Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorAndres Jara-OsegueraThe University of Texas at Austin, Austin, United States of America
- Senior EditorKenton SwartzNational Institute of Neurological Disorders and Stroke, Bethesda, United States of America
Reviewer #1 (Public Review):
Gating of Kv10 channels is unique because it involves coupling between non-domain swapped voltage sensing domains, a domain-swapped cytoplasmic ring assembly formed by the N- and C-termini, and the pore domain. Recent structural data suggests that activation of the voltage sensing domain relieves a steric hindrance to pore opening, but the contribution of the cytoplasmic domain to gating is still not well understood. This aspect is of particular importance because proteins like calmodulin interact with the cytoplasmic domain to regulate channel activity. The effects of calmodulin (CaM) in WT and mutant channels with disrupted cytoplasmic gating ring assemblies are contradictory, resulting in inhibition or activation, respectively. The underlying mechanism for these discrepancies is not understood. In the present manuscript, Reham Abdelaziz and collaborators use electrophysiology, biochemistry and mathematical modeling to describe how mutations and deletions that disrupt inter-subunit interactions at the cytoplasmic gating ring assembly affect Kv10.1 channel gating and modulation by CaM. In the revised manuscript, additional information is provided to allow readers to identify within the Kv10.1 channel structure the location of E600R, one of the key channel mutants analyzed in this study. However, the mechanistic role of the cytoplasmic domains that this study focuses on, as well as the location of the ΔPASCap deletion and other perturbations investigated in the study remain difficult to visualize without additional graphical information. This can make it challenging for readers to connect the findings presented in the study with a structural mechanism of channel function.
The authors focused mainly on two structural perturbations that disrupt interactions within the cytoplasmic domain, the E600R mutant and the ΔPASCap deletion. By expressing mutants in oocytes and recording currents using Two Electrode Voltage-Clamp (TEV), it is found that both ΔPASCap and E600R mutants have biphasic conductance-voltage (G-V) relations and exhibit activation and deactivation kinetics with multiple voltage-dependent components. Importantly, the mutant-specific component in the G-V relations is observed at negative voltages where WT channels remain closed. The authors argue that the biphasic behavior in the G-V relations is unlikely to result from two different populations of channels in the oocytes, because they found that the relative amplitude between the two components in the G-V relations was highly reproducible across individual oocytes that otherwise tend to show high variability in expression levels. Instead, the G-V relations for all mutant channels could be well described by an equation that considers two open states O1 and O2, and a transition between them; O1 appeared to be unaffected by any of the structural manipulations tested (i.e. E600R, ΔPASCap, and other deletions) whereas the parameters for O2 and the transition between the two open states were different between constructs. The O1 state is not observed in WT channels and is hypothesized to be associated with voltage sensor activation. O2 represents the open state that is normally observed in WT channels and is speculated to be associated with conformational changes within the cytoplasmic gating ring that follow voltage sensor activation, which could explain why the mutations and deletions disrupting cytoplasmic interactions affect primarily O2.
Severing the covalent link between the voltage sensor and pore reduced O1 occupancy in one of the deletion constructs. Although this observation is consistent with the hypothesis that voltage-sensor activation drives entry into O1, this result is not conclusive. Structural as well as functional data has established that the coupling of the voltage sensor and pore does not entirely rely on the S4-S5 covalent linker between the sensor and the pore, and thus the severed construct could still retain coupling through other mechanisms, which is consistent with the prominent voltage dependence that is observed. If both states O1 and O2 require voltage sensor activation, it is unclear why the severed construct would affect state O1 primarily, as suggested in the manuscript, as opposed to decreasing occupancy of both open states. In line with this argument, the presence of Mg2+ in the extracellular solution affected both O1 and O2. This finding suggests that entry into both O1 and O2 requires voltage-sensor activation because Mg2+ ions are known to stabilize the voltage sensor in its most deactivated conformations.
Activation towards and closure from O1 is slow, whereas channels close rapidly from O2. A rapid alternating pulse protocol was used to take advantage of the difference in activation and deactivation kinetics between the two open components in the mutants and thus drive an increasing number of channels towards state O1. Currents activated by the alternating protocol reached larger amplitudes than those elicited by a long depolarization to the same voltage. This finding is interpreted as an indication that O1 has a larger macroscopic conductance than O2. In the revised manuscript, the authors performed single-channel recordings to determine why O1 and O2 have different macroscopic conductance. The results show that at voltages where the state O1 predominates, channels exhibited longer open times and overall higher open probability, whereas at more depolarized voltages where occupancy of O2 increases, channels exhibited more flickery gating behavior and decreased open probability. These results are informative but not conclusive because additional details about how experiments were conducted, and group data analysis are missing. Importantly, results showing inhibition of single ΔPASCap channels by a Kv10-specific inhibitor are mentioned but not shown or quantitated - these data are essential to establish that the new O1 conductance indeed represents Kv10 channel activity.
It is shown that conditioning pulses to very negative voltages result in mutant channel currents that are larger and activate more slowly than those elicited at the same voltage but starting from less negative conditioning pulses. In voltage-activated curves, O1 occupancy is shown to be favored by increasingly negative conditioning voltages. This is interpreted as indicating that O1 is primarily accessed from deeply closed states in which voltage sensors are in their most deactivated position. Consistently, a mutation that destabilizes these deactivated states is shown to largely suppress the first component in voltage-activation curves for both ΔPASCap and E600R channels.
The authors then address the role of the hidden O1 state in channel regulation by calmodulation. Stimulating calcium entry into oocytes with ionomycin and thapsigarging, assumed to enhance CaM-dependent modulation, resulted in preferential potentiation of the first component in ΔPASCap and E600R channels. This potentiation was attenuated by including an additional mutation that disfavors deeply closed states. Together, these results are interpreted as an indication that calcium-CaM preferentially stabilizes deeply closed states from which O1 can be readily accessed in mutant channels, thus favoring current activation. In WT channels lacking a conducting O1 state, CaM stabilizes deeply closed states and is therefore inhibitory. It is found that the potentiation of ΔPASCap and E600R by CaM is more strongly attenuated by mutations in the channel that are assumed to disrupt interaction with the C-terminal lobe of CaM than mutations assumed to affect interaction with the N-terminal lobe. These results are intriguing but difficult to interpret in mechanistic terms. The strong effect that calcium-CaM had on the occupancy of the O1 state in the mutants raises the possibility that O1 can be only observed in channels that are constitutively associated with CaM. To address this, a biochemical pull-down assay was carried out to establish that only a small fraction of channels are associated with CaM under baseline conditions. These CaM experiments are potentially very interesting and could have wide physiological relevance. However, the approach utilized to activate CaM is indirect and could result in additional non-specific effects on the oocytes that could affect the results.
Finally, a mathematical model is proposed consisting of two layers involving two activation steps for the voltage sensor, and one conformational change in the cytoplasmic gating ring - completion of both sets of conformational changes is required to access state O2, but accessing state O1 only requires completion of the first voltage-sensor activation step in the four subunits. The model qualitatively reproduces most major findings on the mutants. Although the model used is highly symmetric and appears simple, the mathematical form used for the rate constants in the model adds a layer of complexity to the model that makes mechanistic interpretations difficult. In addition, many transitions that from a mechanistic standpoint should not depend on voltage were assigned a voltage dependence in the model. These limitations diminish the overall usefulness of the model which is prominently presented in the manuscript. The most important mechanistic assumptions in the model are not addressed experimentally, such as the proposition that entry into O1 depends on the opening of the transmembrane pore gate, whereas entry into O2 involves gating ring transitions - it is unclear why O2 would require further gating ring transitions to conduct ions given that the gating ring can already support permeation by O1 without any additional conformational changes.
