Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorDion DickmanUniversity of Southern California, Los Angeles, United States of America
- Senior EditorLu ChenStanford University, Stanford, United States of America
Reviewer #1 (Public review):
Koesters and colleagues investigated the role of the small GTPase Rab3A in homeostatic scaling of miniature synaptic transmission in primary mouse cortical cultures using electrophysiology and immunohistochemistry. The major finding is that TTX incubation for 48 hours does not induce an increase in the amplitude of excitatory synaptic miniature events in neuronal cultures derived from Rab3A KO and Rab3A Earlybird mutant mice. NASPM application had comparable effects on mEPSC amplitude in control and after TTX, implying that Ca2+-permeable glutamate receptors are unlikely modulated during synaptic scaling. Immunohistochemical analysis revealed no significant changes in GluA2 puncta size, intensity, and integral in control and Rab3A KO cultures. Finally, they provide evidence that loss of Rab3A in neurons, but not astrocytes, blocks homeostatic scaling. Based on these data, the authors propose a model in which neuronal Rab3A is required for homeostatic scaling of synaptic transmission through GluA2-dependent and independent mechanisms.
While the title of the manuscript is mostly supported by data of solid quality, many conclusions, as well as the final model, cannot be derived from the results presented. Importantly, the data do not support that GluA2 levels change upon TTX treatment in control cultures, rendering conclusions regarding Rab3A's role in TTX-dependent GluA2 modulation spurious. Other aspects of the model, such as a Rab3A-dependent release of a tropic factor, cannot be derived from the data.
The following points should be addressed:
(1) There is no (significant) increase in GluA2 levels (intensity, area, or integral) upon TTX treatment in controls (Fig. 5). Conclusions regarding Rab3As role in TTX-dependent GluA2 modulation should be revised accordingly. Hence, the data shown in Fig. 4 - 7 do not allow drawing conclusions in the context of Rab3A-dependent GluA2 modulation and scaling.
(2) The effects of Rab3A on TTX-induced mini frequency modulation remains unclear, because TTX does not induce a change in mini frequency in the Rab3A+/Ebd control (Fig. 2). The respective conclusions should be revised accordingly (l. 427).
(3) The model is still not supported by the data. In particular, data supporting a negative regulation of Rab3A by APs, Rab3A-dependent release of a tropic factor, or a Rab3A-dependent increase in GluA2 abundance are not presented.
(4) Data points are not overlapping and appear "quantal" in most box plots. How were the data rounded?
Reviewer #2 (Public review):
In the revised manuscript, the authors investigated the role of a presynaptic protein, Rab3A, in the homeostatic synaptic plasticity in cultured cortical neurons. The study was motivated by their previous findings that Rab3A is required for expression of similar homeostatic mechanisms at the neuromuscular junction. The authors first show that untreated WT neurons express homeostatic synaptic plasticity in response to 48h of TTX treatment (upregulation of both mEPSC amplitude and frequency), whereas neurons lacking Rab3A or carrying a dominant negative mutated Rab3A (earlybird) do not. They also demonstrate that only neuronal, but not glial Rab3A is responsible for this defect. Furthermore, they confirm the increased mEPSC amplitudes in WT neurons reflect the addition of GluA2-containing AMPA receptors rather than calcium-permeable ones, as previously reported by multiple labs. However, the increase in mEPSC amplitude is not accompanied by a corresponding upregulation of GluA2 synaptic clusters according to their IHC data (cluster size and intensity trend slightly upwards but not reaching significance). They further show that this modest upward trend is absent in Rab3A KO neurons, and conclude that Rab3A is involved in postsynaptic GluA2 upregulation during homeostatic synaptic plasticity.
When compared to the original version, the authors have done an admirable job in switching to more established ways to assess homeostatic synaptic plasticity by comparing the mean mEPSC amplitude and frequency, which has greatly improved the legibility of the manuscript to the public. Their data in Figures 1,2, and 8 clearly demonstrate that functional Rab3A in cortical neurons is required for the homeostatic regulation of mEPSCs.
However, the authors still have not provided further investigation of the mechanisms behind the role of Rab3A in this form of plasticity, and the revision therefore has added little to the significance of the study. Moreover, the experimental design for the investigation of the mismatch between mEPSC amplitude and GluA2 cluster fluorescence remains questionable, making it difficult to draw any credible conclusions from groups of data that not only look similar to the eye but also show no significance statistically.
A major claim the authors want to make is that Rab3A, although a presynaptic protein, regulates postsynaptic GluA2, and they do this by showing in Figure 5 that the upward trend of GluA2 cluster size and intensity is absent in Rab3A KO neurons. First, it is difficult to convince readers that this upward trend is real in Figures 5B-D without getting more samples. Second, the authors pick GluA2 clusters on the primary dendrites, whereas mEPSC events come from a much larger synapse population (e.g., more distal), therefore it makes sense that these two forms of measurement do not show matching changes, and this caveat could be addressed by sampling more diverse dendritic locations. Without a convincing phenotype in WT neurons, the support for this claim is weak.
Another claim of the authors is that this mismatch between mEPSC amplitude and GluA2 cluster sizes with the same culture suggests there are other factors contributing to the mEPSC amplitude. They do this by comparing results from individual culture dissociations, which greatly suffer from undersampling (Figure 6). In particular, they point out that 2 out of 3 dissociations show "matching" upward trends in mEPSC and GluA2 cluster (figure 6A and 6B) while the third one shows opposite trends, and use this to support their claim. Anyone who has done culture preparation would know the high variability between dissociations, which is why culture data are always pooled for assessment of any population trend. Anything could have happened to this particular dissociation (culture #3, figure 6C), and drawing conclusion from this single incident does little to support this claim. At least, they should double the dissociation numbers, and their claim would be much more convincing if a similar phenomenon occurs again. Besides, as mentioned above, all these mismatching trends could just be due to sampling differences.
In summary, this study establishes that neuronal Rab3A plays a role in homeostatic synaptic plasticity, but so do a number of other molecules that have been implicated in homeostatic synaptic plasticity in the past two decades (only will grow with the new techniques such as RNAseq). Without going beyond this finding and demonstrating how exactly Rab3A participates in the induction and/or expression of this form of plasticity, or maybe the potential Rab3A-mediated functional and behavioral defects in vivo, the contribution of the current study to the field is limited. However, given the presynaptic location of Rab3A, this finding could serve as a starting point for researchers interested in pre-postsynaptic cross-talk during homeostatic plasticity in general.
Reviewer #3 (Public review):
This manuscript presents a number of interesting findings that have the potential to increase our understanding of the mechanism underlying homeostatic synaptic plasticity (HSP). The data broadly support that Rab3A plays a role in HSP, although the site and mechanism of action remain uncertain.
The authors clearly demonstrate the Rab3A plays a role in HSP at excitatory synapses, with substantially less plasticity occurring in the Rab3A KO neurons. There is also no apparent HSP in the Earlybird Rab3A mutation, although baseline synaptic strength seems already elevated. In this context, it is unclear if the plasticity is absent or just occluded by a ceiling effect due the synapses already being strengthened. Occlusion may also occur in the mixed cultures, with Rab3A missing from neurons but not astrocytes. The authors do appropriately discuss both options. There are also differences in genetic background between the Rab3A KO and Earlybird mutants that could also impact the results, which are also noted. The authors have solid data showing that Rab3A is unlikely to be active in astrocytes, Finally, they attempt to study the linkage between synaptic strength during HSP and AMPA receptor trafficking and conclude that trafficking may not be solely responsible for the changes in synaptic strength.
