 48 hours, and then subjected to subsequent study.
Transfection of Hadhb, Eif4a1, and Anp32 siRNA
siRNA for Hadhb (siHadhb), Eif4a1(siEif4a1), Anp32(siAnp32) and a scrambled negative control RNA (siNC) were synthesized by Generalbiol (Chuzhou, Anhui, China). These siRNAs were transfected at a final concentration of 100 nM into NMVMs using the X-treme GENE Transfection Reagent (Roche, Indianapolis, USA) according to the manufacturer’s protocols. The cardiomyocytes were collected for total RNA isolation or protein purification. The sequences are: siHadhb, sense 5′- GCUCUCAGAUCUUCUAUAATT-3′ and antisense 5′- UUAUAGAAGAUCUGAGAGCTT-3′; siEif4a1, sense 5′- GCUCACCGAGAAGAUGCAUTT-3′ and antisense 5′- UUAUAGAAGAUCUGAGAGCTT-3′; siAnp32, sense 5′- GCCUCACCUCCAUUUCCAATT-3′ and antisense 5′- UUGGAAAUGGAGGUGAGGCTT-3′.
Induction of ventricular arrhythmia
C57BL/6 mice were anesthetized with 2,2,2-tribromoethanol (200 mg/kg, i.p.). An octapolar electrophysiological catheter (1.1F, SciSense Inc., Canada) was inserted into the right ventricle via the jugular vein. Intracardiac pacing was performed using an automated stimulator interfaced with the data acquisition system (GY6000; HeNan HuaNan Medical Science & Technology Ltd., Zhengzhou, China). The surface recording electrode was fixed on LV epicardium to record pseudo-ECG. Inducibility of ventricular tachycardia (VT) was determined by applying a train of ten consecutive electrical pulses with a coupling interval of 80 ms (S1), followed by two extra stimuli (S2 and S3) at coupling intervals of 2 ms, respectively. Successful induction of VT was defined as the appearance of rapid nonsinus rhythm ventricular activations lasting for three beats or more.
Optical mapping recording
Mice were heparinized and euthanized by 2,2,2-tribromoethanol (200 mg/kg, intraperitoneal injection; Sigma, St Louis, MO, USA). The heart was isolated and langendorff perfused with Tyrode’s solution (NaCl 128.2 mM, CaCl2•2H2O 1.3 mM, KCl 4.7 mM, MgCl2•6H2O 1.85 mM, NaH2PO4•2H2O 1.19 mM, Na2CO3 20 mM, and glucose 11.1 mM; pH 7.35) at 37 °C. After 10 min of stabilization, the hearts were stained with RH237 (10 μM) for membrane voltage (Vm) mapping. The dye was excited at 710 nm using monochromatic light-emitting device. The fluorescence was filtered and recorded simultaneously with a MiCAM05 CMOS camera (SciMedia, USA) at 1ms/frame and 100x100 pixels with a spatial resolution of 0.35×0.35 mm2 per pixel. Blebbistatin (10 μM, Selleckchem, Houston, TX, USA) was used to inhibit motion artifact during optical mapping.
Experimental protocol of optical mapping
A pair of hook bipolar electrodes was inserted into apex of heart for pacing. A pseudo-ECG was obtained with widely spaced bipolar electrodes to determine ventricular rhythm. The ventricles were initially paced at a constant pacing cycle length (PCL) of 200ms. The PCLs were progressively shortened (200, 100, 60, 40, 30, 20ms) with a duration of 1-2 s until ventricular tachycardia (VT) was induced or the loss of 1:1 capture of the ventricles. Optical recording was performed after 20 beats of stable pacing at each PCL. Optical recordings were then performed during VT.
Patch-clamp recording
Whole-cell configuration of the patch-clamp technique was used to record peak INa and the inward rectifier K+ current (IK1). INa recordings were performed at room temperature (22~23°C) by using a MultiClamp 700B (Alembic Instruments) amplifier. Pipettes (tip resistance 1 to 2 MΩ) were filled with a solution containing (in mM): NaCl 5, CaCl2 2, MgCl2 2, CsCl 130, HEPES 10, EGTA 15 and MgATP 4 (pH 7.2 with CsOH). Myocytes were bathed with a solution containing (in mM): NaCl 25, CaCl2 2, MgCl2 2.5, CsCl 108.5, HEPES 10, CoCl2 2.5 and glucose 10 (pH 7.4 with CsOH). IK1 was measured as Ba2+-sensitive steady-state current and treated with 300 μmol/L Ba2+ recording at 37 C using a patch clamp amplifier (MultiClamp 700B). The external solution for recording IK1 contained (in mM): NaCl 136, KCl 5.4, NAH2PO4 0.33, MgCl2·6H2O 1, CaCl2 1.8, HEPES 5 and glucose 10 (pH 7.37 with NaOH). The pipette solution for recording IK1 contained (in mM): KCl 20, K-aspartate 110, HEPES 5, MgCl2 1, Na2ATP 5 and EGTA 10 (pH 7.20 with KOH).
Differentiation of human induced pluripotent stem cells(hiPSCs) to cardiomyocytes