Reviewer #3 (Public Review):
In the present manuscript, Abdelaziz and colleagues interrogate the gating mechanisms of Kv10.1, an important voltage-gated K+ channel in cell cycle and cancer physiology. At the molecular level, Kv10.1 is regulated by voltage and Ca-CaM. Structures solved using Cryo-EM for Kv10.1 as well as other members of the KCNH family (Kv11 and Kv12) show channels that do not contain a structured S4-S5 linker imposing therefore a non-domain swapped architecture in the transmembrane region. However, the cytoplasmatic N- and C- terminal domains interact in a domain swapped manner forming a gating ring. The N-terminal domain (PAS domain) of one subunit is located close to the intracellular side of the voltage sensor domain and interacts with the C-terminal domain (CNBHD domain) of the neighbor subunit. Mutations in the intracellular domains has a profound effect in the channel gating. The complex network of interactions between the voltage-sensor and the intracellular domains makes the PAS domain a particularly interesting domain of the channel to study as responsible for the coupling between the voltage sensor domains and the intracellular gating ring.
The coupling between the voltage-sensor domain and the gating ring is not fully understood and the authors aim to shed light into the details of this mechanism. In order to do that, they use well established techniques such as site-directed mutagenesis, electrophysiology, biochemistry and mathematical modeling. In the present work, the authors propose a two open state model that arises from functional experiments after introducing a deletion on the PAS domain (ΔPAS Cap) or a point mutation (E600R) in the CNBHD domain. The authors measure a bi-phasic G-V curve with these mutations and assign each phase as two different open states, one of them not visible on the WT and only unveiled after introducing the mutations. The hypothesis proposed by the authors could change the current paradigm in the current understanding for Kv10.1 and it is quite extraordinary; therefore, it requires extraordinary evidence to support it.
STRENGTHS: The authors use adequate techniques such as electrophysiology and site-directed mutagenesis to address the gating changes introduced by the molecular manipulations. They also use appropriate mathematical modeling to build a Markov model and identify the mechanism behind the gating changes.
WEAKNESSES: The results presented by the authors do not fully support their conclusions since they could have alternative explanations. The authors base their primary hypothesis on the bi-phasic behavior of a calculated G-V curve that do not match the tail behavior, the experimental conditions used in the present manuscript introduce uncertainties, weakening their conclusions and complicating the interpretation of the results. Therefore, their experimental conditions need to be revisited
I have some concerns related to the following points:
(1) Biphasic gating behavior
The authors use the TEVC technique in oocytes extracted surgically from Xenopus Leavis frogs. The method is well established and is adequate to address ion channel behavior. The experiments are performed in chloride-based solutions which present a handicap when measuring outward rectifying currents at very depolarizing potentials due to the presence of calcium activated chloride channel expressed endogenously in the oocytes; these channels will open and rectify chloride intracellularly adding to the outward rectifying traces during the test pulse.
The authors calculate their G-V curves from the test pulse steady-state current instead of using the tail currents. The conductance measurements are normally taken from the 'tail current' because tails are measured at a fix voltage hence maintaining the driving force constant. Calculating the conductance from the traces should not be a problem, however, in the present manuscript, the traces and the tail currents do not agree. The tail traces shown in Fig1E do not show an increasing current amplitude in the voltage range from +50mV to +120mV, they seem to have reached a 'saturation state', suggesting that the traces from the test pulse contain an inward chloride current contamination. In addition, this second component identified by the authors as a second open state appears after +50mV and seems to never saturate. The normalization to the maximum current level during the test pulse, exaggerates this second component on the calculated G-V curve. It's worth noticing that the ΔPASCap mutant experiments on Fig 5 in Mes based solutions do not show that second component on the G-V.
Because these results are the foundation for their two open state hypotheses, I will strongly suggest the authors to repeat all their Chloride-based experiments in Mes-based solutions to eliminate the undesired chloride contribution to the mutants current and clarify the contribution of the mutations to the Kv10.1 gating.
(2) Two step gating mechanism.
The authors interpret the results obtained with the ΔPASCap and the E600R as two step gating mechanisms containing two open states (O1 and O2) and assign them to the voltage sensor movement and gating ring rotation respectively. It is not clear, however how the authors assign the two open states.
The results show how the first component is conserved amongst mutations; however, the second one is not. The authors attribute the second component, hence the second open state to the movement of the gating ring. This scenario seems unlikely since there is a clear voltage-dependence of the second component that will suggest an implication of a voltage-sensing current.
The split channel experiment is interesting but needs more explanation. I assume the authors expressed the 2 parts of the split channel (1-341 and 342-end), however Tomczak et al showed in 2017 how the split presents a constitutively activated function with inward currents that are not visible here, this point needs clarification.
Moreover, the authors assume that the mutations introduced uncover a new open state, however the traces presented for the mutations suggest that other explanations are possible. Other gating mechanisms like inactivation from the closed state, can be introduced by the mutations. The traces presented for ΔPASCap but specially E600R present clear 'hooked tails', a direct indicator of a populations of inactive channels during the test pulse that recover from inactivation upon repolarization (Tristani-Firouzi M, Sanguinetti MC. J Physiol. 1998). The results presented by the authors can be alternatively explained with a change in the equilibrium between the close to inactivated/recovery from inactivation to the open state. Finally, the authors state that they do not detect "cumulative inactivation after repeated depolarization" but that is considering inactivation only from the open state and ignoring the possibility of the existence of close state inactivation or, that like in hERG, that the channel inactivates faster that what it activates (Smith PL, Yellen G. J Gen Physiol. 2002).
(3) Single channel conductance.
The single channels experiments are a great way to assess the different conductance of single channel openings, unfortunately the authors cannot measure accurately different conductances for the two proposed open states. The Markov Model built by the authors, disagrees with their interpretation of the experimental results assigning the exact same conductance to the two modeled open states. To interpret the mutant data, it is needed to add data with the WT for comparison and in presence of specific blockers.
Author response:
The following is the authors’ response to the current reviews.
Gating of Kv10 channels is unique because it involves coupling between non-domain swapped voltage sensing domains, a domain-swapped cytoplasmic ring assembly formed by the N- and C-termini, and the pore domain. Recent structural data suggests that activation of the voltage sensing domain relieves a steric hindrance to pore opening, but the contribution of the cytoplasmic domain to gating is still not well understood. This aspect is of particular importance because proteins like calmodulin interact with the cytoplasmic domain to regulate channel activity. The effects of calmodulin (CaM) in WT and mutant channels with disrupted cytoplasmic gating ring assemblies are contradictory, resulting in inhibition or activation, respectively. The underlying mechanism for these discrepancies is not understood. In the present manuscript, Reham Abdelaziz and collaborators use electrophysiology, biochemistry and mathematical modeling to describe how mutations and deletions that disrupt inter-subunit interactions at the cytoplasmic gating ring assembly affect Kv10.1 channel gating and modulation by CaM. In the revised manuscript, additional information is provided to allow readers to identify within the Kv10.1 channel structure the location of E600R, one of the key channel mutants analyzed in this study. However, the mechanistic role of the cytoplasmic domains that this study focuses on, as well as the location of the ΔPASCap deletion and other perturbations investigated in the study remain difficult to visualize without additional graphical information. This can make it challenging for readers to connect the findings presented in the study with a structural mechanism of channel function.