Strengths:
This work adds another player into the mechanisms underlying an important form of synaptic plasticity. The plasticity is likely only reduced, suggesting Rab3A is only partially required and perhaps multiple mechanisms contribute. The authors speculate about some possible novel mechanisms.
However, the conclusions on the partial dissociation of AMPAR trafficking and synaptic response are made from somewhat weaker data. On average, across 3 culture sets, they saw similar magnitude of change in mEPSC amplitude and GluA2 cluster area and integral, but the GluA2 data was not significant. This is likely due to the nature of the datasets. Their imaging method involves only assessing puncta pairs (GluA2/VGlut1) clearly associated with a MAP2 labeled dendrite. This is a small subset of synapses, with usually less than 20 synapses per neuron analyzed (as stated by the authors). The mEPSC recordings will be averaging across several hundred events, which likely represent a hundred or more synapses given reasonable expectations on release probability. It has been reported, in work from this lab as well as by direct monitoring of tagged AMPARs during HSP (Wang, et al., 2019), that individual synapses are quite variable in their response. So there will almost necessarily be higher variability in the imaging data due to the smaller number of synapses sampled. The overall trends, though, are in alignment with previous data implicating receptor trafficking as the mechanism for HSP. However, the authors go on to evaluate each of the individual cultures, where 2 show similar changes between the mEPSC data and GluA2 clusters, and 1 culture showing little/no change in GluA2 clusters. The n's are very low here, and none of the datasets are significant. They want to conclude for this culture, there was a change in mEPSC amplitude that was not accompanied by a change in GluA2 at synaptic sites. But these data are collected from different coverslips, and due to the low n's, the potential under-sampling of the GluA2 clusters, and neuron-to-neuron variability, it is very hard to distinguish if this apparent difference is a methodological issue rather than a biological one. Much stronger data would be necessary to conclude that additional factors beyond receptor trafficking are required for HSP.
Other questions arise from the NASPM experiments, used to justify looking at GluA2 (and not GluA1) in the immunostaining. First, there is a frequency effect that is unclear in origin. One would expect NASPM to merely block some fraction of the post-synaptic current, and not affect pre-synaptic release or block whole synapses. However the change in frequency seems to argue (as the authors do) that some synapses only have CP-AMPARs, while the rest of the synapses have few or none. Another possibility is that there are pre-synaptic NASPM-sensitive receptors that influence release probability. Further, the amplitude data show a strong trend towards smaller amplitude following NASPM treatment (Fig 3B). The p value for both control and TTX neurons was 0.08 - it is very difficult to argue that there is no effect. The decrease on average is larger in the TTX neurons, and some cells show a strong effect. It is possible there is some heterogeneity between neurons on whether GluA1/A2 heteromers or GluA1 homomers are added during HSP. This would impact the weakly supported conclusions about the GluA2 imaging vs mEPSC amplitude data.
Unaddressed issues that would greatly increase the impact of the paper:
(1) Is Rab3A acting pre-synaptically, post-synaptically or both? The authors provide good evidence that Rab3A is acting within neurons and not astrocytes. But where it is acting (pre or post) would aid substantially in understanding its role. They could use sparse knock-down of Rab3A, or simply mix cultures from KO and WT mice (with appropriate tags/labels). The general view in the field has been that HSP is regulated post-synaptically via regulation of AMPAR trafficking, and considerable evidence supports this view. The more support for their suggestion of a pre-synaptic site of control, the better.
(2) Rab3A is also found at inhibitory synapses. It would be very informative to know if HSP at inhibitory synapses is similarly affected. This is particularly relevant as at inhibitory synapses, one expects a removal of GABARs (ie the opposite of whatever is happening at excitatory synapses). If both processes are regulated by Rab3A, this might suggest a role for this protein more upstream in the signaling; an effect only at excitatory synapses would argue for a more specific role just at these synapses.
Author response:
The following is the authors’ response to the original reviews.
The detailed, thorough critique provided by the three reviewers is very much appreciated. We believe the manuscript is greatly improved by the changes we have made based on those reviews. The major changes are described below, followed by a point by point response.
Major Changes:
(1) We revised our model (old Fig. 10; new Fig. 9) to keep the explanation focused on the data shown in the current study. Specifically, references to GTP/GDP states of Rab3A and changes in the presynaptic quantum have been removed and the mechanisms depicted are confined to pre- or post-synaptic Rab3A participating in either controlling release of a trophic factor that regulates surface GluA2 receptors (pre- or postsynaptic) or directly affecting fusion of GluA2-receptor containing vesicles (postsynaptic).
(2) We replaced all cumulative density function plots and ratio plots, based on multiple quantile samples per cell, with box plots of cell means. This affects new Figures 1, 2, 3, 5, 6, 7 and 8. All references to “scaling,” “divergent scaling,” or “uniform scaling,” have been removed. New p values for comparison of means are provided above every box plot in Figures 1, 2, 3, 5, 6, 7 and 8. The number of cultures is provided in the figure legends.
(3) We have added frequency to Figures 1, 2 and 8. Frequency values overall are more variable, and the effect of activity blockade less robust, than for mEPSC amplitudes. We have added text indicating that the increase in frequency after activity blockade was significant in neurons from cultures prepared from WT in the Rab3A+/- colony but not cultures prepared from KO mice (Results, lines 143 to 147, new Fig. 1G. H). The TTX-induced increase in frequency was significant in the NASPM experiments before NASPM, but not after NASPM (Results, lines 231 to 233, new Fig. 3, also cultures from WT in Rab3A+/- colony). The homeostatic plasticity effect on frequency did not reach significance in WT on WT glia cultures or
WT on KO glia cultures, possibly due to the variability of frequency, combined with smaller sample sizes (Results, lines 400 to 403, new Fig. 8). In the cultures prepared from WT mice in the Rab3A+/Ebd colony, there was a trend towards higher frequency after TTX that did not reach statistical significance, and in cultures prepared from mutant mice, the p value was large, suggesting disruption of the effect, which appears to be due to an increase in frequency in untreated cultures, similar to the behavior of mEPSC amplitudes in neurons from mutant mice (Results, lines 161-167). In sum, the effect of activity on frequency requires Rab3A and Ca2+-permeable receptors, and is mimicked by the presence of the Rab3A Earlybird mutant. We have also added a discussion of these results (Discussion, lines 427-435).
(4) In the revised manuscript we have added analysis of VGLUT1 levels for the same synaptic sites that we previously analyzed GluA2 levels, and these data are described in Results, lines 344 to 371, and appear in new Table 2. In contrast to previous studies, we did not find any evidence for an increase in VGLUT1 levels after activity blockade. We reviewed those studies to determine whether there might be differences in the experimental details that could explain the lack of effect we observed. In (De Gois et al., 2005), the authors measured mRNA and performed western blots to show increases in VGLUT1 after TTX treatment in older rat cortical cultures (DIV 19). The study performs immunofluorescence imaging of VGLUT1 but only after bicuculline treatment (it decreases), not after TTX treatment. In (Wilson et al.,
2005), the hippocampal cultures are treated with AP5, not TTX, and the VGLUT1 levels in immunofluorescence images are reported relative to synapsin I. That the type of activity blockade matters is illustrated by the failure of Wilson and colleagues to observe a consistent increase in VGLUT1/Synapsin ratio in cultures treated with AMPA receptor blockade (NBQX; supplementary information). These points have been added to the Discussion, lines 436 to 447.)
