Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorHenry ColecraftColumbia University, New York, United States of America
- Senior EditorKenton SwartzNational Institute of Neurological Disorders and Stroke, Bethesda, United States of America
Reviewer #1 (Public Review):
Summary:
In this study, the authors show that a long-non coding RNA lncDACH1 inhibits sodium currents in cardiomyocytes by binding to and altering the localization of dystrophin. The authors use a number of methodologies to demonstrate that lncDACH1 binds to dystrophin and disrupt its localization to the membrane, which in turn downregulates NaV1.5 currents. Knockdown of lncDACH1 upregulates NaV1.5 currents. Furthermore, in heart failure, lncDACH1 is shown to be upregulated which suggests that this mechanism may have pathophysiological relevance.
Strengths:
(1) This study presents a novel mechanism of Na channel regulation which may be pathophysiologically important.
(2) The experiments are comprehensive and systematically evaluate the physiological importance of lncDACH1.
Reviewer #2 (Public Review):
This manuscript by Xue et al. describes the effects of a long noncoding RNA, lncDACH1, on the localization of Nav channel expression, the magnitude of INa, and arrhythmia susceptibility in the mouse heart. Because lncDACH1 was previously reported to bind and disrupt membrane expression of dystrophin, which in turn is required for proper Nav1.5 localization, much of the findings are inferred through the lens of dystrophin alterations.
The results report that cardiomyocyte-specific transgenic overexpression of lncDACH1 reduces INa in isolated cardiomyocytes; measurements in whole heart show a corresponding reduction in conduction velocity and enhanced susceptibility to arrhythmia. The effect on INa was confirmed in isolated WT mouse cardiomyocytes infected with a lncDACH1 adenoviral construct. Importantly, reducing lncDACH1 expression via either a cardiomyocyte-specific knockout or using shRNA had the opposite effect: INa was increased in isolated cells, as was conduction velocity in heart. Experiments were also conducted with a fragment of lnDACH1 identified by its conservation with other mammalian species. Overexpression of this fragment resulted in reduced INa and greater proarrhythmic behavior. Alteration of expression was confirmed by qPCR.
The mechanism by which lnDACH1 exerts its effects on INa was explored by measuring protein levels from cell fractions and immunofluorescence localization in cells. In general, overexpression was reported to reduce Nav1.5 and dystrophin levels and knockout or knockdown increased them.
The strengths of this manuscript include convincing evidence of a link between lncDACH1 and Na channel function. The identification of a lncDACH1 segment conserved among mammalian species is compelling. The observation that lncDACH1 is increased in a heart failure model and provides a plausible hypothesis for disease mechanism.
One limitation of the fractionation approach is the uncertain disposition of Na channel protein deemed "cytoplasmic." It seems likely that the membrane fraction includes ER membrane. The signal may reasonably be attributed to Na channel protein in stalled transport vesicles, or alternatively in stress granules, but this was not directly addressed.
Reviewer #3 (Public Review):
Summary:
In this manuscript, the authors report the first evidence of Nav1.5 regulation by a long noncoding RNA, LncRNA-DACH1, and suggest its implication in the reduction in sodium current observed in heart failure. Since no direct interaction is observed between Nav1.5 and the LncRNA, they propose that the regulation is via dystrophin and targeting of Nav1.5 to the plasma membrane.
Strengths:
(1) First evidence of Nav1.5 regulation by a long noncoding RNA.
(2) Implication of LncRNA-DACH1 in heart failure and mechanisms of arrhythmias.
(3) Demonstration of LncRNA-DACH1 binding to dystrophin.
(4) Potential rescuing of dystrophin and Nav1.5 strategy.
Weaknesses:
(1) The fact that the total Nav1.5 protein is reduced by 50% which is similar to the reduction in the membrane reduction questions the main conclusion of the authors implicating dystrophin in the reduced Nav1.5 targeting. The reduction in membrane Nav1.5 could simply be due to the reduction in total protein.
Author response:
The following is the authors’ response to the original reviews.
eLife assessment
This study presents a valuable contribution to cardiac arrhythmia research by demonstrating long noncoding RNA Dachshund homolog 1 (lncDACH1) tunes sodium channel functional expression and affects cardiac action potential conduction and rhythms. Whereas the evidence for functional impact of lncDACH1 expression on cardiac sodium currents and rhythms is convincing, biochemical experiments addressing the mechanism of changes in sodium channel expression and subcellular localization are incomplete.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
In this study, the authors show that a long-non coding RNA lncDACH1 inhibits sodium currents in cardiomyocytes by binding to and altering the localization of dystrophin. The authors use a number of methodologies to demonstrate that lncDACH1 binds to dystrophin and disrupts its localization to the membrane, which in turn downregulates NaV1.5 currents. Knockdown of lncDACH1 upregulates NaV1.5 currents. Furthermore, in heart failure, lncDACH1 is shown to be upregulated which suggests that this mechanism may have pathophysiolgoical relevance.
Strengths:
(1) This study presents a novel mechanism of Na channel regulation which may be pathophysiologically important.
(2) The experiments are comprehensive and systematically evaluate the physiological importance of lncDACH1.
Weaknesses:
(1). What is indicated by the cytoplasmic level of NaV1.5, a transmembrane protein? The methods do not provide details regarding how this was determined. Do you authors means NaV1.5 retained in various intracellular organelles?
Thank you for the good suggestion. Our study showed that Nav1.5 was transferred to the cell membrane by the scaffold protein Dystropin in response to the regulation of LncDACH1, but not all Nav1.5 in the cytoplasm was transferred to the cell membrane. Therefore, the cytoplasmic level of Nav1.5 represents the Nav1.5 protein that is not transferred to the cell membrane but stays in the cytoplasm and various organelles within the cytoplasm when Nav1.5 is regulated by LncDACH1
(2) What is the negative control in Fig. 2b, Fig. 4b, Fig. 6e, Fig. 7c? The maximum current amplitude in these seem quite different. -40 pA/pF in some, -30 pA/pF in others and this value seems to be different than in CMs from WT mice (<-20 pA/pF). Is there an explanation for what causes this variability between experiments and/or increase with transfection of the negative control? This is important since the effect of lncDACH1 is less than 50% reduction and these could fall in the range depending on the amplitude of the negative control.