The authors focused mainly on two structural perturbations that disrupt interactions within the cytoplasmic domain, the E600R mutant and the ΔPASCap deletion. By expressing mutants in oocytes and recording currents using Two Electrode Voltage-Clamp (TEV), it is found that both ΔPASCap and E600R mutants have biphasic conductance-voltage (G-V) relations and exhibit activation and deactivation kinetics with multiple voltage-dependent components. Importantly, the mutant-specific component in the G-V relations is observed at negative voltages where WT channels remain closed. The authors argue that the biphasic behavior in the G-V relations is unlikely to result from two different populations of channels in the oocytes, because they found that the relative amplitude between the two components in the G-V relations was highly reproducible across individual oocytes that otherwise tend to show high variability in expression levels. Instead, the G-V relations for all mutant channels could be well described by an equation that considers two open states O1 and O2, and a transition between them; O1 appeared to be unaffected by any of the structural manipulations tested (i.e. E600R, ΔPASCap, and other deletions) whereas the parameters for O2 and the transition between the two open states were different between constructs. The O1 state is not observed in WT channels and is hypothesized to be associated with voltage sensor activation. O2 represents the open state that is normally observed in WT channels and is speculated to be associated with conformational changes within the cytoplasmic gating ring that follow voltage sensor activation, which could explain why the mutations and deletions disrupting cytoplasmic interactions affect primarily O2.
Severing the covalent link between the voltage sensor and pore reduced O1 occupancy in one of the deletion constructs. Although this observation is consistent with the hypothesis that voltage-sensor activation drives entry into O1, this result is not conclusive. Structural as well as functional data has established that the coupling of the voltage sensor and pore does not entirely rely on the S4-S5 covalent linker between the sensor and the pore, and thus the severed construct could still retain coupling through other mechanisms, which is consistent with the prominent voltage dependence that is observed. If both states O1 and O2 require voltage sensor activation, it is unclear why the severed construct would affect state O1 primarily, as suggested in the manuscript, as opposed to decreasing occupancy of both open states. In line with this argument, the presence of Mg2+ in the extracellular solution affected both O1 and O2. This finding suggests that entry into both O1 and O2 requires voltage-sensor activation because Mg2+ ions are known to stabilize the voltage sensor in its most deactivated conformations.
We agree with the reviewer that access to both states requires a conformational change in the voltage sensor. This was stated in our revised article: “In contrast, to enter O2, all subunits must complete both voltage sensor transitions and the collective gating ring transition.” We interpret the two gating steps as sequential; the effective rotation of the intracellular ring would happen only once the sensor is in its fully activated position.
We also agree that the S4-S5 segment cannot be the only interaction mechanism, as we demonstrated in our earlier work (Lörinczi et al., 2015; Tomczak et al., 2017).
Activation towards and closure from O1 is slow, whereas channels close rapidly from O2. A rapid alternating pulse protocol was used to take advantage of the difference in activation and deactivation kinetics between the two open components in the mutants and thus drive an increasing number of channels towards state O1. Currents activated by the alternating protocol reached larger amplitudes than those elicited by a long depolarization to the same voltage. This finding is interpreted as an indication that O1 has a larger macroscopic conductance than O2. In the revised manuscript, the authors performed single-channel recordings to determine why O1 and O2 have different macroscopic conductance. The results show that at voltages where the state O1 predominates, channels exhibited longer open times and overall higher open probability, whereas at more depolarized voltages where occupancy of O2 increases, channels exhibited more flickery gating behavior and decreased open probability. These results are informative but not conclusive because additional details about how experiments were conducted, and group data analysis are missing. Importantly, results showing inhibition of single ΔPASCap channels by a Kv10-specific inhibitor are mentioned but not shown or quantitated - these data are essential to establish that the new O1 conductance indeed represents Kv10 channel activity.
We observed the activity of a channel compatible with Kv10.1 ΔPAS-Cap (long openings at low-moderate potentials, very short flickery activity at strong depolarizations) in 12 patches from oocytes obtained from different frog operations over a period of two and a half months once the experimental conditions could be established. As stated in the text, we did not proceed to generate amplitude histograms because we could not resolve clear single-channel events at strong depolarizations. Astemizole abolished the activity and (remarkably) strongly reduced the noise in traces at strong depolarizations, which we interpret as partially caused by flicker openings.
Author response image 1.
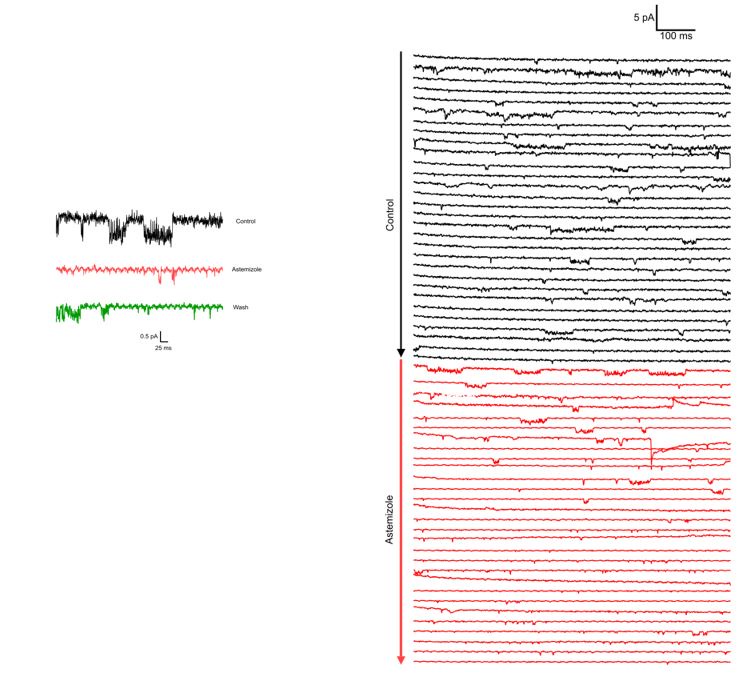
We include two example recordings of Astemizole application (100µM) on two different patches. Both recordings are performed at -60 mV (to decrease the likelihood that the channel visits O2) with 100 mM internal and 60 mM external K+. In both cases, the traces in Astemizole are presented in red.
It is shown that conditioning pulses to very negative voltages result in mutant channel currents that are larger and activate more slowly than those elicited at the same voltage but starting from less negative conditioning pulses. In voltage-activated curves, O1 occupancy is shown to be favored by increasingly negative conditioning voltages. This is interpreted as indicating that O1 is primarily accessed from deeply closed states in which voltage sensors are in their most deactivated position. Consistently, a mutation that destabilizes these deactivated states is shown to largely suppress the first component in voltage-activation curves for both ΔPASCap and E600R channels.
The authors then address the role of the hidden O1 state in channel regulation by calmodulation. Stimulating calcium entry into oocytes with ionomycin and thapsigarging, assumed to enhance CaM-dependent modulation, resulted in preferential potentiation of the first component in ΔPASCap and E600R channels. This potentiation was attenuated by including an additional mutation that disfavors deeply closed states. Together, these results are interpreted as an indication that calcium-CaM preferentially stabilizes deeply closed states from which O1 can be readily accessed in mutant channels, thus favoring current activation. In WT channels lacking a conducting O1 state, CaM stabilizes deeply closed states and is therefore inhibitory. It is found that the potentiation of ΔPASCap and E600R by CaM is more strongly attenuated by mutations in the channel that are assumed to disrupt interaction with the C-terminal lobe of CaM than mutations assumed to affect interaction with the N-terminal lobe. These results are intriguing but difficult to interpret in mechanistic terms. The strong effect that calcium-CaM had on the occupancy of the O1 state in the mutants raises the possibility that O1 can be only observed in channels that are constitutively associated with CaM. To address this, a biochemical pull-down assay was carried out to establish that only a small fraction of channels are associated with CaM under baseline conditions. These CaM experiments are potentially very interesting and could have wide physiological relevance. However, the approach utilized to activate CaM is indirect and could result in additional nonspecific effects on the oocytes that could affect the results.