Reviewer #1:
(1) (model…is not supported by the data), (2) (The analysis of mEPSC data using quantile sampling…), (3) (…statistical analysis of CDFs suffers from n-inflation…), (4) (How does recording noise and the mEPSC amplitude threshold affect “divergent scaling?”) (5) (…justification for the line fits of the ratio data…), (7) (A comparison of p-values between conditions….) and (10) (Was VGLUT intensity altered in the stainings presented in the manuscript?)
The major changes we made, described above, address Reviewer #1’s points. The remaining points are addressed below.
(6) TTX application induces a significant increase in mEPSC amplitude in Rab3A-/- mice in two out of three data sets (Figs. 1 and 9). Hence, the major conclusion that Rab3A is required for homeostatic scaling is only partially supported by the data.
The p values based on CDF comparisons were problematic, but the point we were making is that they were much larger for amplitudes measured in cultures prepared from Rab3A-/- mice (Fig. 1, p = 0.04) compared to those from cultures prepared from Rab3A+/+ mice (Fig. 1, p = 4.6 * 10-4). Now that we are comparing means, there are no significant TTX-induced effects on mEPSC amplitudes for Rab3A-/- data. However, acknowledging that some increase after activity blockade remains, we describe homeostatic plasticity as being impaired or not significant, rather than abolished, by loss of Rab3A, (Abstract, lines 37 to 39; Results, lines 141 to 143; Discussion, lines 415 to 418).
(8) There is a significant increase in baseline mEPSC amplitude in Rab3AEbd/Ebd (15 pA) vs. Rab3AEbd/+ (11 pA) cultures, but not in Rab3A-/- (13.6 pA) vs. Rab3A+/- (13.9 pA). Although the nature of scaling was different between Rab3AEbd/Ebd vs. Rab3AEbd/+ and Rab3AEbd/Ebd with vs. without TTX, the question arises whether the increase in mEPSC amplitude in Rab3AEbd/Ebd is Rab3A dependent. Could a Rab3A independent mechanism occlude scaling?
The Reviewer is concerned that the increase in mEPSC amplitude in the presence of the Rab3A point mutant may be through a ‘non-Rab3A’ mechanism (a concern raised by the lack of such effect in cultures from the Rab3A-/- mice), and secondly, that the already large mEPSC cannot be further increased by the homeostatic plasticity mechanism. It must always be considered that a mutant with an altered genetic sequence may bind to novel partners, causing activities that would not be either facilitated or inhibited by the original molecule. We have added this caveat to Results, lines 180 to 186 We added that a number of other manipulations, implicating individual molecules in the homeostatic mechanism, have caused an increase in mEPSC amplitude at baseline, potentially nonspecifically occluding the ability of activity blockade to induce a further increase (Results lines 186 to 189). Still, it is a strong coincidence that the novel activity of the mutant Rab3A would affect mEPSC amplitude, the same characteristic that is affected by activity blockade in a Rab3A dependent manner, a point which we added to Results, lines 189 to 191.
(9) Figure 4: NASPM appears to have a stronger effect on mEPSC frequency in the TTX condition vs. control (-40% vs -15%). A larger sample size might be necessary to draw definitive conclusions on the contribution of Ca2+-permeable AMPARs.
Our results, even with the modest sample size of 11 cells, are clear: NASPM does not disrupt the effect of TTX treatment on mEPSC amplitude (new Fig. 3A). It also looks like there is a greater magnitude effect of NAPSM on frequency in TTX-treated cells; we note this, but point out that nevertheless, these mEPSCs are not contributing to the increase in mEPSC amplitude (Results, lines 238-241).
(11) The change in GluA2 area or fluorescence intensity upon TTX treatment in controls is modest. How does the GluA2 integral change?
We had reported that GluA2 area showed the most prominent increase following activity blockade, with intensity changing very little. When we examined the integral, it closely matched the change in area. We have added the values for integral to new Fig. 5 D, H; new Fig. 6 A-C; new Fig. 7 A-C and new Table 1 (for GluA2) and new Table 2 (for VGLUT1). These results are described in the text in the following places: Results, lines 289-292; 298-299; 311-319; 328-324). For VGLUT1, both area and intensity changed modestly, and the integral appeared to be a combination of the two, being higher in magnitude and resulting in smaller p values than either area or intensity (Results, lines 344-348; 353-359; new Table 2).
(12) The quantitative comparison between physiology and microscopy data is problematic. The authors report a mismatch in ratio values between the smallest mEPSC amplitudes and the smallest GluA2 receptor cluster sizes (l. 464; Figure 8). Is this comparison affected by the fluorescence intensity threshold? What was the rationale for a threshold of 400 a.u. or 450 a.u.? How does this threshold compare to the mEPSC threshold of 3 pA.
This concern is partially addressed by no longer comparing the rank ordered mEPSC amplitudes with the rank ordered GluA2 receptor characteristics. We had used multiple thresholds in the event that an experiment was not analyzable with the chosen threshold (this in fact happened for VGLUT1, see end of this paragraph). We created box plots of the mean GluA2 receptor cluster size, intensity and integral, for experiments in which we used all three thresholds, to determine if the effect of activity blockade was different depending on which threshold was applied, and found that there was no obvious difference in the results (Author response image 1). Nevertheless, since there is no need to use a different threshold for any of the 6 experiments (3 WT and 3KO), for new Figures 5, 6 and 7 we used the same threshold for all data, 450; described in Methods, lines 746 to 749. For VGLUT1 levels, it was necessary to use a different threshold for Rab3A+/+ Culture #1 (400), but a threshold of 200 for the other five experiments (Methods, lines 751-757). The VGLUT1 immunofluorescent sites in Culture #1 had higher levels overall, and the low threshold caused the entire AOI to be counted as the synapse, which clearly included background levels outside of the synaptic site. Conversely, to use a threshold of 400 on the other experiments meant that the synaptic site found by the automated measurement tool was much smaller that what was visible by eye. In our judgement it would have been meaningless to adhere to a single threshold for VGLUT1 data.
Author response image 1.
Using different thresholds does not substantially alter GluA2 receptor cluster size data. A) Rab3A+/+ Culture #1, size data for three different thresholds, depicted above each graph. B) Rab3A+/+ Culture #2, size data for three different thresholds, depicted above each graph. Note scale bar in A is different from B, to highlight differences for different thresholds. (Culture #3 was only analyzed with 450 threshold).
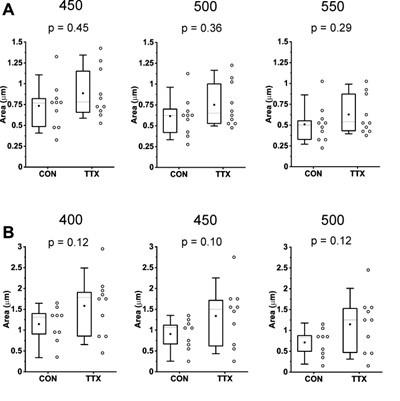
The conclusion that an increase in AMPAR levels is not fully responsible for the observed mEPSC increase is mainly based on the rank-order analysis of GluA2 intensity, yielding a slope of ~0.9. There are several points to consider here: (i) GluA2 fluorescence intensity did increase on average, as did GluA2 cluster size.
(ii) The increase in GluA2 cluster size is very similar to the increase in mEPSC amplitude (each approx. 1820%). (iii) Are there any reports that fluorescence intensity values are linearly reporting mEPSC amplitudes (in this system)? Antibody labelling efficiency, and false negatives of mEPSC recordings may influence the results. The latter was already noted by the authors.