Thank you for the insightful comment. The negative control in Fig. 2b, Fig. 4b, Fig. 6e are primary cardiomyocytes transfected with empty plasmids. The negative control in Fig.7c are cardiomyocytes of wild-type mice injected with control virus. When we prepare cells before the patch-clamp experiments, the transfection efficiency of the transfection reagent used in different batches of cells, as well as the different cell sizes, ultimately lead to differences in CMS.
(3) NaV1.5 staining in Fig. 1E is difficult to visualize and to separate from lncDACH1. Is it possible to pseudocolor differently so that all three channels can be visualized/distinguished more robustly?
Thank you for the good suggestion. We have re-added color to the original image to distinguish between the three channels.
Author response image 1.
(4) The authors use shRNA to knockdown lncDACH1 levels. It would be helpful to have a scrambled ShRNA control.
Thank you for the insightful comment. The control group we used was actually the scrambled shRNA, but we labeled the control group as NC in the article, maybe this has caused you to misunderstand.
(5) Is there any measurement on the baseline levels of LncDACH1 in wild-type mice? It seems quite low and yet is a substantial increase in NaV1.5 currents upon knocking down LncDACH1. By comparison, the level of LncDACH1 seems to be massively upregulated in TAC models. Have the authors measured NaV1.5 currents in these cells? Furthermore, does LncDACH1 knockdown evoke a larger increase in NaV1.5 currents?
Thank you for the insightful comment.
(1).The baseline protein levels of LncDACH1 in wild-type mice and LncDACH1-CKO mice has been verified in a previously published article(Figure 3).(Hypertension. 2019;74:00-00. DOI: 10.1161/HYPERTENSIONAHA.119.12998.)
Author response image 2.
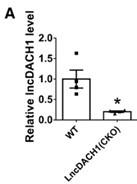
(2). We did not measure the Nav1.5 currents in cardiomyocytes of the TAC model mice in this artical, but in another published paper, we found that the Nav1.5 current in the TAC model mice was remarkably reduced than that in wild-type mice(Figure 4).(Gene Ther. 2023 Feb;30(1-2):142-149. DOI: 10.1038/s41434-022-00348-z)
Author response image 3.
This is consistent with our results in this artical, and our results show that LncDACH1 levels are significantly upregulated in the TAC model, then in the LncDACH1-TG group, the Nav1.5 current is significantly reduced after the LncDACH1 upregulation(Figure 3).
Author response image 4.
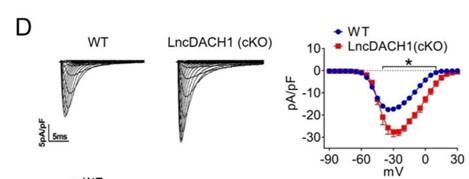
(6) What do error bars denote in all bar graphs, and also in the current voltage relationships?
Thank you for the good comment. All the error bars represent the mean ± SEM. They represent the fluctuation of all individuals of a set of data based on the average value of this set of data, that is, the dispersion of a set of data.
Reviewer #2 (Public Review):
This manuscript by Xue et al. describes the effects of a long noncoding RNA, lncDACH1, on the localization of Nav channel expression, the magnitude of INa, and arrhythmia susceptibility in the mouse heart. Because lncDACH1 was previously reported to bind and disrupt membrane expression of dystrophin, which in turn is required for proper Nav1.5 localization, much of the findings are inferred through the lens of dystrophin alterations.
The results report that cardiomyocyte-specific transgenic overexpression of lncDACH1 reduces INa in isolated cardiomyocytes; measurements in whole heart show a corresponding reduction in conduction velocity and enhanced susceptibility to arrhythmia. The effect on INa was confirmed in isolated WT mouse cardiomyocytes infected with a lncDACH1 adenoviral construct. Importantly, reducing lncDACH1 expression via either a cardiomyocyte-specific knockout or using shRNA had the opposite effect: INa was increased in isolated cells, as was conduction velocity in heart. Experiments were also conducted with a fragment of lnDACH1 identified by its conservation with other mammalian species. Overexpression of this fragment resulted in reduced INa and greater proarrhythmic behavior. Alteration of expression was confirmed by qPCR.
The mechanism by which lnDACH1 exerts its effects on INa was explored by measuring protein levels from cell fractions and immunofluorescence localization in cells. In general, overexpression was reported to reduce Nav1.5 and dystrophin levels and knockout or knockdown increased them.
Thank you for summarizing our work and thank you very much for your appreciation on our work.
Reviewer #3 (Public Review):
Summary:
In this manuscript, the authors report the first evidence of Nav1.5 regulation by a long noncoding RNA, LncRNA-DACH1, and suggest its implication in the reduction in sodium current observed in heart failure. Since no direct interaction is observed between Nav1.5 and the LncRNA, they propose that the regulation is via dystrophin and targeting of Nav1.5 to the plasma membrane.
Strengths:
(1) First evidence of Nav1.5 regulation by a long noncoding RNA.
(2) Implication of LncRNA-DACH1 in heart failure and mechanisms of arrhythmias.
(3) Demonstration of LncRNA-DACH1 binding to dystrophin.
(4) Potential rescuing of dystrophin and Nav1.5 strategy.
Thank you very much for your appreciation on our work.
Weaknesses:
(1) Main concern is that the authors do not provide evidence of how LncRNA-DACH1 regulates Nav1.5 protein level. The decrease in total Nav1.5 protein by about 50% seems to be the main consequence of the LncRNA on Nav1.5, but no mechanistic information is provided as to how this occurs.
Thank you for the insightful comment.