Finally, a mathematical model is proposed consisting of two layers involving two activation steps for the voltage sensor, and one conformational change in the cytoplasmic gating ring - completion of both sets of conformational changes is required to access state O2, but accessing state O1 only requires completion of the first voltage-sensor activation step in the four subunits. The model qualitatively reproduces most major findings on the mutants. Although the model used is highly symmetric and appears simple, the mathematical form used for the rate constants in the model adds a layer of complexity to the model that makes mechanistic interpretations difficult. In addition, many transitions that from a mechanistic standpoint should not depend on voltage were assigned a voltage dependence in the model. These limitations diminish the overall usefulness of the model which is prominently presented in the manuscript. The most important mechanistic assumptions in the model are not addressed experimentally, such as the proposition that entry into O1 depends on the opening of the transmembrane pore gate, whereas entry into O2 involves gating ring transitions - it is unclear why O2 would require further gating ring transitions to conduct ions given that the gating ring can already support permeation by O1 without any additional conformational changes.
In essence, we agree with the reviewer; we already have addressed these points in our revised article:
Regarding the voltage dependence we write “the κ/λ transition could reasonably be expected to be voltage independent because we related it to ring reconfiguration, a process that should occur as a consequence of a prior VSD transition. We have made some attempts to treat this transition as voltage independent but state-specific with upper-layer bias for states on the right and lower-layer bias for states on the left. This is in principle possible, as can already be gleaned from the similar voltage ranges of the left-right transition (α/β) and the κL/λ transition. However, this approach leads to a much larger number of free, less well constrained kinetic parameters and drastically complicated the parameter search. ” As you can see, we also formulated a strategy to free the model of the potentially spurious voltage dependence and (in bold here) explained why we did not follow this route in this study.
Regarding the need for gating ring transitions after O1, we wrote, “Thus, the underlying gating events can be separated into two steps: The first gating step involves only the voltage sensor without engaging the ring and leads to a pre-open state, which is non-conducting in the WT but conducting in our mutants. The second gating event operates at higher depolarizations, involves a change in the ring, and leads to an open state both in WT and in the mutants. ”
We interpret your statements such that you expect the conducting state to remain available once O1 is reached. However, the experimental evidence speaks against that the pore availability remains regardless of the further gating steps beyond O1. The description of model construction is informative here: “... we could exclude many possible [sites at which O1 connects to closed states] because the attachment site must be sufficiently far away from the conventional open state [O2]. Otherwise, the transition from "O1 preferred" to "O2 preferred" via a few closed intermediate states is very gradual and never produces the biphasic GV curves [that we observed]. ”
In other words, voltage-dependent gating steps beyond the state that offers access to O1 appear to close the pore, after it was open. That might occur because only then (for states in which at least one voltage sensor exceeded the intermediate position) the ring is fixed in a particular state until all sensors completed activation. In the WT, closing the pore in deactivated states might rely on an interaction that is absent in the mutant because, at least in HERG: “the interaction between the PAS domain and the C-terminus is more stable in closed than in open KV11.1 (HERG) channels, and a single chain antibody binding to the interface between PAS domain and CNBHD can access its epitope in open but not in closed channels, strongly supporting a change in conformation of the ring during gating ”
Reviewer #3 (Public Review):
In the present manuscript, Abdelaziz and colleagues interrogate the gating mechanisms of Kv10.1, an important voltage-gated K+ channel in cell cycle and cancer physiology. At the molecular level, Kv10.1 is regulated by voltage and Ca-CaM. Structures solved using CryoEM for Kv10.1 as well as other members of the KCNH family (Kv11 and Kv12) show channels that do not contain a structured S4-S5 linker imposing therefore a non-domain swapped architecture in the transmembrane region. However, the cytoplasmatic N- and C- terminal domains interact in a domain swapped manner forming a gating ring. The N-terminal domain (PAS domain) of one subunit is located close to the intracellular side of the voltage sensor domain and interacts with the C-terminal domain (CNBHD domain) of the neighbor subunit. Mutations in the intracellular domains has a profound effect in the channel gating. The complex network of interactions between the voltage-sensor and the intracellular domains makes the PAS domain a particularly interesting domain of the channel to study as responsible for the coupling between the voltage sensor domains and the intracellular gating ring.
The coupling between the voltage-sensor domain and the gating ring is not fully understood and the authors aim to shed light into the details of this mechanism. In order to do that, they use well established techniques such as site-directed mutagenesis, electrophysiology, biochemistry and mathematical modeling. In the present work, the authors propose a two open state model that arises from functional experiments after introducing a deletion on the PAS domain (ΔPAS Cap) or a point mutation (E600R) in the CNBHD domain. The authors measure a bi-phasic G-V curve with these mutations and assign each phase as two different open states, one of them not visible on the WT and only unveiled after introducing the mutations.
The hypothesis proposed by the authors could change the current paradigm in the current understanding for Kv10.1 and it is quite extraordinary; therefore, it requires extraordinary evidence to support it.
STRENGTHS: The authors use adequate techniques such as electrophysiology and sitedirected mutagenesis to address the gating changes introduced by the molecular manipulations. They also use appropriate mathematical modeling to build a Markov model and identify the mechanism behind the gating changes.
WEAKNESSES: The results presented by the authors do not fully support their conclusions since they could have alternative explanations. The authors base their primary hypothesis on the bi-phasic behavior of a calculated G-V curve that do not match the tail behavior, the experimental conditions used in the present manuscript introduce uncertainties, weakening their conclusions and complicating the interpretation of the results. Therefore, their experimental conditions need to be revisited.
We respectfully disagree. We think that your suggestions for alternative explanations are addressed in the current version of the article. We will rebut them once more below, but we feel the need to point out that our arguments are already laid out in the revised article.
I have some concerns related to the following points:
(1) Biphasic gating behavior
The authors use the TEVC technique in oocytes extracted surgically from Xenopus Leavis frogs. The method is well established and is adequate to address ion channel behavior. The experiments are performed in chloride-based solutions which present a handicap when measuring outward rectifying currents at very depolarizing potentials due to the presence of calcium activated chloride channel expressed endogenously in the oocytes; these channels will open and rectify chloride intracellularly adding to the outward rectifying traces during the test pulse. The authors calculate their G-V curves from the test pulse steady-state current instead of using the tail currents. The conductance measurements are normally taken from the 'tail current' because tails are measured at a fix voltage hence maintaining the driving force constant.
We respectfully disagree. In contrast to other channels, like HERG, a common practice for Kv10 is not to use tail currents. It is long known that in this channel, tail currents and test-pulse steady-state currents can appear to be at odds because the channels deactivate extremely rapidly, at the border of temporal resolution of the measurements and with intricate waveforms. This complicates the estimation of the instantaneous tail current. Therefore, the outward current is commonly used to estimate conductance (Terlau et al., 1996; Schönherr et al., 1999; Schönherr et al., 2002; Whicher and MacKinnon, 2019), while the latter authors also use the extreme of the tail for some mutants.
Due to their activation at very negative voltage, the reversal potential in our mutants can be measured directly; we are, therefore, more confident with this approach. Nevertheless, we have determined the initial tail current in some experiments. The behavior of these is very similar to the average that we present in Figure 1. The biphasic behavior is unequivocally present.
Author response image 2.
Calculating the conductance from the traces should not be a problem, however, in the present manuscript, the traces and the tail currents do not agree.
The referee’s observation is perfectly in line with the long-standing experience of several labs working with KV10: tail current amplitudes in KV10 appear to be out of proportion for the WT open state (O2). Importantly, this is due to the rapid closure, which is not present in O1. As a consequence, the initial amplitude of tail currents from O1 are easier to estimate correctly, and they are much more obvious in the graphs. Taken together, these differences between O1 and O2 explain the misconception the reviewer describes next.