Our comparison between mEPSC amplitude and GluA2 receptor cluster characteristics has been reexamined in the revised version using means rather than rank-ordered data in rank-order plots or ratio plots. Importantly, all of these methods revealed that in one out of three WT cultures (Culture #3) GluA2 receptor cluster size (old Fig. 8, old Table 1; new Fig. 6, new Table 1), intensity and integral (new Fig. 6, new Table 1) values decreased following activity blockade while in the same culture, mEPSC amplitudes increased. It is based on this lack of correspondence that we conclude that increases in mEPSC amplitude are not fully explained by increases in GluA2 receptors, and suggest there may be other contributors. These points are made in the Abstract (lines 108-110); Results (lines 319 to 326; 330337; 341-343) and the Discussion (lines 472 to 474). To our knowledge, there are not any reports that quantitatively compare receptor levels (area, intensity or integrals) to mEPSC amplitudes in the same cultures. We examined the comparisons very closely for 5 studies that used TTX to block activity and examined receptor levels using confocal imaging at identified synapses (Hou et al., 2008; Ibata et al., 2008; Jakawich et al., 2010a; Xu and Pozzo-Miller, 2017; Dubes et al., 2022). We were specifically looking for whether the receptor data were more variable than the mEPSC amplitude data, as we found. However, for 4 of the studies, sample sizes were very different so that we cannot simply compare the p values. Below is a table of the comparisons.
Author response table 1.
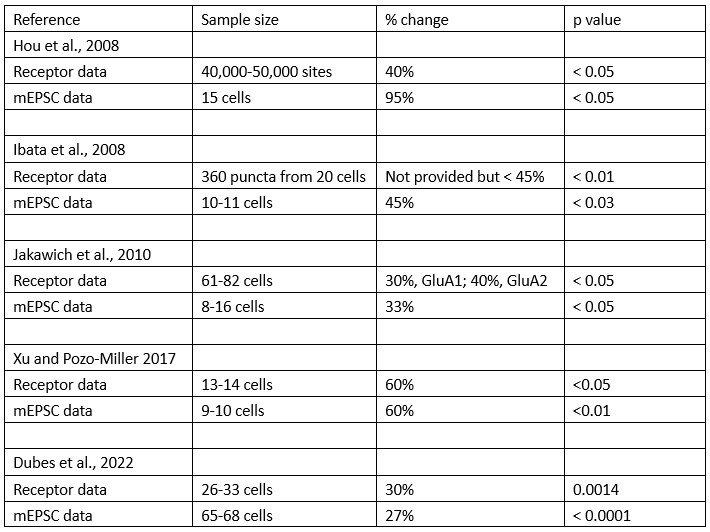
In Xu 2017 the sample sizes are close enough that we feel comfortable concluding that the receptor data were slightly more variable (p < 0.05) than mEPSC data (p<0.01) but recognize that it is speculative to say our finding has been confirmed. A discussion of these articles is in Discussion, lines 456-474.
(iv) It is not entirely clear if their imaging experiments will sample from all synapses. Other AMPAR subtypes than GluA2 could contribute, as could kainite or NMDA receptors.
While our imaging data only examined GluA2, we used the application of NASPM to demonstrate Ca2+permeable receptors did not contribute quantitatively to the increase in mEPSC amplitude following TTX treatment. Since GluA3 and GluA4 are also Ca2+-permeable, the findings in new Figure 3 (old Fig. 4) likely rule out these receptors as well. There are also reports that Kainate receptors are Ca2+-permeable and blocked by NASPM (Koike et al., 1997; Sun et al., 2009), suggesting the NASPM experiment also rules out the contribution of Kainate receptors. Finally, given our recording conditions, which included normal magnesium levels in the extracellular solution as well as TTX to block action-potential evoked synaptic transmission, NMDA receptors would not be available to contribute currents to our recordings due to block by magnesium ions at resting Vm. These points have been added to the Methods section, lines 617 to 677 (NMDA); 687-694 (Ca2+-permeable AMPA receptors and Kainate receptors).
Furthermore, the statement “complete lack of correspondence of TTX/CON ratios” is not supported by the data presented (l. 515ff). First, under the assumption that no scaling occurs in Rab3A-/-, the TTX/CON ratios show a 20-30% change, which indicates the variation of this readout. Second, the two examples shown in Figure 8 for Rab3A+/+ are actually quite similar (culture #1 and #2, particularly when ignoring the leftmost section of the data, which is heavily affected by the raw values approaching zero.
We are no longer presenting ratio plots in the revised manuscript, so we do not base our conclusion that mEPSC amplitude data is not always corresponding to GluA2 receptor data on the difference in behavior of TTX/CON ratio values, but only on the difference in direction of the TTX effect in one out of three cultures. We agree with the reviewer that the ratio plots are much more sensitive to differences between control and treated values than the rank order plot, and we feel these differences are important, for example, there is still a homeostatic increase in the Rab3A-/- cultures, and the effect is still divergent rather than uniform. But the comparison of ratio data will be presented elsewhere.
(13) Figure 7A: TTX CDF was shifted to smaller mEPSC amplitude values in Rab3A-/- cultures. How can this be explained?
While this result is most obvious in CDF plots, we still observe a trend towards smaller mEPSC amplitudes after TTX treatment in two of three individual cultures prepared from Rab3A-/- mice when comparing means (new Fig. 7, Table 1) which did not reach statistical significance for the pooled data (new Fig. 5, new Table 1). There was not any evidence of this decrease in the larger data set (new Fig. 1) nor for Rab3A-/- neurons on Rab3A+/+ glia (new Fig. 8). Given that this effect is not consistent, we did not comment on it in the revised manuscript. It may be that there is a non-Rab3A-dependent mechanism that results in a decrease in mEPSC amplitude after activity blockade, which normally pulls down the magnitude of the activity-dependent increase typically observed. But studying this second component would be difficult given its magnitude and inconsistent presentation.
Reviewer #1 (Recommendations For the Authors):
(1) Abstract, last sentence: The conclusion of the present manuscript should be primarily based on the results presented. At present, it is mainly based on a previous publication by the authors.
We have revised the last sentence to reflect actual findings of the current study (Abstract, lines 47 to 49).
(2) Line 55: “neurodevelopmental”
This phrase has been removed.
(3) Line 56: “AMPAergic” should be replaced by AMPAR-mediated
This sentence was removed when all references to “scaling” were removed; no other instances of “AMPAergic” are present.
(4) Figure 9: The use of BioRender should be disclosed in the Figure Legend.
We used BioRender in new Figures 3, 7 and 8, and now acknowledge BioRender in those figure legends.
(5) Figure legends and results: The number of cultures should be indicated for each comparison.
Number of cultures has been added to the figure legends.
(6) Line 289: A comparison of p-values between conditions does not allow any meaningful conclusions.
Agreed, therefore we have removed CDFs and the KS test comparison p values. All comparisons in the revised manuscript are for cell means.
(7) Line 623ff: The argument referring to NMJ data is weak, given that different types of receptors are involved.
We still think it is valid to point out that Rab3A is required for the increase in mEPC at the NMJ but that ACh receptors do not increase (Discussion, lines 522 to 525). We are not saying that postsynaptic receptors do not contribute in cortical cultures, only that there could be another Rab3A-dependent mechanism that also affects mEPSC amplitude.