Undifferentiated hiPS cells (AC-iPSC) were purchased from NC5 (Help Stem Cell Innovations, NC2001) and cultured on Matrigel-coated plates in an E8 medium (CA1001500, CELLAPY). Differentiation basal medium composed of RPMI1640 medium (C11875500BT, Thermo Fisher Scientific) and B27 minus insulin (A1895601, Thermo Fisher Scientific) was used to induce cardiomyocyte differentiation. Specifically, the 70~80% confluent hiPSCs were incubated in differentiation basal medium added with CHIR-99021 (HY-10182, MCE) for 1 day and Wnt-C59 (S7037, Selleck Chemicals) for 2 days. Then, the cells were cultured in RPMI1640 basal medium containing B27 (17504044, Thermo Fisher Scientific), which was replaced with fresh medium every 1~2 days. Beating cells were observed after 8 days of differentiation induction and used for further study.
Construction of truncated LncDACH1 fragment plasmids
The sequence of lncDACH1 was divided into five fragments. The cDNA of each fragment was inserted into the pCDNA3.1, respectively. The first 417 nts of the entire sequences was cut off and constructed as fragment-a (418-2085 nts). Another 417 nts was cut off to generate fragment-b (835-2085 nts). The third 417 nts was cut off to generate fragment-c (1251-2085 nts). Fragment-d is from 835-1668 nts, and fragment-e is from 835-1251 nts.
Isolation of cardiac myocytes
Hearts were rapidly excised, cannulated, and perfused with Ca2+ -free Tyrode solution (in mM): NaCl 137, KCl 5.4, NaH2PO4 0.16, glucose 10, CaCl2 1.8, MgCl2 0.5, HEPES 5.0, and NaHCO3 3.0 (pH 7.4 adjusted with NaOH) for 5 min. The heart was then perfused with a solution containing collagenases B and D (Roche) and protease XIV (Sigma) until digestion was complete. Tissue was dissociated using forceps, and dissociated left ventricular cardiomyocytes were gradually exposed to Ca2+ (from 50 to 500 μM over 40 min) and plated in culture chambers for further studies.
Immunocytochemistry of isolated mouse ventricular myocytes
Cardiomyocytes were fixed for 10 min with 4% paraformaldehyde in PBS, and then washed in PBS for 10 min (2 times). The cells were permeabilized with 0.5% Tween 20 for 30 min. After washed out with PBS for 10 min (3 times), cardiomyocytes were incubated with primary antibodies against Nav1.5 (ASC005, Alomone, 1;200) and dystrophin (MANDYS8, SIGMA, 1;300) overnight at 4°C. Following washout with PBS (10 min, 3 times), cells were incubated with secondary antibodies for 1h and washed with PBS (10 min, 3 times). The cover slips were mounted onto frosted slides in a solution composed of 90% FluorSave Reagent (Calbiochem, La Jolla, CA, USA) and 10% 10X PBS.
Fluorescent in situ hybridization (FISH)
In situ hybridization was performed with a Fluorescent in Situ Hybridization (FISH) Kit (RiboBio, Guangzhou, China). Briefly, isolated cardiomyocytes were fixed in 4% formaldehyde at 4°C for 10 min and dried out on the slides at room temperature (RT). The slides were rinsed and permeabilized with 0.5% Triton-100 in PBS at RT for 30 min, washed with PBS solution, and prehybridized at 37°C for 30 min before hybridization. The prehybridized slides were then incubated with lncRNA-probe in hybridization solution at 37°C for 16 h. After hybridization, the slides were washed six times with prewarmed wash buffer and PBS solution. Finally, the slides were counterstained with DAPI and visualized using a confocal laser-scanning microscope (Zeiss 800, Germany).
Quantitative Real-time RT-PCR
Total RNA was extracted by using Trizol reagent (Invitrogen, USA) according to manufacturer’s protocol. Total RNA (0.5μg) was reverse transcribed by using the TransScript reverse transcriptase (GMO technology, Beijing) to obtain cDNA. The RNA levels were determined using SYBR Green I incorporation method on ABI 7500 fast Real Time PCR system (Applied Biosystems, USA), The expression levels of mRNA were calculated using the comparative cycle threshold (Ct) method (2−ΔΔCt). Each data point was then normalized to ACTIN as an internal control in each sample. The final results are expressed as fold changes by normalizing the data to the values from control subjects. The primers are: Mus lncDACH1 Forward: 5′-AAGATAGGATGTTGGGGCAG-3′ Mus lncDACH1 Reverse: 5′-ACCATAGCACAAACACTTCC-3′ Mus Dystrophin Forward:
5′- CGGGTTGGCTTTGAATGCTC -3′; Mus Dystrophin Reverse:
5′- AGTCTTTGGGTGGCTGAGTG -3′; Mus DACH1 Forward:5′-AACCGCAAGAGACAGCATCG-3′; Mus DACH1 Reverse: 5′-GGACAGGCCATCAGGAAACAG-3′; Mus SCN5a Forward:
5′- GAAGGAACGCAGCACAGACAG-3′; Mus SCN5a Reverse:
5′- CATCGCCCTTGACCCATACTA -3′; Mus Hadhb Forward:
5′- CCAAGAAGGCACAGGATGAAG -3′; Mus Hadhb Reverse:
5′- CCAGTGAGGAAGGACGGATG -3′; Mus klhl1 Forward:
5′- CCGCTTGGTTGTTCTCATAATC-3′; Mus klhl1 Reverse: 5′-TCTGCCTCTTGCTATCATCAC-3′; Mus aurora kinase A Forward:5′-TTTAGTAATCTGGAGGGAAGGTTC-3′; Mus aurora kinase A Reverse :5′- CACACTCTGGCACTTGAAGG-3′; Mus MZT1 Forward:5′-TCGTCTGCTGGTCAGTAGTC-3′; Mus MZT1 Reverse:5′-CTTGGAAGAGATGCCTGTAATAG-3′; Mus DIS3 Forward: 5′- GCGGCTATGAATGATGATATTACC- 3′; Mus DIS3 Reverse: 5′-CATGTGGAATCGGATCTCTGG-3′.
Western blot
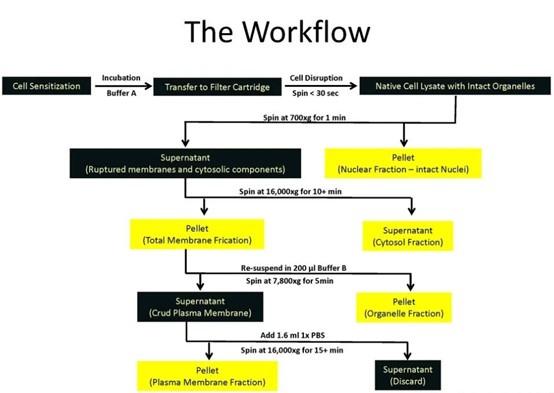