The tail traces shown in Fig1E do not show an increasing current amplitude in the voltage range from +50mV to +120mV, they seem to have reached a 'saturation state', suggesting that the traces from the test pulse contain an inward chloride current contamination.
As stated in the text and indicated in Author response image 3, the tail currents In Figure 1E increase in amplitude between +50 and +120 mV, as can be seen in the examples below from different experiments (+50 is presented in black, +120 in red). As stated above, the increase is not as evident as in traces from other mutants because the predominance of O2 also implies a much faster deactivation.
Author response image 3.
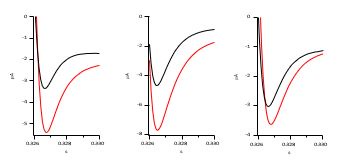
We are aware that Ca2+-activated Cl- currents can represent a problem when interpreting electrophysiological data in oocytes. In fact, we show in Supplement 1 to Figure 8 that this can be the case during the Ca2+-CaM experiments, where the increase in Ca2+ would certainly augment Cl- contribution to the outward current. This is why we performed these experiments in Cl--free solutions. As we show in Figure 8, the biphasic behavior was also present in those experiments.
Importantly, Cl- free bath solutions would not correct contamination during the tail, since this would correspond to Cl- exiting the oocyte. Yet, if there would be contamination of the outward currents by Cl-, one would expect it to increase with larger depolarizations as the typical Ca2+activated Cl- current in oocytes does. As the reviewer states, this does not seem to be the case.
In addition, this second component identified by the authors as a second open state appears after +50mV and seems to never saturate. The normalization to the maximum current level during the test pulse, exaggerates this second component on the calculated G-V curve.
We agree that this second component continues to increase; the reviewer brought this up in the first review, and we have already addressed this in our reply and in the discussion of the revised version: “This flicker block might also offer an explanation for a feature of the mutant channels, that is not explained in the current model version: the continued increase in current amplitude, hundreds of milliseconds into a strong depolarization (Supp. 4 to Fig. 9). If the relative stability of O2 and C2 continued to change throughout depolarization, such a current creep-up could be reproduced. However, this would require either the introduction of further layers of On ↔Cn states, or a non-Markovian modification of the model’s time evolution.” With non-Markovian, we mean a Langevin-type diffusive process.
It's worth noticing that the ΔPASCap mutant experiments on Fig 5 in Mes based solutions do not show that second component on the G-V.
For the readers of this conversation, we would like to clarify that the reviewer likely refers to experiments shown in Fig. 5 of the initial submission but shown in Fig. 6 of the revised version (“Hyperpolarization promotes access to a large conductance, slowly activating open state.” Fig. 5 deals with single channels). We agree that these data look different, but this is because the voltage protocols are completely different (compare Fig. 6A (fixed test pulse, varied prepulse) and Fig. 2A (varied test pulse, fixed pre-pulse). Therefore, no biphasic behavior is expected.
Because these results are the foundation for their two open state hypotheses, I will strongly suggest the authors to repeat all their Chloride-based experiments in Mes-based solutions to eliminate the undesired chloride contribution to the mutants current and clarify the contribution of the mutations to the Kv10.1 gating.
In summary, we respectfully disagree with all concerns raised in point (1). Our detailed arguments rebutting them are given above, but there is a more high-level concern about this entire exchange: the referee casts doubt on observations that are not new. Several labs have reported for a group of mutant KCNH channels: non-monotonic voltage dependence of activation (see, e.g., Fig. 6D in Zhao et al., 2017), multi-phasic tail currents (see e.g. Fig. 4A in Whicher and MacKinnon, 2019, in CHO cells where Cl- contamination is not a concern), and activation by high [Ca2+]i (Lörinczi et al., 2016). Our study replicates those observations and hypothesizes that the existence of an additional conducting state can alone explain all previously unexplained observations. We highlight the potency of this hypothesis with a Markov model that qualitatively reproduces all phenomena. We not only factually disagree with the individual points raised, but we also think that they don't touch on the core of our contribution
(2) Two step gating mechanism.
The authors interpret the results obtained with the ΔPASCap and the E600R as two step gating mechanisms containing two open states (O1 and O2) and assign them to the voltage sensor movement and gating ring rotation respectively. It is not clear, however how the authors assign the two open states.
The results show how the first component is conserved amongst mutations; however, the second one is not. The authors attribute the second component, hence the second open state to the movement of the gating ring. This scenario seems unlikely since there is a clear voltagedependence of the second component that will suggest an implication of a voltage-sensing current.
We do not suggest that the gating ring motion is not voltage dependent. We would like to point out that voltage dependence can be conveyed by voltage sensor coupling to the ring; this is the widely accepted theory of how the ring can be involved. Should the reviewer mean it in a narrow sense, that the model should be constructed such that all voltage-dependent steps occur before and independently of ring reconfiguration and that only then an additional step that reflects the (voltage-independent) reconfiguration solely, we would like to point the reviewer to the article, where we write: “the κ/λ transition could reasonably be expected to be voltage independent because we related it to ring reconfiguration, a process that should occur as a consequence of a prior VSD transition. We have made some attempts to treat this transition as voltage independent but state-specific with upper-layer bias for states on the right and lower-layer bias for states on the left. This is in principle possible, as can already be gleaned from the similar voltage ranges of the left-right transition (α/β) and the κL/λ transition. However, this approach leads to a much larger number of free, less well constrained kinetic parameters and drastically complicated the parameter search. ” As you can see, we also formulated a strategy to free the model from the potentially spurious voltage dependence and (in bold here) explained why we did not follow this route in this study.
The split channel experiment is interesting but needs more explanation. I assume the authors expressed the 2 parts of the split channel (1-341 and 342-end), however Tomczak et al showed in 2017 how the split presents a constitutively activated function with inward currents that are not visible here, this point needs clarification.
As stated in the panel heading, the figure legend, and the main text, we did not use 1-341 and 342-end as done in Tomczak et al. Instead, “we compared the behavior of ∆2-10 and ∆210.L341Split,”. Evidently, the additional deletion (2-10) causes a shift in activation that explains the difference you point out. However, as we do not compare L341Split and ∆210.L341Split but ∆2-10 and ∆2-10.L341Split, our conclusion remains that “As predicted, compared to ∆2-10, ∆2-10.L341Split showed a significant reduction in the first component of the biphasic GV (Fig. 2C, D).” Remarkably, the behavior of the ∆3-9 L341Split described in Whicher and MacKinnon, 2019 (Figure 5) matches that of our ∆2-10 L341Split, which we think reinforces our case.
Moreover, the authors assume that the mutations introduced uncover a new open state, however the traces presented for the mutations suggest that other explanations are possible. Other gating mechanisms like inactivation from the closed state, can be introduced by the mutations. The traces presented for ΔPASCap but specially E600R present clear 'hooked tails', a direct indicator of a populations of inactive channels during the test pulse that recover from inactivation upon repolarization (Tristani-Firouzi M, Sanguinetti MC. J Physiol. 1998).
There is a possibility that we are debating nomenclature here. In response to the suggestion that all our observations could be explained by inactivation, we attempted a disambiguation of terms in the reply and the article. As the argument is brought up again without reference to our clarification attempts, we will try to be more explicit here:
If, starting from deeply deactivated states, an open state is reached first, and then, following further activation steps, closed states are reached, this might be termed “inactivation”. In such a reading, our model features many inactivated states. The shortest version of such a model is C-O-I. It is for instance used by Raman and Bean (2001; DOI: 10.1016/S00063495(01)76052-3) to explain NaV gating in Purkinje neurons. If “inactivation” is meant in the sense that a gating transition exists, which is orthogonal to an activation/deactivation axis, and that after this orthogonal transition, an open state cannot be reached anymore, then all of the upper floor in our model is inactivated with respect to the open state O1. Finally, the state C2 is an inactivated state to O2. In this view, “inactivation” explains the observed phenomena.