(8) Plotting data points outside of the ranges should be avoided (e.g., Fig. 2Giii, 7F).
These two figures are no longer present in the revised manuscript. In revising figures, we made sure no other plots have data points outside of the ranges.
(9) The rationale for investigating Rab3AEbd/Ebd remains elusive and should be described.
A rationale for investigating Rab3AEbd/Ebd is that if the results are similar to the KO, it strengthens the evidence for Rab3A being involved in homeostatic synaptic plasticity. In addition, since its phenotype of early awakening was stronger than that demonstrated in Rab3A KO mice (Kapfhamer et al., 2002), it was possible we would see a more robust effect. These points have been added to the Results, lines 118 to 126.
(10) Figures 3 and 4, as well as Figure 5 and 6 could be merged.
In the revised version, Figure 3 has been eliminated since its main point was a difference in scaling behavior. Figure 4 has been expanded to include a model of how NASPM could reduce frequency (new Fig. 3.) Images of the pyramidal cell body have been added to Figure 5 (new Fig. 4), and Figure 6 has been completely revised and now includes pooled data for both Rab3A+/+ and Rab3A-/- cultures, for mEPSC amplitude, GluA2 receptor cluster size, intensity and integral.
(11) Figure 5: The legend refers to MAP2, but this is not indicated in the figure.
MAP2 has now been added to the labels for each image and described in the figure legend (new Fig. 4).
Reviewer #2:
Technical concerns:
(1) The culture condition is questionable. The authors saw no NMDAR current present during spontaneous recordings, which is worrisome since NMDARs should be active in cultures with normal network activity (Watt et al., 2000; Sutton et al., 2006). It is important to ensure there is enough spiking activity before doing any activity manipulation. Similarly it is also unknown whether spiking activity is normal in Rab3AKO/Ebd neurons.
In the studies cited by the reviewer, NMDA currents were detected under experimental conditions in which magnesium was removed. In our recordings, we have normal magnesium (1.3 mM) and also TTX, which prevents the necessary depolarization to allow inward current through NMDA receptors. This point has been added to our Methods, lines 674 to 677. We acknowledge we do not know the level of spiking in cultures prepared from Rab3A+/+, Rab3A-/- or Rab3A_Ebd/Ebd_ mice. Given the similar mEPSC amplitude for untreated cultures from WT and KO studies, we think it unlikely that activity was low in the latter, but it remains a possibility for untreated cultures from Rab3A_Ebd/Ebd_ mice, where mEPSC amplitude was increased. These points are added to the Methods, lines 615 to 622.
(2) Selection of mEPSC events is not conducted in an unbiased manner. Manually selecting events is insufficient for cumulative distribution analysis, where small biases could skew the entire distribution. Since the authors claim their ratio plot is a better method to detect the uniformity of scaling than the well-established rank-order plot, it is important to use an unbiased population to substantiate this claim.
We no longer include any cumulative distributions or ratio plot analysis in the revised version. We have added the following text to Methods, lines 703 to 720:
“MiniAnalysis selects many false positives with the automated feature when a small threshold amplitude value is employed, due to random fluctuations in noise, so manual re-evaluation of the automated process is necessary to eliminate false positives. If the threshold value is set high, there are few false positives but small amplitude events that visually are clearly mEPSCs are missed, and manual re-evaluation is necessary to add back false negatives or the population ends up biased towards large mEPSC amplitudes. As soon as there is a manual step, bias is introduced. Interestingly, a manual reevaluation step was applied in a recent study that describes their process as ‘unbiased (Wu et al., 2020). In sum, we do not believe it is currently possible to perform a completely unbiased detection process. A fully manual detection process means that the same criterion (“does this look like an mEPSC?”) is applied to all events, not just the false positives, or the false negatives, which prevents the bias from being primarily at one end or the other of the range of mEPSC amplitudes. It is important to note that when performing the MiniAnalysis process, the researcher did not know whether a record was from an untreated cell or a TTX-treated cell.”
(3) Immunohistochemistry data analysis is problematic. The authors only labeled dendrites without doing cell-fills to look at morphology, so it is questionable how they differentiate branches from pyramidal neurons and interneurons. Since glutamatergic synapse on these two types of neuron scale in the opposite directions, it is crucial to show that only pyramidal neurons are included for analysis.
We identified neurons with a pyramidal shape and a prominent primary dendrite at 60x magnification without the zoom feature. This should have been made clear in the description of imaging. We have added an image of the two selected cells to our figure of dendrites (old Fig. 5, new Fig. 4), and described this process in the Methods, lines 736 to 739, and Results, lines 246 to 253. Given the morphology of the neurons selected it is highly unlikely that the dendrites we analyzed came from interneurons.
Conceptual Concerns
The only novel finding here is the implicated role for Rab3A in synaptic scaling, but insights into mechanisms behind this observation are lacking. The authors claim that Rab3A likely regulates scaling from the presynaptic side, yet there is no direct evidence from data presented. In its current form, this study’s contribution to the field is very limited.
We have demonstrated that loss of Rab3A and expression of a Rab3A point mutant disrupt homeostatic plasticity of mEPSC amplitudes, and that in the absence of Rab3A, the increase in GluA2 receptors at synaptic sites is abolished. Further, we show that this effect cannot be through release of a factor, like TNFα, from astrocytes. In the new version, we add the finding that VGLUT1 is not increased after activity blockade, ruling out this presynaptic factor as a contributor to homeostatic increases in mEPSC amplitude. We show for the first time by examining mEPSC amplitudes and GluA2 receptors in the same cultures that the increases in GluA2 receptors are not as consistent as the increases in mEPSC amplitude, suggesting the possibility of another contributor to homeostatic increases in mEPSC amplitude. We first proposed this idea in our previous study of Rab3A-dependent homeostatic increases in mEPC amplitudes at the mouse neuromuscular junction. In sum, we dispute that there is only one novel finding and that we have no insights into mechanism. We acknowledge that we have no direct evidence for regulation from the presynaptic side, and have removed this claim from the revised manuscript. We have retained the Discussion of potential mechanisms affecting the presynaptic quantum and evidence that Rab3A is implicated in these mechanisms (vesicle size, fusion pore kinetics; Discussion, lines 537 to 563). One way to directly show that the amount of transmitter released for an mEPSC has been modified after activity blockade is to demonstrate that a fast off-rate antagonist has become less effective at inhibiting mEPSCs (because the increased glutamate released out competes it; see (Liu et al., 1999) and (Wilson et al., 2005) for example experiments). This set of experiments is underway but will take more time than originally expected, because we are finding surprisingly large decreases in frequency, possibly the result of mEPSCs with very low glutamate concentration that are completely inhibited by the dose used. Once mEPSCs are lost, it is difficult to compare the mEPSC amplitude before and after application of the antagonist. Therefore we intend to include this experiment in a future report, once we determine the reason for the frequency reduction, or, can find a dose where this does not occur.
(1) Their major argument for this is that homeostatic effects on mEPSC amplitudes and GluA2 cluster sizes do not match. This is inconsistent with reports from multiple labs showing that upscaling of mEPSC amplitude and GluA2 accumulation occur side by side during scaling (Ibata et al., 2008; Pozo et al., 2012; Tan et al., 2015; Silva et al., 2019). Further, because the acquisition and quantification methods for mEPSC recordings and immunohistochemistry imaging are entirely different (each with its own limitations in signal detection), it is not convincing that the lack of proportional changes must signify a presynaptic component.
