Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorToby AllenRMIT University, Melbourne, Australia
- Senior EditorQiang CuiBoston University, Boston, United States of America
Reviewer #1 (Public review):
Summary:
The authors investigate ligand and protein-binding processes in GPCRs (including dimerization) by the multiple walker supervised molecular dynamics method. The paper is interesting and it is very well written.
Strengths:
The authors' method is a powerful tool to gain insight on the structural basis for the pharmacology of G protein-coupled receptors.
Reviewer #2 (Public review):
The study by Deganutti and co-workers is a methodological report on an adaptive sampling approach, multiple walker supervised molecular dynamics (mwSuMD), which represents an improved version of the previous SuMD.
Case-studies concern complex conformational transitions in a number of G protein Coupled Receptors (GPCRs) involving long time-scale motions such as binding-unbinding and collective motions of domains or portions. GPCRs are specialized GEFs (guanine nucleotide exchange factors) of heterotrimeric Gα proteins of the Ras GTPase superfamily. They constitute the largest superfamily of membrane proteins and are of central biomedical relevance as privileged targets of currently marketed drugs.
MwSuMD was exploited to address:
a) binding and unbinding of the arginine-vasopressin (AVP) cyclic peptide agonist to the V2 vasopressin receptor (V2R);
b) molecular recognition of the β2-adrenergic receptor (β2-AR) and heterotrimeric GDP-bound Gs protein;
c) molecular recognition of the A1-adenosine receptor (A1R) and palmotoylated and geranylgeranylated membrane-anchored heterotrimeric GDP-bound Gi protein;
d) the whole process of GDP release from membrane-anchored heterotrimeric Gs following interaction with the glucagon-like peptide 1 receptor (GLP1R), converted to the active state following interaction with the orthosteric non-peptide agonist danuglipron.
The revised version has improved clarity and rigor compared to the original also thanks to the reduction in the number of complex case studies treated superficially.
The mwSuMD method is solid and valuable, has wide applicability and is compatible with the most world-widely used MD engines. It may be of interest to the computational structural biology community.
The huge amount of high-resolution data on GPCRs makes those systems suitable, although challenging, for method validation and development.
While the approach is less energy-biased than other enhanced sampling methods, knowledge, at the atomic detail, of binding sites/interfaces and conformational states is needed to define the supervised metrics, the higher the resolution of such metrics is the more accurate the outcome is expected to be. Definition of the metrics is a user- and system-dependent process.
Reviewer #3 (Public review):
Summary:
In the present work Deganutti et al. report a structural study on GPCR functional dynamics using a computational approach called supervised molecular dynamics.
Strengths:
The study has potential to provide novel insight into GPCR functionality. Example is the interaction between D344 and R385 identified during the Gs coupling by GLP-1R. However, validation of the findings, even computationally through for instance in silico mutagenesis study, is advisable.
Weaknesses:
No significant advance of the existing structural data on GPCR and GPCR/G protein coupling is provided. Most of the results are reproductions of the previously reported structures.
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
The authors investigate ligand and protein-binding processes in GPCRs (including dimerization) by the multiple walker supervised molecular dynamics method. The paper is interesting and it is very well written.
Strengths:
The authors' method is a powerful tool to gain insight into the structural basis for the pharmacology of G protein-coupled receptors.
Weaknesses:
Cholesterol may play a fundamental role in GPCR dimerization (as cited by the authors, Prasanna et al, "Cholesterol-Dependent Conformational Plasticity in GPCR Dimers"). Yet they do not use cholesterol in their simulations of the dimerization.
We thank Reviewer #1 for the positive comment on mwSuMD.
In the revised version of the manuscript, the section about the A2A/D2 receptors dimerization has been removed because largely speculative. We agree that the lack of cholesterol in those simulations added uncertainty to the presented results.
Reviewer #2 (Public Review):
The study by Deganutti and co-workers is a methodological report on an adaptive sampling approach, multiple walker supervised molecular dynamics (mwSuMD), which represents an improved version of the previous SuMD.
Case-studies concern complex conformational transitions in a number of G protein Coupled Receptors (GPCRs) involving long time-scale motions such as binding-unbinding and collective motions of domains or portions. GPCRs are specialized GEFs (guanine nucleotide exchange factors) of heterotrimeric Gα proteins of the Ras GTPase superfamily. They constitute the largest superfamily of membrane proteins and are of central biomedical relevance as privileged targets of currently marketed drugs.