However, we must disagree if the referee means that a parsimonious explanation exists in which a single conducting state is the only source for all observed currents.
There is a high-level reason: we found a single assumption that explains three different phenomena, while the inactivation hypothesis with one conducting state cannot explain one of them (the increase of the first component under raised CaM). But there is also a low-level reason: the tails in Tristani-Firouzi and Sanguinetti 1998 are fundamentally different from what we report herein in that they lack a third component. Thus, those tails are consistent with recovery from inactivation through a single open state, while a three-component tail is not. In the framework of a Markov model, the time constants of transitions from and to a given state (say O2), cannot change unless the voltage changes. During the tail current, the voltage does not change, yet we observe:
i) a rapid decrease with a time constant of at most a few milliseconds (Fig 9 S2, 1-> 2), ii) a slow increase in current, peaking after approximately 25 milliseconds and iii) a relaxation to zero current with a time constant of >50 ms.
According to the reviewer’s suggestion, these processes on three timescales should all be explained by depopulating and repopulating the same open state while all rates are constant. There might well be a complicated multi-level state diagram with a single open state with different variants, like (open and open inactivated) that could produce triphasic tails with these properties if the system had not reached a steady state distribution at the end of the test pulse. It cannot, however, achieve it from an equilibrated system, and certainly, it cannot at the same time produce “biphasic activation” and “activation by CaM”.
The results presented by the authors can be alternatively explained with a change in the equilibrium between the close to inactivated/recovery from inactivation to the open state.
Again, we disagree. The model construction explains in detail that the transition from the first to the second phase is not gradual. Shifting equilibria cannot reproduce this. We have extensively tested that idea and can exclude this possibility.
Finally, the authors state that they do not detect "cumulative inactivation after repeated depolarization" but that is considering inactivation only from the open state and ignoring the possibility of the existence of close state inactivation or, that like in hERG, that the channel inactivates faster that what it activates (Smith PL, Yellen G. J Gen Physiol. 2002).
We respectfully disagree. We explicitly model an open state that inactivates faster (O2->C2) than it activates. Once more, this is stated in the revised article, which we point to for details. Again, this alternative mechanism does not have the potential to explain all three effects. As discussed above about the chloride contamination concerns, this inactivation hypothesis was mentioned in the first review round and, therefore, addressed in our reply and the revised article. We also explained that “inactivation” has no specific meaning in Markov models. In the absence of O1, all transitions towards the lower layer are effectively “inactivation from closed states”, because they make access to the only remaining open state less likely”. But this is semantics. What is relevant is that no network of states around a single open state can reproduce the three effets in a more parsimonious way than the assumption of the second open state does.
(3) Single channel conductance.
The single channels experiments are a great way to assess the different conductance of single channel openings, unfortunately the authors cannot measure accurately different conductances for the two proposed open states. The Markov Model built by the authors, disagrees with their interpretation of the experimental results assigning the exact same conductance to the two modeled open states. To interpret the mutant data, it is needed to add data with the WT for comparison and in presence of specific blockers.
We respectfully disagree. As previously shown, the conductance of the flickering wild-type open state is very difficult to resolve. Our recordings do not show that the two states have different single-channel conductances, and therefore the model assumes identical singlechannel conductance.
The important point is that the single-channel recordings clearly show two different gating modes associated with the voltage ranges in which we predict the two open states. One has a smaller macroscopic current due to rapid flickering (aka “inactivation”). These recordings are another proof of the existence of two open states because the two gating modes occur. Wild-type data can be found in Bauer and Schwarz, (2001, doi:10.1007/s00232-001-0031-3) or Pardo et al., (1998, doi:10.1083/jcb.143.3.767) for comparison.
We appreciate the effort editors and reviewers invested in assessing the revised manuscript. Yet, we think that the demanded revision of experimental conditions and quantification methods contradicts the commonly accepted practice for KV10 channels. Some of the reviewer comments are skeptical about the biphasic behavior, which is an established and replicated finding for many mutants and by many researchers. The alternative explanations for these disbelieved findings are either “semantics” or cannot quantitatively explain the measurements. Therefore, only the demand for more explanations and unprecedented resolution in singlechannel recordings remains. We share these sentiments.
———— The following is the authors’ response to the original reviews.
(1) The authors must show that the second open state is not just an artifact of endogenous activity but represents the activity of the same EAG channels. I suggest that the authors repeat these experiments in Mes-based solutions.
(2) Along the same lines, it is necessary to show that these currents can be blocked using known EAG channel blockers such as astemizole. Ultimately, it will be important to demonstrate using single-channel analysis that these do represent two distinct open states separated by a closed state.
We have addressed these concerns using several approaches. The most substantial change is the addition of single-channel recordings on ΔPASCap. In those experiments, we could provide evidence of the two types of events in the same patch, and the presence of an outward current at -60 mV, 50 mV below the equilibrium potential for chloride. The channels were never detected in uninjected oocytes, and Astemizole silenced the activity in patches containing multiple channels. These observations, together with the maintenance of the biphasic behavior that we interpret as evidence of the presence of O1 in methanesulfonate-based solutions, strongly suggest that both O1 and O2 obey the expression of KV10.1 mutants.
(3) Currents should be measured by increasing the pulse lengths as needed in order to obtain the true steady-state G-V curves.
We agree that the endpoint of activation is ill-defined in the cases where a steady-state is not reached. This does indeed hamper quantitative statements about the relative amplitude of the two components. However, while the overall shape does change, its position (voltage dependence) would not be affected by this shortcoming. The data, therefore, supports the claim of the “existence of mutant-specific O1 and its equal voltage dependence across mutants.”
(4) A more clear and thorough description should be provided for how the observations with the mutant channels apply to the behavior of WT channels. How exactly does state O1 relate to WT behavior, and how exactly do the parameters of the mathematical model differ between WT and mutants? How can this be interpreted at a structural level? What could be the structural mechanism through which ΔPASCap and E600R enable conduction through O1? It seems contradictory that O1 would be associated exclusively with voltage-sensor activation and not gating ring transitions, and yet the mutations that enable cation access through O1 localize at the gating ring - this needs to be better clarified.
We have undertaken a thorough rewriting of all sections to clarify the structural correlates that may explain the behavior of the mutants. In brief, we propose that when all four voltage sensors move towards the extracellular side, the intracellular ring maintains the permeation path closed until it rotates. If the ring is altered, this “lock” is incompetent, and permeation can be detected (page 34). By fixing the position of the ring, calmodulin would preclude permeation in the WT and promote the population of O1 in the mutants.
(5) Rather than the t80% risetime, exponential fits should be performed to assess the kinetics of activation.
We agree that the assessment of kinetics by a t80% is not ideal. We originally refrained from exponential fits because they introduce other issues when used for processes that are not truly exponential (as is the case here). We had planned to perform exponential fits in this revised version, but because the activation process is not exponential, the time constants we could provide would not be accurate, and the result would remain qualitative as it is now. In the experiments where we did perform the fits (Fig. 3), the values obtained support the statement made.
(6) It is argued based on the G-V relations in Figure 2A that none of the mutations or deletions introduced have a major effect on state O1 properties, but rather affect state O2. However, the occupancy of state O2 is undetermined because activation curves do not reach saturation. It would be interesting to explore the fitting parameters on Fig.2B further to test whether the data on Fig 2A can indeed only be described by fits in which the parameters for O1 remain unchanged between constructs.