Within the analyses in the revised manuscript, which are now based only on comparison of cell/dendrite means, we find a very good match in the magnitude of increase for the pooled data of mEPSC amplitudes and GluA2 receptor cluster sizes (+19.7% and +20.0% respectively; new Table 1). However, when looking at individual cultures, we had one of three WT cultures in which mEPSC amplitude increased 17.2% but GluA2 cluster size decreased 9.5%. This result suggests that while activity blockade does lead to an increase in GluA2 receptors after activity blockade, the effect is more variable than that for mEPSC amplitude. We went back to published studies to see if this has been previously observed, but found that it was difficult to compare because the sample sizes were different for the two characteristics (see Author response table 1). We included these particular 5 studies because they use the same treatment (TTX), examine receptors using imaging of identified synaptic sites, and record mEPSCs in their cultures (although the authors do not indicate that imaging and recordings are done simultaneously on the same cultures.) Only one of the studies listed by the Reviewer is in our group (Ibata et al., 2008). The study by (Tan et al., 2015) uses western blots to measure receptors; the study by (Silva et al., 2019) blocks activity using a combination of AMPA and NMDA receptor blockers; the study by (Pozo et al., 2012) correlates mEPSC amplitude changes with imaging but not in response to activity blockade, instead for changing the expression of GluA2. While it may seem like splitting hairs to reject studies that use other treatment protocols, there is ample evidence that the mechanisms of homeostatic plasticity depend on how activity was altered, see the following studies for several examples of this (Sutton et al., 2006; Soden and Chen, 2010; Fong et al., 2015). A discussion of the 5 articles we selected is in the revised manuscript, Discussion, lines 456 to 474. In sum, we provide evidence that activity blockade is associated with an overall increase in GluA2 receptors; what we propose is that this increase, being more variable, does not fully explain the increase in mEPSC amplitude. However, we acknowledge that the disparity could be explained by the differences in limitations of the two methods (Discussion, lines 469-472).
(2) The authors also speculate in the discussion that presynaptic Rab3A could be interacting with retrograde BDNF signaling to regulate postsynaptic AMPARs. Without data showing Rab3A-dependent presynaptic changes after TTX treatment, this argument is not compelling. In this retrograde pathway, BDNF is synthesized in and released from dendrites (Jakawich et al., 2010b; Thapliyal et al., 2022), and it is entirely possible for postsynaptic Rab3A to interfere with this process cell-autonomously.
We have added the information that Rab3A could control BDNF from the postsynaptic cell and included the two references provided by the reviewer, Discussion, lines 517 to 518. We have added new evidence, recently published, that the Rab3 family has been shown to regulate targeting of EGF receptors to rafts (among other plasma membrane molecules), with Rab3A itself clearly present in nonneuronal cells (Diaz-Rohrer et al., 2023) (added to Discussion, lines 509 to 515).
(3) The authors propose that a change in AMPAR subunit composition from GluA2-containing ones to GluA1 homomers may account for the distinct changes in mEPSC amplitudes and GluA2 clusters. However, their data from the NASPM wash-in experiments clearly show that the GluA1 homomer contributions have not changed before and after TTX treatment.
We have revised this section in the Discussion, lines 534 to 536, to clarify that any change due to GluA1 homomers should have been detectable by a greater ability of NASPM to reverse the TTX-induced increase.
Reviewer #2 (Recommendations for the Authors):
For authors to have more convincing arguments in general, they will need to clarify/improve certain details in their data collection by addressing the above technical concerns. Additionally, the authors should design experiments to test whether Rab3A regulates scaling from pre- or post-synaptic site. For example, they could sparsely knock out Rab3A in WT neurons to test the postsynaptic possibility. On the other hand, their argument for a presynaptic role would be much more compelling if they could show whether there are clear functional changes such as in vesicle sizes and release probability in the presynaptic terminal of Rab3AKO neurons.
An important next step is to identify whether Rab3A is acting pre- or post-synaptically (Discussion, lines 572 to 573), but these experiments will be undertaken in the future. It would not add much to simply show vesicle size is altered in the KO (and we do not necessarily expect this since mEPSC amplitude is normal in the KO). It will be very difficult to establish that vesicle size is changing with activity blockade and that this change is prevented in the Rab3A KO, because we are looking for a ~25% increase in vesicle volume, which would correspond to a ~7.5% increase in diameter. Finally, we do not believe demonstrating changes in release probability tell us anything about a presynaptic role for Rab3A in regulating the size of the presynaptic quantum.
Reviewer #3 (Public Review)
Weaknesses: However, the rather strong conclusions on the dissociation of AMPAR trafficking and synaptic response are made from somewhat weaker data. The key issue is the GluA2 immunostaining in comparison with the mEPSC recordings. Their imaging method involves only assessing puncta clearly associated with a MAP2 labeled dendrite. This is a small subset of synapses, judging from the sample micrographs (Fig. 5). To my knowledge, this is a new and unvalidated approach that could represent a particular subset of synapses not representative of the synapses contributing to the mEPSC change (they are also sampling different neurons for the two measurements; an additional unknown detail is how far from the cell body were the analyzed dendrites for immunostaining.) While the authors acknowledge that a sampling issue could explain the data, they still use this data to draw strong conclusions about the lack of AMPAR trafficking contribution to the mEPSC amplitude change. This apparent difference may be a methodological issue rather than a biological one, and at this point it is impossible to differentiate these. It will unfortunately be difficult to validate their approach. Perhaps if they were to drive NMDAdependent LTD or chemLTP, and show alignment of the imaging and ephys, that would help. More helpful would be recordings and imaging from the same neurons but this is challenging. Sampling from identified synapses would of course be ideal, perhaps from 2P uncaging combined with SEP-labeled AMPARs, but this is more challenging still. But without data to validate the method, it seems unwarranted to make such strong conclusions such as that AMPAR trafficking does not underlie the increase in mEPSC amplitude, given the previous data supporting such a model.
In the new version, we soften our conclusion regarding the mismatch between GluA2 receptor levels and mEPSC amplitudes, now only stating that receptors may not be the sole contributor to the TTX effect on mEPSC amplitude (Discussion, lines 472 to 474). With our analysis in the new version focusing on comparisons of cell means, the GluA2 receptor cluster size and the mEPSC amplitude data match well in magnitude for the data pooled across the 3 matched cultures (20.0% and 19.7%, respectively, see new Table 1). However, in one of the three cultures the direction of change for GluA2 receptors is opposite that of mEPSC amplitudes (Table 1, Culture #3, -9.5% vs +17.2%, respectively).
It is unlikely that the lack of matching of homeostatic plasticity in one culture, but very good matching in two other cultures, can be explained by an unvalidated focus on puncta associated with MAP2 positive dendrites. We chose to restrict analysis of synaptic GluA2 receptors to the primary dendrite in order to reduce variability, reasoning that we are always measuring synapses for an excitatory pyramidal neuron, synapses that are relatively close to the cell body, on the consistently identifiable primary dendrite. We measured how far this was for the two cells depicted in old Figure 5 (new Fig. 4). Because we always used the 5X zoom window which is a set length, and positioned it within ~10 microns of the cell body, these cells give a ball park estimate for the usual distances. For the untreated cell, the average distance from the cell body was 38.5 ± 2.8 µm; for the TTX-treated cell, it was 42.4 ± 3.2 µm (p = 0.35, KruskalWallis test). We have added these values to the Results, lines 270 to 274.