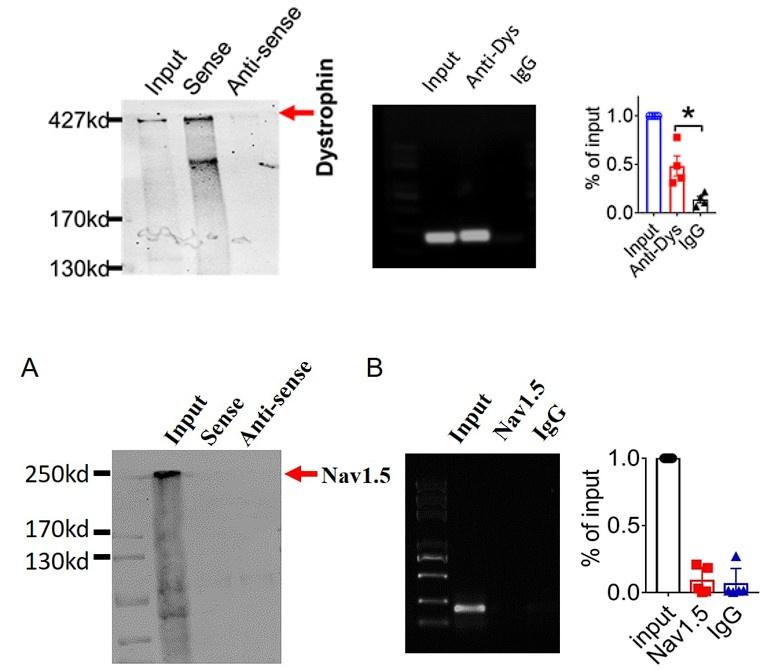
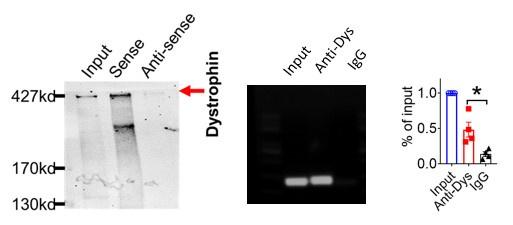
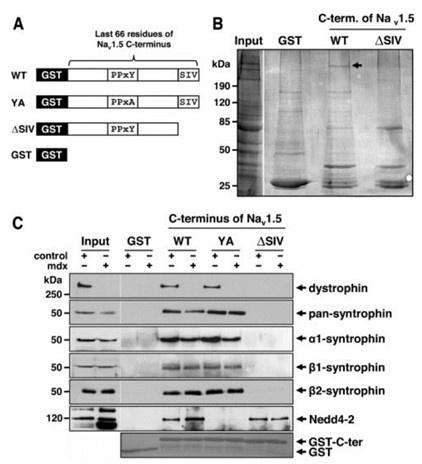
(1) The mechanism of the whole article is as mentioned in the discussion at the end of the article: LncDACH1 binds to dystrophin and thus inhibits membrane trafficking of Nav1.5, Dystrophin is a well-characterized Nav1.5 partner protein. It indirectly interacts with Nav1.5 via syntrophin, which binds with the C-terminus of dystrophin and with the SIV motif on the C-terminus of Nav1.5(Circ Res. 2006;99:407-414. doi: 10.1161/01.RES.0000237466.13252.5e)(Circulation.2014;130:147-160.doi:10.1161/CIRCULATIONAHA.113.007852).
And we performed pulldown and RNA immunoprecipitation experiments to verify it (Figure 1).
Author response image 5.
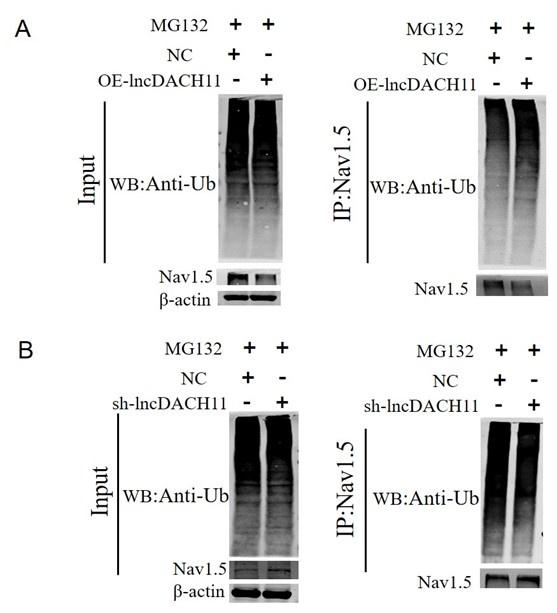
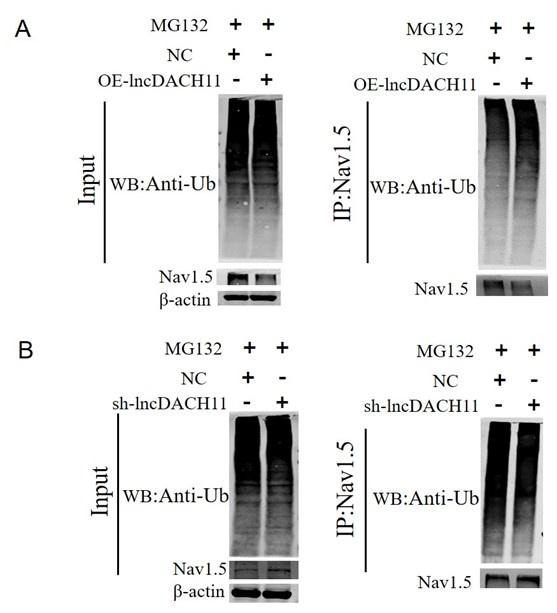
2) Then we found that overexpression of lncDACH1 increased the ubiquitination of Nav1.5, which explains the downregulation of total Nav1.5 protein (Online Supplementary Figure 12).
Author response image 6.