We agree that the absolute occupancy of O2 cannot be properly determined if a steady state is not reached. This is, however, a feature of the channel. During very long depolarizations in WT, the current visually appears to reach a plateau, but a closer look reveals that the current keeps increasing after very long depolarizations (up to 10 seconds; see, e.g., Fig. 1B in Garg et al., 2013, Mol Pharmacol 83, 805-813. DOI: 10.1124/mol.112.084384). Interestingly, although the model presented here does not account for this behavior, we propose changes in the model that could. “If the relative stability of O2 and C2 continued to change throughout the depolarization such a current creep-up could be reproduced. However, this would require either the introduction of further layers of On↔Cn states or a non-Markovian modification of the model’s evolution.” Page 34.
(7) The authors interpret the results obtained with the mutants DPASCAP and E600R -tested before by Lorinczi et al. 2016, to disrupt the interactions between the PASCap and cNBHD domains- as a two-step gating mechanism with two open states. All the results obtained with the E600R mutant and DPASCap could also be explained by inactivation/recovery from inactivation behavior and a change in the equilibrium between the closed states closed/inactivated states and open states. Moreover, the small tails between +90 to +120 mV suggest channels accumulate in an inactive state (Fig 1E). It is not convincing that the two open-state model is the mechanism underlying the mutant's behavior.
We respectfully disagree with the notion that a single open state can provide a plausible explanation for "All the results obtained with the E600R mutant and DPASCap". We think that our new single channel results settle the question, but even without this direct evidence, a quantitative assessment of the triphasic tail currents all but excludes the possibility of a single open state. We agree that it is, in principle, possible to obtain some form of a multiphasic tail with a single open state using the scheme suggested in this comment: at the end of the test pulse, a large fraction of the channels must be accumulated in inactive states, and a few are in the open state. The hyperpolarization to -100mV then induces a rapid depopulation of the open state, followed by slower replenishments from the inactive state. Exactly this process occurs in our model, when C2 empties through O2 (Supp. 5 to Fig 9, E600R model variant). However, this alone is highly unlikely to quantitatively explain the measured tail currents, because of the drastically different time scales of the initial current decay (submillisecond to at most a few milliseconds lifetime) and the much slower transient increase in current (several tens of milliseconds) and the final decay with time constants of >100 ms (see for instance data in Fig. 1 E for E600R +50 to +120mV test pulse). To sustain the substantial magnitude of slowly decaying current by slow replenishment of an open state with a lifetime of 1 ms requires vast amounts of inactivated channels. A rough estimation based on the current integral of the initial decay and the current integral of the slowly decaying current suggests that at the end of the test pulse, the ratio inactivated/open channels would have to be 500 to 1500 for this mechanism to quantitatively explain the observed tail currents. To put this in perspective: This would suggest that without inactivation all the expressed channels in an oocyte would provide 6 mA current during the +100 mV test pulse. While theoretically possible, we consider this a less likely explanation than a second open state.
(8) Different models should be evaluated to establish whether the results in Figure 4 can also be explained by a model in which states O1 and O2 have the same conductance. It would be desirable if the conductance of both states were experimentally determined - noise analysis could be applied to estimate the conductance of both states.
In the modified model, O1 and O2 have the same single-channel conductance. The small conductance combined with the fast flickering did not allow an accurate determination, but we can state that there is no evidence that the single-channel conductance of the states is different.
(9) Although not included, it looks like the model predicts some "conventional inactivation" This can be appreciated in Fig 8, and in the traces at -60mV. Interestingly, the traces obtained in the absence of Cl- also undergo slow inactivation, or 'conventional inactivation' as referred to by the authors. Please revise the following statement "Conventional inactivation was never detected in any mutants after repeated or prolonged depolarization. In the absence of inactivation, the pre-pulse dependent current increase at +40 mV could be related to changes in the relative occupancy of the open states".
We have carefully edited the manuscript to address this concern. The use of the term inactivation admittedly represents a challenge. We agree that the state that results from the flickering block (C2) could be defined as “inactivated” because it is preceded by an open state. Yet, in that case, the intermediate states that the channel travels between O1 and O2 would also be sensu stricto “inactivated”, but only in the mutants. We have made this clear in page 17.
Recommendations for improving the writing and presentation.
(1) Methods section: Please state the reversal potential calculated for the solution used. It looks like the authors used an Instantaneous I-V curve method to calculate the reversal potential; if that's correct, please show the I-V and the traces together with the protocol used.
We have provided the calculated reversal potentials for excised patches. We cannot predict the reversal potential in whole oocytes because we have no control over the intracellular solution. The reversal potential was determined in the mutants through the current at the end of the stimulus because the mutants produced measurable inward currents. The differences in reversal potential were not significant among mutants.
Pulse protocols have been added to the figures.
(2) Figure 1 suggestion: Combine the two panels in panel D and move the F panel up so the figure gets aligned in the lower end.
Thank you, this has been done.
(3) Please clarify the rationale for using the E600R-specific mutant. I assume it is based on the Lorinzci et al. 2016 effect and how this is similar to the DPASCap phenotype, or is it due to the impact of this mutation in the interactions between the N-term and the cNBHD?
We have explained the rationale for the use of E600R explicitly on page 6.
(4) Fig S1A is not present in the current version of the manuscript. Include a cartoon as well as a structural figure clearly depicting the perturbations introduced by E600R, ΔPASCap, and the other deletions that are tested. Additional structural information supporting the discussion would also be helpful to establish clearer mechanistic links between the experimental observations described here and the observed conformational changes between states in Kv10 channel structures.
We have corrected this omission, thank you for pointing it out.
(5) It would be informative to see the traces corresponding to the I-V shown in Fig 7 A and B at the same indicated time points (0, 60, 150, and 300s). Did the authors monitor the Ca2+ signal rise after the I&T treatment to see if it coincides with the peak in the 60s?
In Figure 7 (now Figure 8) we used voltage ramps instead of discrete I-V protocols because of the long time required for recording the latter. This is stated on page 19. Ca2+ was monitored through Cl- current after ionomycin/thapsigargin. The duration of the Ca2+ increase was reproducible among oocytes and in good agreement with the changes observed in the biphasic behavior of the mutants (Supplement 1 to Figure 8).
(6) Fig 4. Please state in the legend what the different color traces correspond to in E600R and DPASCap. Is there a reason to change the interpulse on DPASCap to -20mV and not allow this mutant to close? Please state. How do the authors decide the 10 ms interval for the experiments in Fig 2?
Thank you for pointing this out, we have added the description. We have explained why we use a different protocol for ΔPASCap and the reason for using 10 ms interval (we believe the referee means Figure 4) on page 12.
(7) Fig. 5. Since the pre-pulse is supposed to be 5s, but the time scale doesn't correspond with a pre-pulse of 5 s before the test pulse to +40mV. Has the pre-pulse been trimmed for representation purposes? If so, please state.
The pre-pulse was 5s, but as the reviewer correctly supposed, the trace is trimmed to keep the +40 mV stimulus visible. This has now been clearly stated in the legend.
(8) The mutant L322H is located within the S4 helix according to the Kv10.1 structure (PDB 5K7L), not in the 'S3-S4 linker'; please correct.
This has been done, thank you.
The introduction of this mutant should also shift the voltage dependence toward more hyperpolarizing potentials (around 30mV, according to Schoenherr et al. 1999). It looks like that shift is present within the first component of the G-V. Still, since the max amplitude from the second component could be contaminated by endogenous Cl- currents, this effect is minimized. Repeating these experiments in the no Cl- solutions will help clarify this point and see the effect of the DPASCap and E600R in the background of a mutation that accelerates the transitions between the closed states (see Major comment 1). Did the authors record L322H alone for control purposes?