We did not mean to propose that AMPA receptor levels do not contribute at all to mEPSC amplitude, and we acknowledge there are clear cases where the two characteristics change in parallel (for example, in the study cited by Reviewer #2, (Pozo et al., 2012), increases in GluA2 receptors due to exogenous expression are closely matched by increases in mEPSC amplitudes.) What our matched culture experiments demonstrate is that in the case of TTX treatment, both GluA2 receptors and mEPSC amplitudes increase on average, but sometimes mEPSC amplitudes can increase in the absence of an increase in GluA2 receptors (Culture #3, Rab3A+/+ cultures), and sometimes mEPSC amplitudes do not increase even though GluA2 receptor levels do increase (Culture #3, Rab3A-/- cultures). Therefore, it would not add anything to our argument to examine receptors and mEPSCs in NMDA-dependent LTP, a different plasticity paradigm in which changes in receptors and mEPSCs may more closely align. It has been demonstrated that mEPSCs of widely varying amplitude can be recorded from a single synaptic site (Liu and Tsien, 1995), so we would need to measure a large sample of individual synapse recordings to detect a modest shift in average values due to activity blockade. In addition, it would be essential to express fluorescent AMPA receptors in order to correlate receptor levels in the same cells we record from (or at the same synapses). And yet, even after these heroics, one is still left with the issue that the two methods, electrophysiology and fluorescent imaging, have distinct limitations and sources of variability that may obscure any true quantitative correlation.
Other questions arise from the NASPM experiments, used to justify looking at GluA2 (and not GluA1) in the immunostaining. First, there is a frequency effect that is quite unclear in origin. One would expect NASPM to merely block some fraction of the post-synaptic current, and not affect pre-synaptic release or block whole synapses. It is also unclear why the authors argue this proves that NASPM was at an effective concentration (lines 399-400). Further, the amplitude data show a strong trend towards smaller amplitude. The p value for both control and TTX neurons was 0.08 – it is very difficult to argue that there is no effect. And the decrease is larger in the TTX neurons. Considering the strong claims for a presynaptic locus and the use of this data to justify only looking at GluA2 by immunostaining, these data do not offer much support of the conclusions. Between the sampling issues and perhaps looking at the wrong GluA subunit, it seems premature to argue that trafficking is not a contributor to the mEPSC amplitude change, especially given the substantial support for that hypothesis. Further, even if trafficking is not the major contributor, there could be shifts in conductance (perhaps due to regulation of auxiliary subunits) that does not necessitate a pre-synaptic locus. While the authors are free to hypothesize such a mechanism, it would be prudent to acknowledge other options and explanations.
We have created a model cartoon to explain how NASPM could reduce mEPSC frequency (new Fig. 3D). mEPSCs that arise from a synaptic site that has only Ca2+-permeable AMPA receptors will be completely blocked by NASPM, if the NASPM concentration is maximal. The reason we conclude that we have sufficient NASPM reaching the cells is that the frequency is decreased, as expected if there are synaptic sites with only Ca2+-permeable AMPA receptors. We previously were not clear that there is an effect of NASPM on mEPSC amplitude, although it did not reach statistical significance (new Fig. 3B). Where there is no effect is on the TTX-induced increase in mEPSC amplitude, which remains after the acute NASPM application (new Fig. 3A). We have revised the description of these findings in Results, lines 220 to 241. In reviewing the literature further, we could find no previous studies demonstrating an increase in conductance in GluA2 or Ca2+-impermeable receptors, only in GluA1 homomers. In other words, any conductance change would have been due to a change in GluA1 homomers, and should have been visible as a disruption of the homeostatic plasticity by NASPM application. We have added text to Results, lines 211 to 217; 236-241; Discussion, lines 420 to 422; 526-536 and Methods, lines 685 to 695 regarding this point.
The frequency data are missing from the paper, with the exception of the NASPM dataset. The mEPSC frequencies should be reported for all experiments, particularly given that Rab3A is generally viewed as a pre-synaptic protein regulating release. Also, in the NASPM experiments, the average frequency is much higher in the TTX treated cultures. Is this statistically above control values?
This comment is addressed by the major change #3, above.
Unaddressed issues that would greatly increase the impact of the paper:
(1) Is Rab3A activity pre-synaptically, post-synaptically or both. The authors provide good evidence that Rab3A is acting within neurons and not astrocytes. But where is it acting (pre or post) would aid substantially in understanding its role (and particularly the hypothesized and somewhat novel idea that the amount of glutamate released per vesicle is altered in HSP). They could use sparse knockdown of Rab3A, or simply mix cultures from KO and WT mice (with appropriate tags/labels). The general view in the field has been that HSP is regulated post-synaptically via regulation of AMPAR trafficking, and considerable evidence supports this view. The more support for their suggestion of a pre-synaptic site of control, the better.
This is similar to the request of Reviewer #2, Recommendations to the Authors. An important next step is to identify whether Rab3A is working pre- or postsynaptically. However, it is possible that it is acting pre-synaptically to anterogradely regulate trafficking of AMPAR, as we have depicted in our model, new Fig. 9. To demonstrate that the presynaptic quantum is being altered, we would need to show that vesicle size is increased, or the amount of transmitter being released during an mEPSC is increased after activity blockade. To that end, we are currently performing experiments using a fast off-rate antagonist. As described above in response to Reviewer #2’s Conceptual Concerns, we find dramatic decreases in frequency not explained by the 30-60% inhibition observed for the largest amplitude mEPSCs, which suggests the possibility that small mEPSCs are more sensitive than large mEPSCs and therefore may have less transmitter. Due to these complexities and the delay while we test other antagonists to see if the effect is specific to fast-off rate antagonists, we are not including these results here.
(2) Rab3A is also found at inhibitory synapses. It would be very informative to know if HSP at inhibitory synapses is similarly affected. This is particularly relevant as at inhibitory synapses, one expects a removal of GABARs and/or a decrease of GABA-packaging in vesicles (ie the opposite of whatever is happening at excitatory synapses.). If both processes are regulated by Rab3A, this might suggest a role for this protein more upstream in the signaling, an effect only at excitatory synapses would argue for a more specific role just at these synapses.
It will be important to determine if homeostatic synaptic plasticity at inhibitory synapses on excitatory neurons is sensitive to Rab3A deletion, especially in light of the fact that unlike many of the other molecules implicated in homeostatic increases in mEPSCS, Rab3A is not a molecule known to be selective for glutamate receptor trafficking (in contrast to Arc/Arg3.1 or GRIP1, for example). Such a study would warrant its own publication.
Reviewer #3 (Recommendations for the Authors):
There are a number of minor points or suggestions for the authors:
Is RIM1 part of this pathway (or expected to be)? Some discussion of this would be nice.
RIM, Rab3-interacting molecule, has been implicated at the drosophila neuromuscular junction in a presynaptic form of homeostatic synaptic plasticity in which evoked release is increased after block of postsynaptic receptors (Muller et al., 2012), a plasticity that also requires Rab3-GAP (Muller et al., 2011). To our knowledge there is no evidence that RIM is involved in the homeostatic plasticity of mEPSC amplitude after activity blockade by TTX. The Rim1a KO does not have a change in mEPSC amplitude relative to WT (Calakos et al., 2004), but that is not unexpected given the normal mEPSC amplitude in neurons from cultures prepared from Rab3A-/- mice in the current study. It would be interesting to look at homeostatic plasticity in cortical cultures prepared from Rim1a or other RIM deletion mice, but we have not added these points to the revised manuscript since there are a number of directions one could go in attempting to define the molecular pathway and we feel it is more important to discuss the potential location of action and physiological mechanisms.