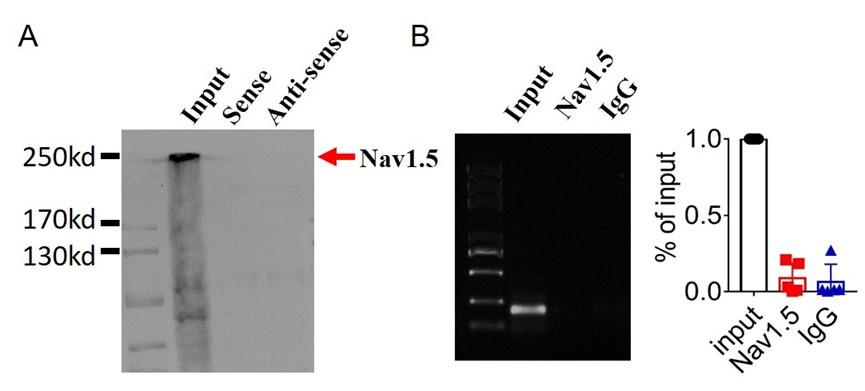
3). Lastly,we found that lncDACH1 failed to pulldown Nav1.5 and anti-Nav1.5 did not precipitate lncDACH1( Supplementary Fig. 1).
Author response image 7.
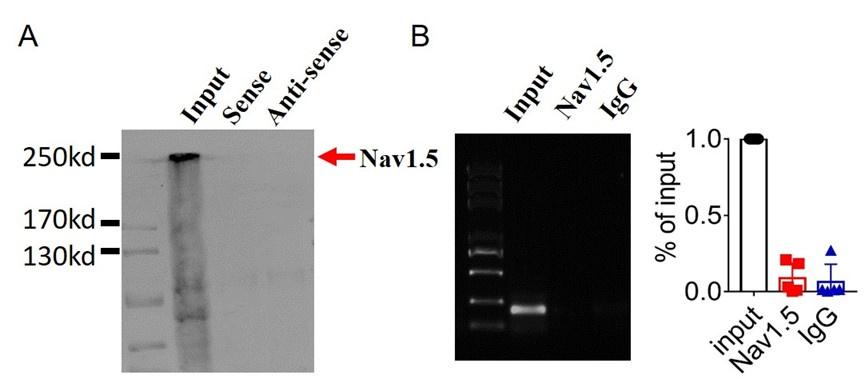
These data indicated that lncDACH does not interact with Nav1.5 directly. It participates in the regulation of Nav1.5 by binding to dystrophin.Cytoplasmic Nav1.5 that failed to target on plasma membrane may be quickly distinguished and then degraded by these ubiquitination enzymes.
(2) The fact that the total Nav1.5 protein is reduced by 50% which is similar to the reduction in the membrane reduction questions the main conclusion of the authors implicating dystrophin in the reduced Nav1.5 targeting. The reduction in membrane Nav1.5 could simply be due to the reduction in total protein.
Thank you for the insightful comment. We do not rule out the possibility that the reduction in membrane Nav1.5 maybe be due to the reduction in total protein, but we don't think this is the main mechanism. Our data indicates that the membrane and total protein levels of Nav1.5 were reduced by 50%. However, the cytoplasmic Nav1.5 increased in the hearts of lncDACH1-TG mice than WT controls rather than reduced like membrane and total protein(Figure 1).
Author response image 8.
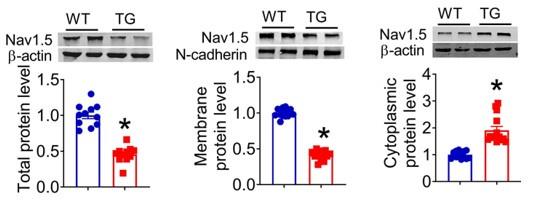
Therefore, we think the mian mechanism of the whole article is as mentioned in the discussion at the end of the article: LncDACH1 binds to dystrophin and thus inhibits membrane trafficking of Nav1.5.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
(1) In Fig. 6E the error bars are only in one direction for cF-lncDACH1. It seems that this error overlaps for NC and cF-lncDACH1 at several voltages, yet it is marked as statistically significant. Also in Fig. 7C, what statistical test was used? Do the authors account for multiple comparisons?
Thank you for the insightful comment.
(1) We have recalculated the two sets of data and confirmed that there are indeed statistically significant between the two sets of data for NC and cF-lncDACH1 at In Fig. 6E, The overlaps in the picture may only be visually apparent.
(2) The data in Fig. 7C are expressed as mean ± SEM. Statistical analysis was performed using unpaired Student’s t test or One-Way Analysis of Variance (ANOVA) followed by Tukey’s post-hoc analysis.
(2) line 57, "The Western blot" remove "The"
Sorry for the mistake. We have corrected it.
(3) line 61, "The opposite data were collected" It is unclear what is meant by opposite.
Sorry for the mistake. We have corrected it.
(4) Lines 137-140. This sentence is complex, I would simplify as two sentences.
Sorry for the mistake. We have corrected it.
(5) Line 150, "We firstly validated" should be "we first validated"
Sorry for the mistake. We have corrected it.
(6) Line 181, "Consistently, the membrane" Is this statement meant to indicate that the experiments yielded a consistent results or that this statement is consistent with the previous one? In either case, this sentence should be reworded for clarification.
Sorry for the mistake. We have corrected it.
(7) Line 223, "In consistent, the ex vivo" I am not sure what In consistent means here.
Thank you for the good suggestion. We mean that the results of ex vivo is consistent with the results of in vivo. We have corrected it to make it clearer.
(8) Line 285. "a bunch of studies" could be rephrased as "multiple studies"
Sorry for the mistake. We have corrected it.
(9) Line 299 "produced no influence" Do you mean produced no change?
Thank you for the good suggestion.As you put it,we mean it produced no change.
(10) Line 325 "is to interact with the molecules" no need for "the molecules
Sorry for the mistake. We have corrected it.
(11) lines 332-335. This sentence is very confusing.
Thank you for the insightful comment. We have corrected it.
(12) Lines 341-342. It is unnecessary to claim primacy here.
Thank you for the good suggestion. We have removed this sentence.
(13) Line 373. "Sodium channel remodeling is commonly occured in" perhaps rephrase as occurs commonly
Thank you for the insightful comment. We have corrected it.
Reviewer #2 (Recommendations For The Authors):
Critique
(1) Aside from some issues with presentation noted below, these data provide convincing evidence of a link between lncDACH1 and Na channel function. The identification of a lncDACH1 segment conserved among mammalian species is compelling. The observation that lncDACH1 is increased in a heart failure model and provides a plausible hypothesis for disease mechanism.