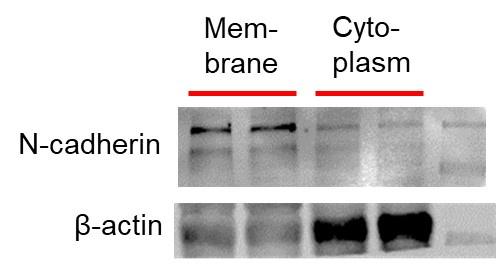
The total, membrane and cytoplasmic protein samples were extracted from cardiac tissues of C57BL/6 mice for immunoblotting analysis. Total protein was collected with the treatment of RIPA lysis buffer (Beyotime, Beijing, China) and a protease inhibitor cocktail (Roche, Basel, Switzerland) at 4°C followed by centrifugation. Extraction of surface and cytoplasmic proteins was conducted using the Surface and Cytoplasmic Protein Reagent Kit (Cat#P0033; Beyotime, Shanghai, China) according to the manufacturer’s instructions. Protein samples were fractionated by SDS-PAGE and then transferred to PVDF membrane. The membranes were blocked in Tris-buffered saline containing 5% milk and then incubated with primary antibodies at 4°C overnight. The primary antibodies include anti-Nav1.5 (ASC005, Alomone, 1:200), anti-dystrophin (MANDYS8, SIGMA, 1:500). The anti- β-actin (1:20000 dilution, 66009-1-Ig, Proteintech), and anti-N-cadherin antibody (Cat#ab76011, 1:5000; Abcam, Cambridge, UK) were used as internal controls. Western blot bands were captured on the Odyssey Infrared Imaging System (LI-COR Biosciences, USA) and quantified with Odyssey v1.2 software by measuring the band intensity (area × OD) in each group. The band intensity was normalized to the internal control. All antibodies were diluted in PBS buffer.
RNA pulldown and immunoblotting
The RNA pull-down was performed as described in the previous study13. Briefly, Biotin-labeled, full length lncDACH1 RNA and antisense RNA were prepared with the Biotin RNA Labeling Mix (Roche) and T7 RNA polymerase (Roche). Biotinylated RNAs were treated with RNase-free DNase I (Invitrogen) and purified on G-50 Sephadex Quick Spin columns (Roche). Biotinylated RNA (17μg) was heated to 65°C for 10 min and slowly cooled to 4°C. Then the RNA was mixed with tissue extracts in pulldown buffer supplemented with tRNA (0.1 μg/μl) and incubated at 4°C for 2 h. Washed Streptavidin agarose beads (60 μl, Invitrogen) were added to each binding reaction and further incubated at 4°C for 1 h. Beads were washed briefly five times in pulldown buffer and boiled in SDS buffer, and the retrieved protein was visualized by immunoblotting.
RNA immunoprecipitation (RIP)
RNA immunoprecipitation (RIP) experiments were performed by using a Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA) as previously reported13. Briefly, heart tissue was pieced and lysed in 220 μl of lysis buffer containing protease inhibitors and RNase Inhibitor and centrifuged at 14,000 × g for 10 min. The supernatants were incubated with anti-dystrophin, anti-hadhb and anti-rabbit IgG antibody for overnight at 4°C with gentle rotation. Protein G magnetic beads (50 μl) were added and incubated at RT with gentle rotation for 3 h. RNA was extracted with 400 μl phenol:chloroform:isoamyl alcohol (125:24:1, pH = 4.3) according to the manufacturer’s instructions before quantitation by RT-qPCR.
Mouse models of heart failure (HF) by transaortic constriction (TAC) and by coronary artery ligation
Mice were randomly divided into sham and TAC groups. In each group, mice were anesthetized with 2,2,2-tribromoethanol (200 mg/kg, i.p.) for TAC model. The animal was orally intubated with 20-gauge tube, and ventilated (mouse ventilator, UGO BASILE, Biological Research Apparatus, Italy) at the respiratory rate of 100 breaths/min with a tidal volume of 0.3 ml. The transverse aorta was constricted by a 6-0 silk suture ligature tied firmly against a 27-gauge needle between the carotid arteries. Then, the needle was promptly removed to yield a constriction of 0.4 mm in diameter. For sham group mice, the animals received the same procedures without aorta constriction.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analysis was performed using unpaired Student’s t test or One-Way Analysis of Variance (ANOVA) followed by Tukey’s post-hoc analysis. A P< 0.05 was considered statistically different.
Acknowledgements
G.Xue, Y.Zhang, J.Yang, performed experiments, analyzed data, and prepared the manuscript. Y.Yang, R.Zhang, D.Li, T.Tian, J.Li, X.Zhang, C.Li, X.Li, J.Yang, K.Shen, Y.Guo, X.Liu and G.Yang helped perform experiments and collect data. B.Yang and Y.Lu oversaw the project and proofread the manuscript. Z.Pan designed the project, oversaw the experiments and prepared the manuscript.
Declarations
Sources of Funding
This work was supported by National Key R&D Program of China (2017YFC1307404 to Z. P.), National Natural Science Foundation of China (82070344, 81870295 to Z. P. 81730012, 81861128022 to B. Y.), Heilongjiang Touyan Innovation Team Program and CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-078 to B. Y.).
Supplemental materials