MwSuMD was exploited to address:
(1) Binding and unbinding of the arginine-vasopressin (AVP) cyclic peptide agonist to the V2 vasopressin receptor (V2R);
(2) Molecular recognition of the β2-adrenergic receptor (β2-AR) and heterotrimeric GDPbound Gs protein;
(3) Molecular recognition of the A1-adenosine receptor (A1R) and palmitoylated and geranylgeranylated membrane-anchored heterotrimeric GDP-bound Gi protein;
(4) The whole process of GDP release from membrane-anchored heterotrimeric Gs following interaction with the glucagon-like peptide 1 receptor (GLP1R), converted to the active state following interaction with the orthosteric non-peptide agonist danuglipron;
(5) The heterodimerization of D2 dopamine and A2A adenosine receptors (D2R and A2AR, respectively) and binding to a bi-valent ligand.
The mwSuMD method is solid and valuable, has wide applicability, and is compatible with the most world-widely used MD engines. It may be of interest to the computational structural biology community.
The huge amount of high-resolution data on GPCRs makes those systems suitable, although challenging, for method validation and development.
While the approach is less energy-biased than other enhanced sampling methods, knowledge, at the atomic detail, of binding sites/interfaces and conformational states is needed to define the supervised metrics, the higher the resolution of such metrics is the more accurate the outcome is expected to be. The definition of the metrics is a user- and system-dependent process.
The too many and ambitious case-studies undermine the accuracy of the output and reduce the important details needed for a methodological report. In some cases, the available CryoEM structures could have been exploited better.
The most consistent example concerns AVP binding/unbinding to V2R. The consistency with CryoEM data decreases with an increase in the complexity of the simulated process and involved molecular systems (e.g. receptor recognition by membrane-anchored G protein and the process of nucleotide exchange starting from agonist recognition by an inactive-state receptor). The last example, GPCR hetero-dimerization, and binding to a bi-valent ligand, is the most speculative one as it does not rely on high-resolution structural data for metrics supervision.
We praise Reviewer #2 for the detailed comment on the manuscript. In this revised version, the hetero-dimerization between A2AR and D2R has been removed. Also, results about GPCR case studies other than GLP-1R have been reduced and downgraded in importance to focus on the fundamental key points of the adaptive sampling method. We agree that the consistency with cryoEM data tends to decrease with an increase in the complexity of the simulated process and involved molecular systems. While it is possible to approximate cryoEM results our unbiased adaptive sampling technique finds its most interesting application in mechanistically unknown out-of-equilibrium processes rather than reproducing known experimental data perfectly. The simulated case studies we present showcase the versatility, speed and consistency of our adaptive method to explore energetically unbiased transitions.
Reviewer #3 (Public Review):
Summary:
In the present work, Deganutti et al. report a structural study on GPCR functional dynamics using a computational approach called supervised molecular dynamics.
Strengths:
The study has the potential to provide novel insight into GPCR functionality. An example is the interaction between loops of GPCR and G proteins, which are not resolved experimentally, or the interaction between D344 and R385 identified during the Gs coupling by GLP-1R. However, validation of the findings, even computationally through for instance in silico mutagenesis study, is advisable.
Weaknesses:
In its current form, the manuscript seems immature and in particular, the described results grasp only the surface of the complex molecular mechanisms underlying GPCR activation. No significant advance of the existing structural data on GPCR and GPCR/G protein coupling is provided. Most of the results are a reproduction of the previously reported structures.
We thank Reviewer #3 for the positive comment on the work. The revised manuscript focuses more on the GLP-1R and Gs case studies. We believe it addresses the weaknesses raised by showing the behaviour of key structural motifs and providing new hypotheses about GDP release.
Reviewer #2 (Recommendations For The Authors):
In this methodological report, Deganutti and co-workers propose an improved version of supervised molecular dynamics (SuMD), named multiple walker SuMD (mwSuMD). Such an adaptive sampling method was challenged in simulations of complex transitions involving GPCRs, which are out of reach by classical MD.