Thank you very much for your appreciation on our work.
(2) Has a causal link between dystrophin and Na channel surface expression has been made, or is it an argument based on correlation? Is it possible to rule out a direct effect of lncDACH1 on Na channel expression? A bit more discussion of the limitations of the study would help here.
Thank you for the insightful comment.
(1). Dystrophin is a well-characterized Nav1.5 partner protein. It indirectly interacts with Nav1.5 via syntrophin, which binds with the C-terminus of dystrophin and with the SIV motif on the C-terminus of Nav1.5(Circ Res. 2006;99:407-414. doi: 10.1161/01.RES.0000237466.13252.5e)(Circulation.2014;130:147-160.doi:10.1161/CIRCULATIONAHA.113.007852).
Author response image 9.
(2).we performed pulldown and RNA immunoprecipitation experiments. The data showed that lncDACH1 failed to pulldown Nav1.5 and anti-Nav1.5 did not precipitate lncDACH1 (Online Supplementary Figure 11). These data indicated that lncDACH does not interact with Nav1.5 directly. ( Supplementary Fig. 1)
Author response image 10.
(3) What normalization procedures were used for qPCR quantification? I could not find these.
Thank you for the good suggestion.The expression levels of mRNA were calculated using the comparative cycle threshold (Ct) method (2−ΔΔCt). Each data point was then normalized to ACTIN as an internal control in each sample. The final results are expressed as fold changes by normalizing the data to the values from control subjects. We have added the normalization procedures in the methods section of the article.
(4) In general, I found the IF to be unconvincing - first, because the reported effects were not very apparent to me, but more importantly, because only exemplars were shown without quantification of a larger sample size.
Thank you for the good suggestion. Accordingly, we quantified the immunostaining data. The data have been included in Supplementary Figure 2- 16.The sample size is labeled in the caption.
Author response image 11.
Fluorescence intensity of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes of lncDACH1-TG mice. a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=9 for dys. N=8 for Nav1.5. P<0.05 versus WT group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=9. P<0.05 versus WT group. e, Fluorescence in situ hybridization (FISH) images of LncDACH1. N=10. *P<0.05 versus WT group. P-values were determined by unpaired t test.
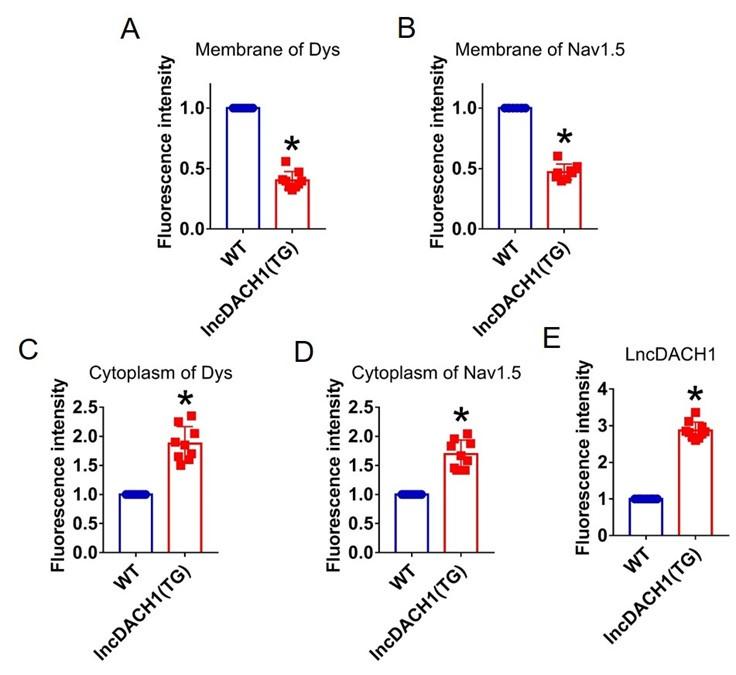
Author response image 12.
Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocyte overexpressing lncDACH1. a,b, Membrane levels of dystrophin and Nav1.5. N=9. P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=9 for dys. N=12 for Nav1.5. P<0.05 versus NC group. P-values were determined by unpaired t test.
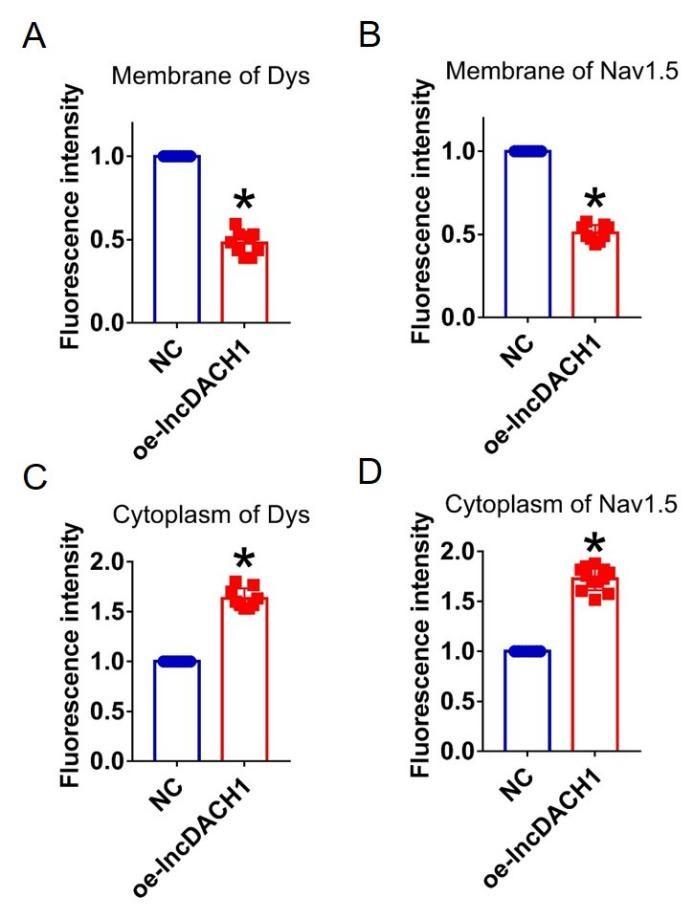
Author response image 13.