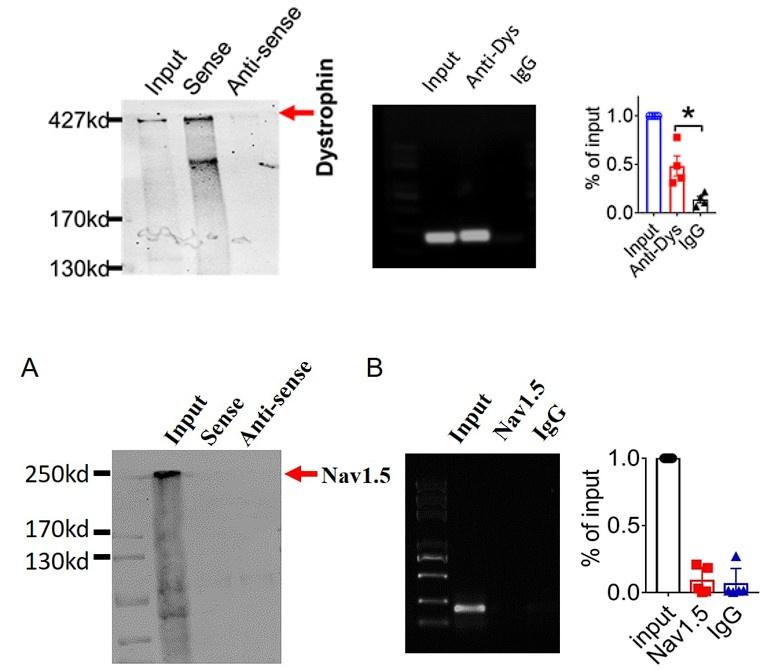
Examination of the potential interaction between lncDACH1 and Nav1.5.
a, Pulldown of Nav1.5 with sense and antisense sequences of lncDACH1. b, RNA immunoprecipitation of lncDACH1 with anti-Nav1.5 antibody. N=4.