Is the Earlybird mutation a GOF? More information about this mutation would help.
We have added a description of how the Earlybird mutation was identified, in a screen for rest:activity mutants (Results, lines 118 to 123). Rab3A Earlybird mice have a shortened circadian period, shifting their wake cycle earlier and earlier. When Rab3A deletion mice were tested in the same activity raster plot measurements, the shift was smaller than that for the Earlybird mutant, suggesting the possibility that it is a dominant negative mutation.
The high K used in the NASPM experiments seems a bit unusual. Have the authors done high K/no drug controls to see if this affects the synapses in any way?
We used the high K based on previous studies that indicated the blocking effect of the Ca2+-permeable receptor blockers was use dependent (Herlitze et al., 1993; Iino et al., 1996; Koike et al., 1997). We reasoned that a modest depolarization would increase the frequency of AMPA receptor mEPSCs and allow access of the NASPM. We have added this point to the Methods, lines 695 to 708.
The NASPM experiments do not show that GluA1 does not contribute (line 401), only that GluA1 homomers are not contributing (much – see above). GluA1/A2 heteromers are quite likely involved. Also, the SEM is missing from the WT pre/post NASPM data.
Imaging of GluA2-positive sites will not distinguish between GluA2 homomers and GluA2-GluA1 heteromers, so we have added this clarification to Results, lines 242 to 246. We have remade the NASPM pre-post line plots so that the mean values and error bars are more visible (new Fig. 3B, C).
It seems odd to speculate based on non-significant findings (line 650-1), with lower significance (p = 0.11) than findings being dismissed in the paper (NASPM on mEPSC amplitude; p = 0.08).
We did not mean to dismiss the effect of NASPM on mEPSC amplitude (new Fig. 3B), rather, we dismiss the effect of NASPM on the homeostatic increase in mEPSC amplitude caused by TTX treatment (new Fig. 3A). We have emphasized this distinction in Results, lines 223 to 225, and Discussion, lines 420 to 422, as well as adding that the stronger effect of NASPM on frequency after TTX treatment suggests an activity-dependent increase in the number of synapses expressing only Ca2+ permeable homomers (Results, lines 236 to 241; Discussion, lines 431 to 435).
Fig. 4 could be labeled better (to make it clear that B is amplitude and C is freq from the same cells).
Fig. 4 has been revised—now the amplitude and frequency plots from the same condition (new Fig. 3, B, C; CON or TTX) are in a vertical line and the figure legend states that the frequency data are from the same cells as in Fig. 3A.
The raw amplitude data seems a bit hidden in the inset panels – I would suggest these data are at least as important as the cumulative distributions in the main panel. Maybe re-organizing the figures would help.
We have removed all cumulative distributions, rank order plots, and ratio plots. The box plots are now full size in new Figures 1, 2, 5, 6, 7 and 8.
I’m not sure I would argue in the paper that 12 cells a day is a limiting issue for experiments. It doesn’t add anything and doesn’t seem like that high a barrier. It is fine to just say it is difficult and therefore there is a limited amount of data meeting the criteria.
We have removed the comment regarding difficulty.
Calakos N, Schoch S, Sudhof TC, Malenka RC (2004) Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron 42:889-896.
De Gois S, Schafer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD (2005) Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci 25:7121-7133.
Diaz-Rohrer B, Castello-Serrano I, Chan SH, Wang HY, Shurer CR, Levental KR, Levental I (2023) Rab3 mediates a pathway for endocytic sorting and plasma membrane recycling of ordered microdomains. Proc Natl Acad Sci U S A 120:e2207461120.
Dubes S, Soula A, Benquet S, Tessier B, Poujol C, Favereaux A, Thoumine O, Letellier M (2022) miR-124dependent tagging of synapses by synaptopodin enables input-specific homeostatic plasticity. EMBO J 41:e109012.
Fong MF, Newman JP, Potter SM, Wenner P (2015) Upward synaptic scaling is dependent on neurotransmission rather than spiking. Nat Commun 6:6339.
Herlitze S, Raditsch M, Ruppersberg JP, Jahn W, Monyer H, Schoepfer R, Witzemann V (1993) Argiotoxin detects molecular differences in AMPA receptor channels. Neuron 10:1131-1140.
Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY (2008) Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci U S A 105:775-780.
Ibata K, Sun Q, Turrigiano GG (2008) Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57:819-826.
Iino M, Koike M, Isa T, Ozawa S (1996) Voltage-dependent blockage of Ca(2+)-permeable AMPA receptors by joro spider toxin in cultured rat hippocampal neurones. J Physiol 496 ( Pt 2):431437.
Jakawich SK, Neely RM, Djakovic SN, Patrick GN, Sutton MA (2010a) An essential postsynaptic role for the ubiquitin proteasome system in slow homeostatic synaptic plasticity in cultured hippocampal neurons. Neuroscience 171:1016-1031.
Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA (2010b) Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68:1143-1158.
Kapfhamer D, Valladares O, Sun Y, Nolan PM, Rux JJ, Arnold SE, Veasey SC, Bucan M (2002) Mutations in Rab3a alter circadian period and homeostatic response to sleep loss in the mouse. Nat Genet 32:290-295.
Koike M, Iino M, Ozawa S (1997) Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res 29:27-36.
Liu G, Tsien RW (1995) Properties of synaptic transmission at single hippocampal synaptic boutons. Nature 375:404-408.
Liu G, Choi S, Tsien RW (1999) Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22:395409.
Muller M, Pym EC, Tong A, Davis GW (2011) Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron 69:749-762.
Muller M, Liu KS, Sigrist SJ, Davis GW (2012) RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J Neurosci 32:16574-16585.
Pozo K, Cingolani LA, Bassani S, Laurent F, Passafaro M, Goda Y (2012) beta3 integrin interacts directly with GluA2 AMPA receptor subunit and regulates AMPA receptor expression in hippocampal neurons. Proc Natl Acad Sci U S A 109:1323-1328.
Silva MM, Rodrigues B, Fernandes J, Santos SD, Carreto L, Santos MAS, Pinheiro P, Carvalho AL (2019) MicroRNA-186-5p controls GluA2 surface expression and synaptic scaling in hippocampal neurons. Proc Natl Acad Sci U S A 116:5727-5736.
Soden ME, Chen L (2010) Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci 30:16910-16921.
Sun HY, Bartley AF, Dobrunz LE (2009) Calcium-permeable presynaptic kainate receptors involved in excitatory short-term facilitation onto somatostatin interneurons during natural stimulus patterns. J Neurophysiol 101:1043-1055.
Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM (2006) Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125:785-799.
Tan HL, Queenan BN, Huganir RL (2015) GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc Natl Acad Sci U S A 112:10026-10031.
Thapliyal S, Arendt KL, Lau AG, Chen L (2022) Retinoic acid-gated BDNF synthesis in neuronal dendrites drives presynaptic homeostatic plasticity. Elife 11.
Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G (2005) Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci 25:62216234.
Wu YK, Hengen KB, Turrigiano GG, Gjorgjieva J (2020) Homeostatic mechanisms regulate distinct aspects of cortical circuit dynamics. Proc Natl Acad Sci U S A 117:24514-24525.
Xu X, Pozzo-Miller L (2017) EEA1 restores homeostatic synaptic plasticity in hippocampal neurons from Rett syndrome mice. J Physiol 595:5699-5712.