Fluorescence intensity of lncDACH1, dystrophin and Nav1.5 in isolated cardiomyocytes of lncDACH1-cKO mice. a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=12 for dys. N=8 for Nav1.5. P<0.05 versus WT group. c,d, Distribution of cytoplasm levels of dystrophin and Nav1.5. N=12. P<0.05 versus WT group. e, Fluorescence in situ hybridization (FISH) images of LncDACH1 expression. N=8. *P<0.05 versus WT group. P-values were determined by unpaired t test.
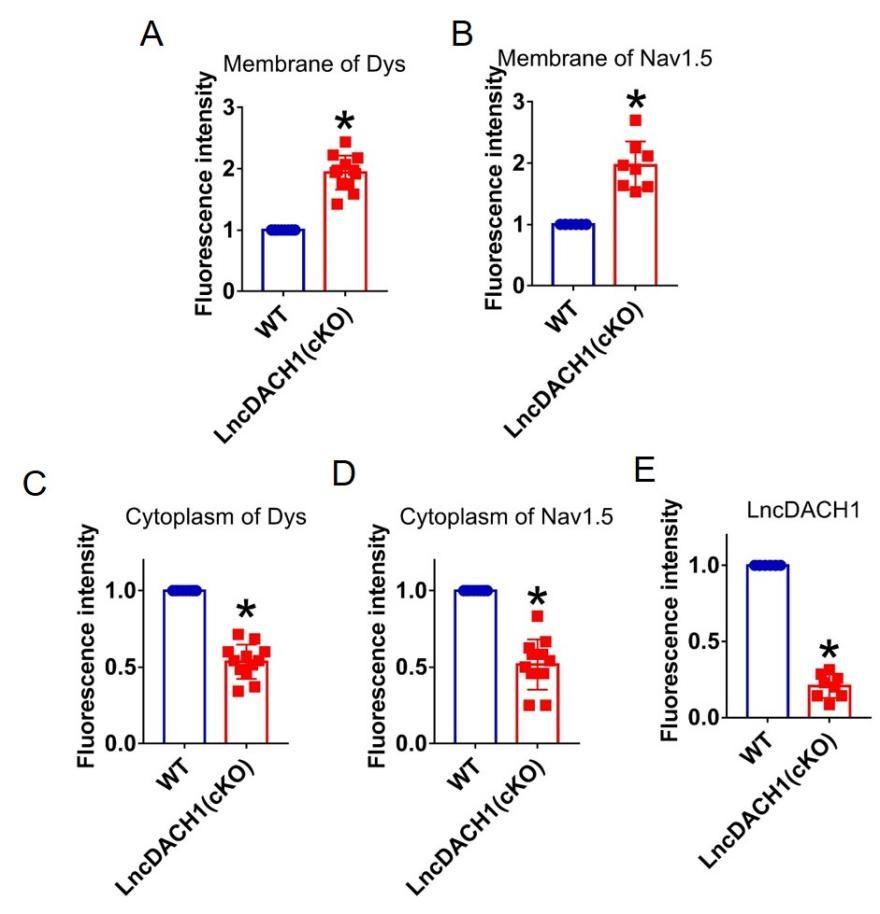
Author response image 14.
Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocytes after knocking down of lncDACH1. a,b, Distribution of membrane levels of dystrophin and Nav1.5. N=11 for dys. N=8 for Nav1.5.P<0.05 versus NC group. c,d, Distribution of cytoplasm levels of dystrophin and Nav1.5. N=12 for dys. N=9 for Nav1.5.P<0.05 versus NC group. P-values were determined by unpaired t test.
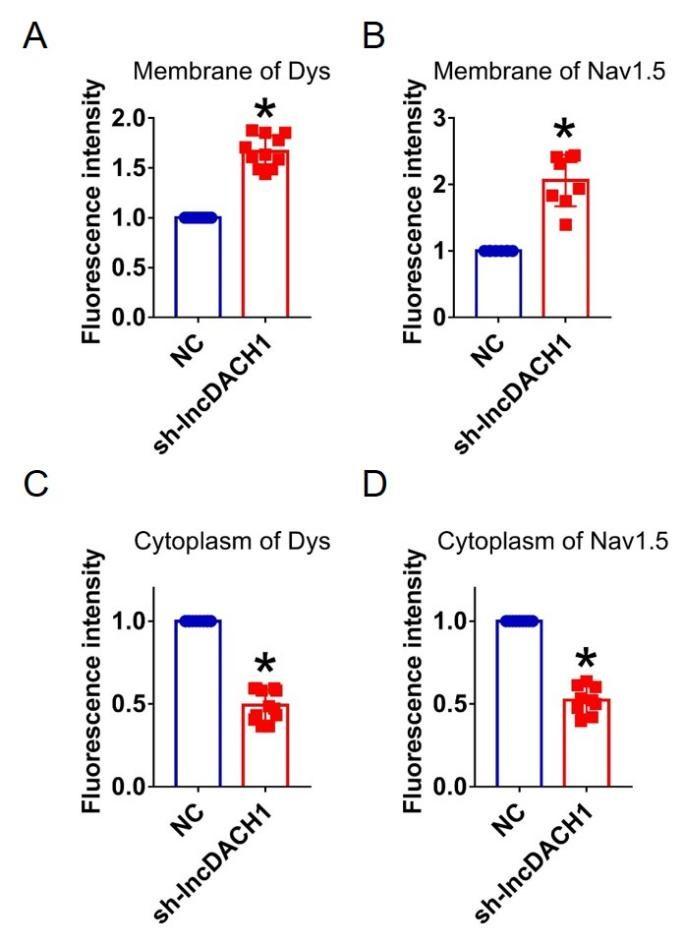
Author response image 15.