Fluorescence intensity of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes of lncDACH1-TG mice.
a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=9 for dys. N=8 for Nav1.5. *P<0.05 versus WT group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=9. *P<0.05 versus WT group. e, Fluorescence in situ hybridization (FISH) images of LncDACH1. N=10. *P<0.05 versus WT group. P-values were determined by unpaired t test.

Overexpression of lncDACH1(oe-lncDACH1) in cultured neonatal cardiomyocytes decreased membrane dystrophin and Nav1.5 expression.
a, The total, membrane and cytoplasm levels of dystrophin by Western blot. N-cadherin is the loading control for membrane extracts. N=10 for total protein; N= 6 for membrane protein; N= 7 for cytoplasm protein. *P<0.05 versus NC group. P-values were determined by unpaired t test. b, The total, membrane and cytoplasm levels of Nav1.5 by Western blot. N-cadherin is the loading control for membrane extracts. N=5 for total protein; N= 5 for membrane protein; N= 9 for cytoplasm protein. *P<0.05 versus WT group. P-values were determined by unpaired t test.

Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocyte overexpressing lncDACH1.
a,b, Membrane levels of dystrophin and Nav1.5. N=9. *P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=9 for dys. N=12 for Nav1.5. *P<0.05 versus NC group. P-values were determined by unpaired t test.

Fluorescence intensity of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes of lncDACH1-cKO mice.
a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=12 for dys. N=8 for Nav1.5. *P<0.05 versus WT group. c,d, Distribution of cytoplasm levels of dystrophin and Nav1.5. N=12. *P<0.05 versus WT group. e, Fluorescence in situ hybridization (FISH) images of LncDACH1 expression. N=8. *P<0.05 versus WT group. P-values were determined by unpaired t test.