We have decided not to measure L322H alone or repeat the measurements in Cl--free solutions because we do not see a way to use the quantitative assessment of the voltage dependence of L322H and the L322H-variants of the eag domain mutants. Like in our answer to main point 3, we base our arguments not on the precise voltage dependence of the second component but on the shape of the G-V curves instead, specifically the consistent appearance of the first component and the local conductance minimum between the first and second components. After the introduction of L322H the first component is essentially absent.
We think that the measurements of the L322H mutants cannot be interpreted as a hyperpolarizing shift in the first component. The peak of the first conductance component occurs around -20 mV in ΔPASCap and E600R (Fig. 7 C, D). After a -30mV shift, in L322H+DPASCap and L322H+E600R, this first peak would still be detected within the voltage range in our experiments, but it is not. A contamination of the second component would have little impact on this observation, which is why we refrain from the suggested measurements.
(9) The authors differentiate between an O1 vs. O2 state with different conductances, and maybe I missed it, but there's no quantitative distinction between the components; how are they different?
Please see the response to the main comments 1 and 2. This has been addressed in singlechannel recordings.
(10) Please state the voltage protocols, holding voltages, and the solutions (K+ concentration and Cl-presence/absence) used for the experiments presented in the legends on the figures. Hence, it's easier to interpret the experiments presented.
Thank you, this has been done.
(11) The authors state on page 7 that "with further depolarizations, the conductance initially declined to rise again in response to strong depolarizations. This finding matches the changes in amplitude of the tail currents, which, therefore, probably reflect a true change in conductance" However, the tails in the strong voltage range (+50 to +120 mV) for the E600R mutant argue against this result. Please review.
The increase in the amplitude of the tail current is also present in E600R, but the relative increase is smaller. We have decided against rescaling these traces because the Figure is already rather complex. We indicated this fact with a smaller arrow and clarified it in the text (page 8).
(12) The authors mention that the threshold of activation for the WT is around -20mV; however, the foot of the G-V is more around -30 or -40mV. Please revise.
Thank you. We have done this.
(13) The authors state on page 9 that the 'second component occurs at progressively more depolarized potentials for increasingly larger N-terminal deletions" However E600R mutant that conserves the N-terminal intact has a shift as pronounced as the DPASCap and larger than the D2-10. How do the authors interpret this result?
We have corrected this statement in page 10 : “…the second component occurs at progressively more depolarized potentials for increasingly larger N-terminal deletions and when the structure of the ring is altered through disruption of the interaction between N- and C-termini (E600R)”.
(14) The equation defined to fit the G-Vs, can also be used to describe the WT currents. If the O1 is conserved and present in the WT, this equation should also fit the WT data properly. The 1-W component shown could also be interpreted as an inactivating component that, in the WT, shifts the voltage-dependence of activation towards depolarizing potentials and is not visible. Still, the mutants do show it as if the transition from closed-inactivated states is controlled by interactions in the gating ring, and disturbing them does affect the transitions to the open state.
Out of the two open states in the mutant, O2 is the one that shares properties with the WT (e.g. it is inaccessible during Ca2+-CaM binding) while O1 is the open state with the voltage dependence that is conserved across the mutants. We, therefore, believe that this question is based on a mix-up of the two open states. We appreciate the core of the question: does the pattern in the mutants’ G-V curves find a continuation in the WT channel?
Firstly, the component that is conserved among mutants does not lead to current in the WT because the corresponding open state (O1) is not observed in WT. However, the gating event represented by this component should also occur in WT and –given its apparent insensitivity to eag domain mutations– this gating step should occur in WT with the same voltage dependence as in all the mutants. This means that this first component sets a hard boundary for the most hyperpolarized G-V curve we can expect in the WT, based on our mutant measurements. Secondly, the second component shows a regular progression across mutants: The more intact the eag domain is, the more hyperpolarized the Vhalf values of transition term (1-W) and O2 activation. In Δ2-10, the transition term already almost coincides with O1 activation (estimated Vhalf values of -33.57 and -33.47 mV). A further shift of (1-W) in the WT is implausible because, if O1 activation is coupled to the earliest VSD displacement, the transition should not occur before O1 activation. Still, the second component might shift to more hyperpolarized values in the WT, depending on the impact of amino acids 2 to 10 on the second VSD transition.
In summary, in WT the G-V should not be more hyperpolarized than the first component of the mutants, and the (1-W)-component probably corresponds to the Δ2-10 (1-W)-component. In WT the second component should be no more depolarized than the second component of Δ2-10. The WT G-V (Fig.1B) meets all these predictions derived from the pattern in the mutant GVs: When we use Eq. 4 to fit the WT G-V with A1=0 (O1 is not present in WT) and the parameters of the transition term (1-W) fixed to the values attained in Δ2-10, we obtain a fit for the O2 component with Vhalf\=+21mV. This value nicely falls into the succession of Vhalf values for Δeag, ΔPASCap, and Δ2-10 (+103mV,+80mV,+52mV) and, at the same time, it is not more hyperpolarized than the conserved first component (Vhalf -34mV). Our measurements therefore support that the O2 component in the mutants corresponds to the single open state in the WT.
(15) Page 15, the authors state that 'The changes in amplitude and kinetics in response to rising intracellular Ca2+ support our hypothesis that Ca-CaM stabilized O1, possibly by driving the channels to deep closed states (Fig 5 and 6)' (pg 15). This statement seems contradictory; I can't quite follow the rationale since Ca2+ potentiates the current (Fig 7), and the addition of the L322H mutant in Fig 7 makes the shift of the first component to negative potentials visible.
Please check the rationale for this section.
We have explained this more explicitly in the discussion (page 32). “Because access to O1 occurs from deep closed states, this could be explained by an increased occupancy of such deactivated states in response to CaM binding. This appears to be the case since CaM induces a biphasic behavior in the mutant channels that show reduced access to deep closed states; thus, L322H mutants behave like the parental variants in the presence of Ca2+-CaM. This implies a mechanistic explanation for the effect of Ca2+-CaM on WT since favoring entry into deep closed states would result in a decrease in current amplitude in the absence of (a permeable) O1”.
Also, Figs 5 and 6 seem miscited here.
Thank you, we have corrected this.
(16) For Figure 5, it would be helpful if each of the current traces corresponding to a particular voltage had a different color. That way, it will be easier to see how the initial holding voltage modulates current.
We have considered this suggestion, and we agree that it would make it easier to follow. Yet, since we have identified the mutants with different colors, it would be inconsistent if we used another color palette for this Figure. Supplement 3 to Figure 9 shows the differences in a clearer way.
(17) Add zero-current levels to all current traces.
We have done this.
(18) The mathematical model should be described better. Particularly, the states from which O1 can be accessed should be described more clearly, as well as whether the model considers any direct connectivity between states O1 and O2. The origin of the voltage-dependence for transitions that do not involve voltage-sensor movements should be discussed. Also, it separation of kappa into kappa-l and kappa-r should be described.
We have extensively rewritten the description of the mathematical model to address these concerns.
(19) Page 4, "reveals a pre-open state in which the transmembrane regions of the channel are compatible with ion permeation, but is still a nonconducting state". Also, page 27, "renders a hydrophobic constriction wider than 8 Å, enough to allow K+ flow, but still corresponds to a non-conducting state". These sentences are confusing - how can the regions be compatible with ion permeation, and still not be conducting? Is cation conductance precluded by a change in the filter, or elsewhere? How is it established that it represents a non-conducting state?
We have rephrased to clarify this apparent inconsistence. Page 4: “(…) in which the transmembrane regions of the channel are compatible with ion permeation (the permeation path is dilated, like in open states) but the intracellular gate is still in the same conformation as in closed states (Zhang et al., 2023).” Page 31: “The presence of an intact intracellular ring would preclude ionic flow in the WT, and its alteration would explain the permeability of this state in the mutants.”