Fluorescence intensity of dystrophin and Nav1.5 in isolated cardiomyocytes overexpressing cF-lncDACH1. a,b, Membrane levels of dystrophin (dys) and Nav1.5. N=9 for dys. N=7 for Nav1.5. P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=6 for dys. N=7 for Nav1.5. P<0.05 versus NC group. P-values were determined by unpaired t test.
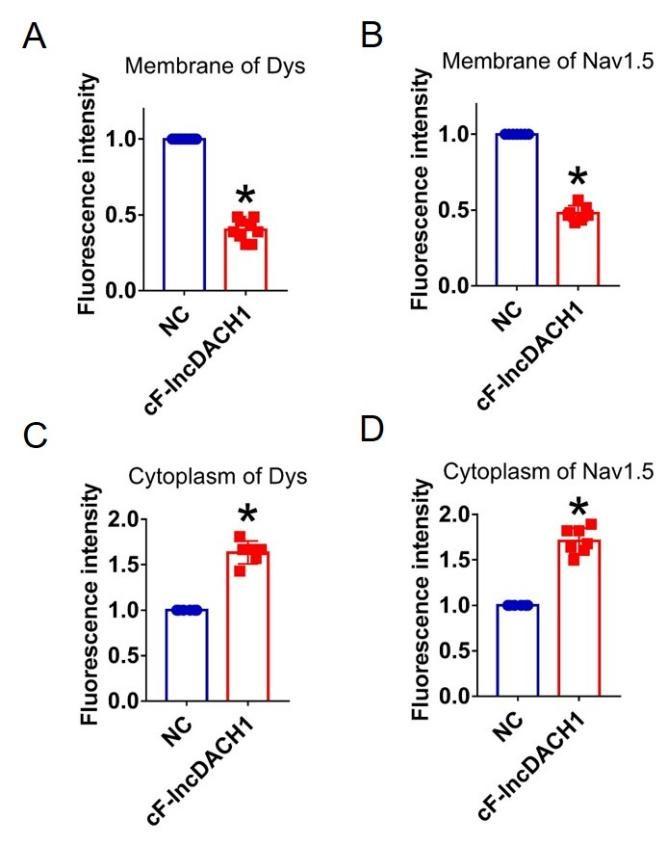
Author response image 16.
Fluorescence intensity of dystrophin and Nav1.5 in cultured neonatal cardiomyocytes overexpressing cF-lncDACH1. a,b, Membrane levels of dystrophin and Nav1.5. N=10 for dys. N=11 for Nav1.5. P<0.05 versus NC group. c,d, Cytoplasm levels of dystrophin and Nav1.5. N=7 for dys. N=6 for Nav1.5.P<0.05 versus NC group. P-values were determined by unpaired t test.
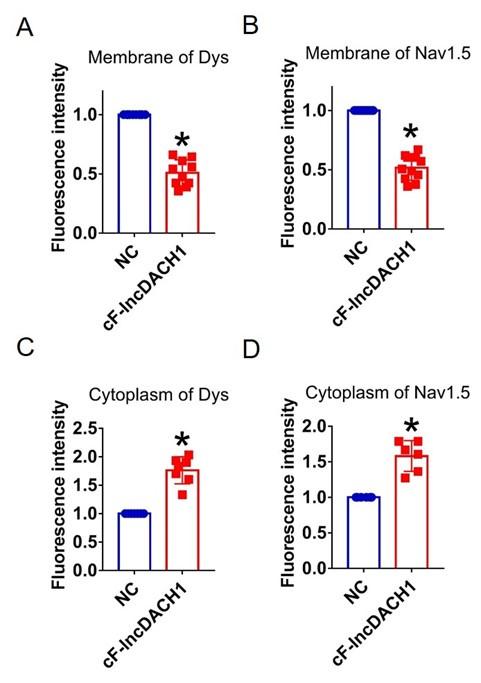
Author response image 17.
Fluorescence intensity of Nav1.5 in human iPS differentiated cardiomyocytes overexpressing cF-lncDACH1. a, Membrane levels of Nav1.5. N=8 for Nav1.5. P<0.05 versus NC group. b, Cytoplasm levels of Nav1.5. N=10 for Nav1.5.P<0.05 versus NC group. P-values were determined by unpaired t test.
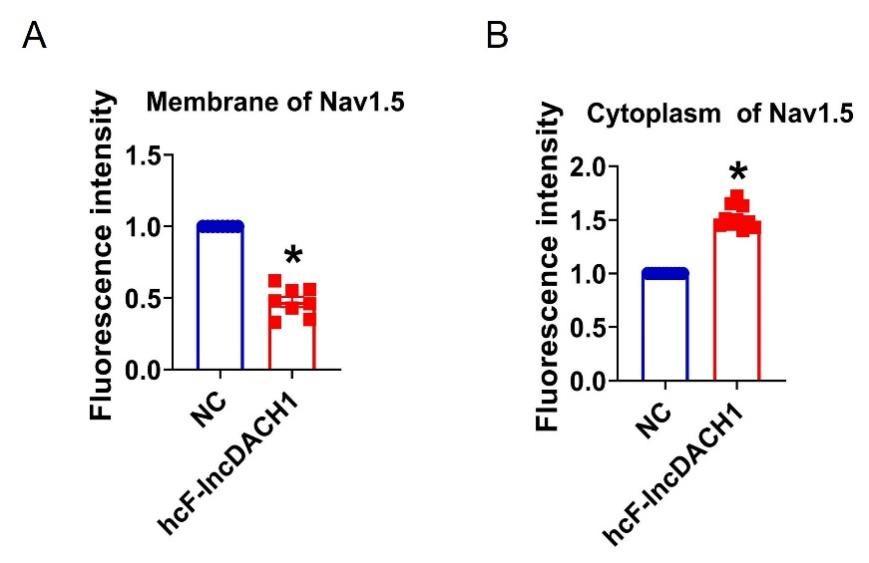
(5) More information on how the fractionation kit works would be helpful. How are membrane v. cytoplasm fractions identified?
a. I presume the ER is part of the membrane fraction? When Nav1.5 is found in the cytoplasmic fraction, what subcompartment is it in - the proteasome?
b. In the middle panel of A - is the dystrophin signal visible on the WB for WT? I assume the selected exemplar is the best of the blots and so this raises concerns. Much is riding on the confidence with which the fractions report "membrane" v "cytoplasm."