Effects of lncDACH1 knockdown with shRNA on the protein expression of dystrophin and Nav1.5 expression in cultured neonatal cardiomyocytes.
a, The total, membrane and cytoplasm levels of dystrophin by Western blot. N-cadherin is the loading control for membrane extracts. N=10 for total protein; N= 7 for membrane protein; N= 5 for cytoplasm protein. *P<0.05 versus NC group. P-values were determined by unpaired t test. b, The total, membrane and cytoplasm levels of Nav1.5 by Western blot. N-cadherin is the loading control for membrane extracts. N=15 for total protein; N= 6 for membrane protein; N= 11 for cytoplasm protein. *P<0.05 versus WT group. P-values were determined by unpaired t test. Dys, dystrophin; NC, negative control; sh-lncDACH1, shRNA for lncDACH1

Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocytes after knocking down of lncDACH1.
a,b, Distribution of membrane levels of dystrophin and Nav1.5. N=11 for dys. N=8 for Nav1.5.*P<0.05 versus NC group. c,d, Distribution of cytoplasm levels of dystrophin and Nav1.5. N=12 for dys. N=9 for Nav1.5.*P<0.05 versus NC group. P-values were determined by unpaired t test.

The mRNA expression of KLHL1, BORA, MZt1, DIS3 and Dach1 in the cardiac tissue of lncDACH1 knockout a, and transgenic overexpression (b) mice.
N=7-12. *P<0.05 versus WT. Data are expressed as mean±SEM. Wt, wild type litter mates; lncDACH1(Tg), lncDACH1 cardiac transgenic overexpression; lncDACH1(CKO), lncDACH1 cardiomyocyte conditional knockout.

Fluorescence intensity of dystrophin and Nav1.5 in isolated cardiomyocytes overexpressing cF-lncDACH1.
a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=9 for dys. N=7 for Nav1.5. *P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=6 for dys. N=7 for Nav1.5. *P<0.05 versus NC group. P-values were determined by unpaired t test.

The conserved fragment of lncDACH1(cF-lncDACH1) inhibited sodium channel function in mice.
a, The mRNA levels of SCN5A and dystrophin. N=4-5. b, Kinetics of INa. N=6-17 cells; N=3 mice of NC; N=4 mice of cF-lncDACH1.

Effects of cF-lncDACH1 overexpression on sodium channel expression and function in cultured neonatal cardiomyocytes.
a, Verification of the expression of cF-lncDACH1 after transfection of adenovirus carrying cF-lncDACH1. N=8 from 3 independent cultures. *P<0.05 versus NC (negative control, empty plasmid). P-values were determined by unpaired t test. b, Peak INa currents, I-V curves and kinetics of INa. N=8-15 cells from 3 independent cultures. *P<0.05 versus NC, P-values were determined by unpaired t test. c, Distribution of Nav1.5 and dystrophin by immunofluorescent staining. d, The mRNA levels of dystrophin and SCN5A. N=8-12 from 3 independent cultures.

Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocytes overexpressing cF-lncDACH1.
a,b, Membrane levels of dystrophin and Nav1.5. N=10 for dys. N=11 for Nav1.5. *P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=7 for dys. N=6 for Nav1.5.*P<0.05 versus NC group. P-values were determined by unpaired t test.

Overexpression of cF-lncDACH1(oe-cF-lncDACH1) in cultured neonatal cardiomyocytes decreased membrane dystrophin and Nav1.5 expression.
a, The total, membrane and cytoplasm levels of dystrophin by Western blot. N-cadherin is the loading control for membrane extracts. N=5 for total protein; N= 8 for membrane protein; N= 6 for cytoplasm protein. *P<0.05 versus NC group. P-values were determined by unpaired t test. b, The total, membrane and cytoplasm levels of Nav1.5 by Western blot. N-cadherin is the loading control for membrane extracts. N=8 for total protein; N= 6 for membrane protein; N= 11 for cytoplasm protein. *P<0.05 versus WT group. P-values were determined by unpaired t test.

a, UCSC snapshot of lncDACH1. b, Conservation of lncdach1 between mice and other species. c, Alignment of murine lncDACH1 and human orthologue. d, Alignment of murine lncDACH1 and sheep orthologue. e, Alignment of murine lncDACH1 and Pig orthologue. f, Alignment of murine lncDACH1 and dog orthologue. g, Alignment of murine lncDACH1 and Rat orthologue

Schematic model for the construction of AAV9 virus carrying the dCas9-SAM system (a) to activate the transcription of dystrophin and tail-vein injection to mice (b).
VP64, VP64 transactivator; dCas9, deactivated CRISPR associated protein 9 nuclease. TSS, transcriptional start point.

Fluorescence intensity of Nav1.5 in human iPS differentiated cardiomyocytes overexpressing cF-lncDACH1.
a, Membrane levels of Nav1.5. N=8 for Nav1.5. *P<0.05 versus NC group. b, Cytoplasm levels of Nav1.5. N=10 for Nav1.5.*P<0.05 versus NC group. P-values were determined by unpaired t test.

Effects of cF-lncDACH1 overexpression on sodium current and Vmax of APD in hiPS-CMs.
a,b, Peak INa currents and I-V curve of INa. N=11-12 from 3 independent cultures. *P<0.05 versus NC. c,d, Rising phase diagram of action potential and Vmax of ascending branch of action potential phase 0, N=9. *P<0.05 versus NC.

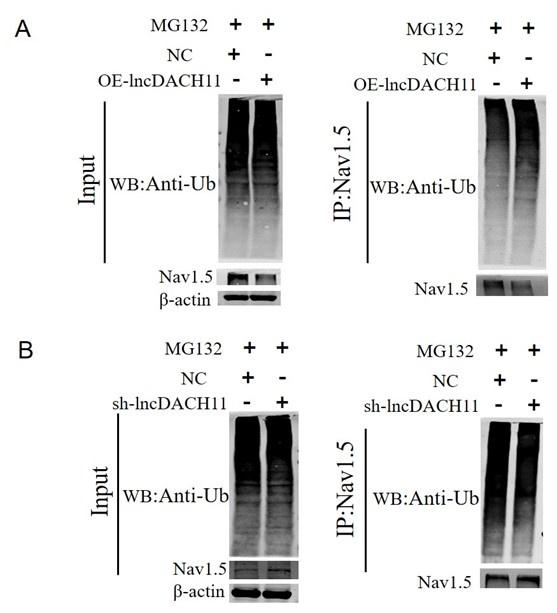
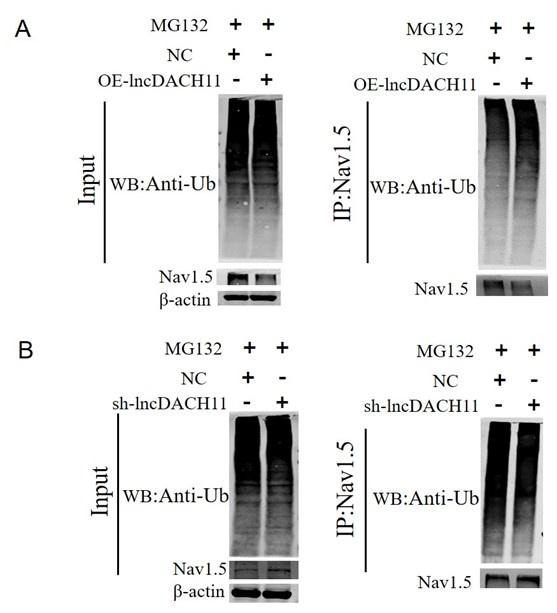
Ubiquitination of Nav1.5 in the Primary cardiomyocytes of lncDACH1 transgenic overexpression and knockout.
a, the ubiquitination of Nav1.5 of overexpression of lncDACH1. b, the ubiquitination of Nav1.5 of Knockdown of lncDACH1.
References
- 1Biology of cardiac sodium channel Nav1.5 expressionCardiovasc Res 93:12–23https://doi.org/10.1093/cvr/cvr252Google Scholar
- 2Regulation of the cardiac Na+ channel NaV1.5 by post-translational modificationsJ Mol Cell Cardiol 82:36–47https://doi.org/10.1016/j.yjmcc.2015.02.013Google Scholar
- 3Sodium channel mutations and arrhythmiasNat Rev Cardiol 6:337–348https://doi.org/10.1038/nrcardio.2009.44Google Scholar
- 4Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac deathCirc Res 116:1971–1988https://doi.org/10.1161/CIRCRESAHA.116.305017Google Scholar
- 5The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemmaCirc Res 94:1023–1031https://doi.org/10.1161/01.RES.0000126574.61061.25Google Scholar
- 6Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophinCirc Res 99:407–414https://doi.org/10.1161/01.RES.0000237466.13252.5eGoogle Scholar
- 7SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytesCirc Res 108:294–304https://doi.org/10.1161/CIRCRESAHA.110.228312Google Scholar
- 8Unique features of long non-coding RNA biogenesis and functionNat Rev Genet 17:47–62https://doi.org/10.1038/nrg.2015.10Google Scholar
- 9Long Noncoding RNAs in Cardiovascular Pathology, Diagnosis, and TherapyCirculation 134:1484–1499https://doi.org/10.1161/CIRCULATIONAHA.116.023686Google Scholar
- 10Long Noncoding RNA Kcna2 Antisense RNA Contributes to Ventricular Arrhythmias via Silencing Kcna2 in Rats With Congestive Heart FailureJ Am Heart Assoc 6https://doi.org/10.1161/JAHA.117.005965Google Scholar
- 11Long noncoding RNA MALAT1 downregulates cardiac transient outward potassium current by regulating miR-200c/HMGB1 pathwayJ Cell Biochem 119:10239–10249https://doi.org/10.1002/jcb.27366Google Scholar
- 12Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular couplingNat Commun 9:4176https://doi.org/10.1038/s41467-018-06637-9Google Scholar
- 13Long Noncoding RNA-DACH1 (Dachshund Homolog 1) Regulates Cardiac Function by Inhibiting SERCA2a (Sarcoplasmic Reticulum Calcium ATPase 2a)Hypertension 74:833–842https://doi.org/10.1161/HYPERTENSIONAHA.119.12998Google Scholar