Thank you for the insightful comment.
(1). How the fractionation kit works:
The kit utilizes centrifuge column technology to obtain plasma membrane structures with native activity and minimal cross-contamination with organelles without the need for an ultracentrifuge and can be used for a variety of downstream assays. Separation principle: cells/tissues are sensitized by Buffer A, the cells pass through the centrifuge column under the action of 16000Xg centrifugation, the cell membrane is cut to make the cell rupture, and then the four components of nucleus, cytoplasm, organelle and plasma membrane will be obtained sequentially through differential centrifugation and density centrifugation, which can be used for downstream detection.
Author response image 18.
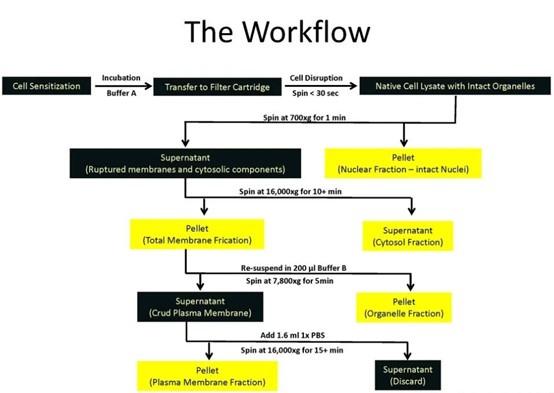
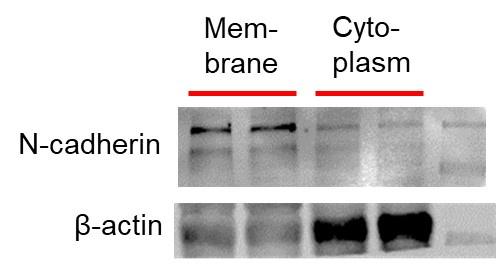
(2). How are membrane v. cytoplasm fractions identified:
The membrane proteins and cytosolic proteins isolated by the kit, and then the internal controls we chose when performing the western blot experiment were :membrane protein---N-cadherin cytosolic protein---β-Actin
Most importantly, when we incubate either the primary antibody of N-cadherin with the PVDF membrane of the cytosolic protein, or the primary antibody of the cytosolic control β-Actin with the PVDF membrane of the membrane protein, the protein bands cannot be obtained in the scan results
Author response image 19.
(6) More detail in Results, figures, and figure legends will assist the reader.
a. In Fig. 5, it would be helpful to label sinus rhythm vs. arrhythmia segments.
Thank you for the good suggestion. We've marked Sinus Rhythm and Arrhythmia segments with arrows
Author response image 20.
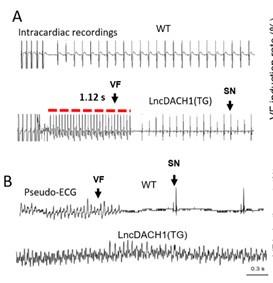
b. Please explain in the figure legend what the red bars in 5A are
Thank you for the insightful comment. We've added the explanation to the figure legend .The red lines in the ECG traces indicate VT duration.
c. In 5C, what the durations pertain to.
Thank you for the good suggestion. 720ms-760ms refers to the duration of one action potential, with 720ms being the peak of one action potential and 760ms being the peak of another action potential.The interval duration is not fixed, in this artical, we use 10ms as an interval to count the phase singularities from the Consecutive phase maps. Because the shorter the interval duration, the larger the sample size and the more convincing the data.
d. In the text, please define "breaking points" and explain what the physiological underpinning is. Define "phase singularity."
Thank you for the insightful comment. Cardiac excitation can be viewed as an electrical wave, with a wavefront corresponding to the action potential upstroke (phase 0) and a waveback corresponding to rapid repolarization (phase 3). Normally, Under normal circumstances, cardiac conduction is composed of a sequence of well-ordered action potentials, and in the results of optical mapping experiments, different colors represent different phases.when a wave propagates through cardiac tissue, wavefront and waveback never touch.when arrhythmias occur in the heart, due to factors such as reenfrant phenomenon, the activation contour will meet the refractory contour and waves will break up, initiating a newly spiral reentry. Corresponding to the optical mapping result graph, different colors representing different time phases (including depolarization and repolarization) come together to form a vortex, and the center of the vortex is defined as the phase singularity.
(7) In reflecting on why enhanced INa is not proarrhythmic, it is noted that the kinetics are not altered. I agree that is key, but perhaps the consequence could be better articulated. Because lncDACH1 does not alter Nav1.5 gating, the late Na current may not be enhanced to the same effect as observed with LQT gain-of-function Nav1.5 mutations, in which APD prolongation is attributed to gating defects that increase late Na current.
Thank you for the good suggestion. Your explanation is very brilliant and important for this article. We have revised the discussion section of the article and added these explanations to it.
Reviewer #3 (Recommendations For The Authors):
(1) Experiments to specifically address the reduction in total Nav1.5 protein should be included.
Thank you for the insightful comment. We examined the ubiquitination of Nav1.5. We found that overexpression of lncDACH1 increased the ubiquitination of Nav1.5, which explains the downregulation of total Nav1.5 protein (Online Supplementary Figure 12).
Author response image 21.
(2) Experiments to convincingly demonstrate that LncRNA-DACH1 regulates Nav1.5 targeting via dystrophin are missing. As it is, total reduction in Nav1.5 seems to be the explanation as to why there is a decrease in membrane Nav1.5.
Thank you for the insightful comment. we performed pulldown and RNA immunoprecipitation experiments. The data showed that lncDACH1 can pulldown dystrophin(Figure 1),but failed to pulldown Nav1.5 and anti-Nav1.5 did not precipitate lncDACH1( Supplementary Fig. 1). These data indicated that lncDACH does not interact with Nav1.5 directly. It participates in the regulation of Nav1.5 by binding to dystrophin.
Author response image 22.