- 14Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarctionCell death and differentiation https://doi.org/10.1038/s41418-020-0492-5Google Scholar
- 15CRISPR RNA-guided activation of endogenous human genesNat Methods 10:977–979https://doi.org/10.1038/nmeth.2598Google Scholar
- 16Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardiaCirculation 112:2386–2396https://doi.org/10.1161/CIRCULATIONAHA.105.534784Google Scholar
- 17Increased late sodium current in myocytes from a canine heart failure model and from failing human heartJ Mol Cell Cardiol 38:475–483https://doi.org/10.1016/j.yjmcc.2004.12.012Google Scholar
- 18Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload modelEur J Heart Fail 11:749–757https://doi.org/10.1093/eurjhf/hfp092Google Scholar
- 19Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heartCardiovasc Res 114:1728–1737https://doi.org/10.1093/cvr/cvy152Google Scholar
- 20Cellular functions of long noncoding RNAsNat Cell Biol 21:542–551https://doi.org/10.1038/s41556-019-0311-8Google Scholar
- 21lincRNAs: genomics, evolution, and mechanismsCell 154:26–46https://doi.org/10.1016/j.cell.2013.06.020Google Scholar
- 22PDZ domain-binding motif regulates cardiomyocyte compartment-specific NaV1.5 channel expression and functionCirculation 130:147–160https://doi.org/10.1161/CIRCULATIONAHA.113.007852Google Scholar
- 23UBC9 regulates cardiac sodium channel Nav1.5 ubiquitination, degradation and sodium current densityJ Mol Cell Cardiol 129:79–91https://doi.org/10.1016/j.yjmcc.2019.02.007Google Scholar
- 24Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitinationCirc Res 95:284–291https://doi.org/10.1161/01.RES.0000136816.05109.89Google Scholar
- 25Ubiquitination-activating enzymes UBE1 and UBA6 regulate ubiquitination and expression of cardiac sodium channel Nav1.5Biochem J 477:1683–1700https://doi.org/10.1042/BCJ20200138Google Scholar
- 26The endosomal trafficking regulator LITAF controls the cardiac Nav1.5 channel via the ubiquitin ligase NEDD4-2J Biol Chem https://doi.org/10.1074/jbc.RA120.015216Google Scholar
- 27Dysfunctional Nav1.5 channels due to SCN5A mutationsExp Biol Med (Maywood) 243:852–863https://doi.org/10.1177/1535370218777972Google Scholar
- 28Cardiac Arrhythmias Related to Sodium Channel DysfunctionHandb Exp Pharmacol 246:331–354https://doi.org/10.1007/164_2017_43Google Scholar
- 29Evolutionary conservation of long non-coding RNAs; sequence, structure, functionBiochim Biophys Acta 1840:1063–1071https://doi.org/10.1016/j.bbagen.2013.10.035Google Scholar
- 30Long non-coding RNAs in development and disease: conservation to mechanismsJ Pathol 250:480–495https://doi.org/10.1002/path.5405Google Scholar
Article and author information
Author information
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Reviewed Preprint version 3:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.89690. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Pan et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.