Although less energy-biased than other enhanced sampling methods, mwSuMD requires knowledge of the atomic detail of the ligand-protein or protein-protein binding site/interfaces and the structural hallmarks of the states whose conversion the method is going to address. Such knowledge is, indeed, necessary to define the supervised metrics (e.g. distances, RMSD, etc), which is a user- and system-dependent process.
We classify mwSuMD as an adaptive, rather than enhanced, sampling method as it does not use any energy bias. We agree with the Reviewer that some knowledge of the system is required to productively set up the simulations, but this is the case for almost any MD advanced methods.
The text requires improvement in the essential methodological details and cleaning of those parts is not properly instrumental in method validation.
While attempting to prove the widest possible applicability of the method, the authors exaggerated the number of examples, which, in spite of the increasing complexity were only summarily described. Please, limit the case studies to AVP binding/unbinding to V2R and the whole process of GDP release from membrane-anchored Gs following activation of GLP1R by danuglipron. The latter case, indeed, involves small ligand binding (danuglipron), small ligand dissociation (GDP), receptor activation, and activated receptor binding to membraneanchored G protein and G protein conformational transition instrumental to nucleotide depletion, which is already too much. In this framework, the cases of Gs-β2AR and Gi-A2R recognition are redundant. Most importantly, the case of D2R-A2AR heterodimerization and binding to a bi-valent ligand must be eliminated. The reason is that the case is not entirely based on the mwSuMD and the biased protein-protein interface does not rely on highresolution data (i.e. no structural model of D2R-A2AR dimer has been determined so far). Last but not least, the high intrinsic flexibility of the bi-valent ligand adds further indetermination to the computational experiment. Being too speculative, the case-study does not serve to model validation.
We thank the Reviewer for the suggestion. In the current revised form, the manuscript focuses on AVP binding/unbinding to V2R and the GLP-1R activation, Gs recognition and GDP release.
While eliminating the three case studies mentioned above, the remaining ones should be described more extensively and clearly, highlighting the most productive setup for each system. Incidentally, listing the performance parameters (e.g. distribution mode and minimum RMSD) of each simulation setting in Table S1 is worth doing.
More accuracy in the methodological description is needed.
As for the supervised metrics, the rationale behind the choice of a particular index and whether it is the outcome of a number of trials must be declared and the selected indices must be better defined. Here there are a few examples.
AVP-V2R case. It is not clear why the AVP centroids were computed on residues C1-Q4 (I suppose the Cα-atoms) and not on the Cα-atoms of the whole cyclic part (C1-C6). Along the same line, the choice of the Cα-atoms of four amino acid residues to compute the receptor binding-site centroids requires justification.
We have amended the text to clarify that all the heavy atoms of AVP residues C1-Q4, which are anticipated to bind deep into V2R, were considered alongside V2R residues part of the peptide binding site (Cα atoms only). From our experience, the choice of including side chains or not for the definition of centroids usually does not affect the supervision output. It should only affect the output of mwSuMD simulations based on the RMSD which considers the specific relative distance from the reference. However, a benchmark of the differences produced by divergent selections is beyond the scope of the present work.
GLP1R case. The statement: "Since the opening of TM1-ECL1 was observed in two replicas out of four, we placed the ligand in a favorable position for crossing that region of GLP-1R" is rather weak as a strategy to manually (?) define the input position of the ligand.
As stated in the manuscript, placing the agonist in that position was driven by preliminary 8 μs of classic MD simulations that pointed out the possible path for binding. We agree with the Reviewer that there is still some degree of arbitrarity in it and for this reason, we have not presented structural details of the F06882961 binding path.
As for the supervised metrics, what does it mean "the distance between the ligand and GLP-1R TM7 residues L3797.34-F3817.36"? Was the distance computed between ligand and L379-F381 centroids? Also: "In the supervised stages, the distance between residues M386-L394 Gas of helix 5 (α5) and the GLP-1R intracellular residues R1762.46, R3486.37, S3526.41, and N4057.60 was monitored" was it an inter-centroid distance? Furthermore, "supervising the distance between AHD residues G70-R199 Gas and K300-L394Gas" was it the distance between the centroid of the AHD and the centroid of the C-terminal half of the Ras-like domain? In general, when more than two atoms are involved in distance calculation, please, specify if the distance is inter-centroid.
Also: "During the third phase, the RMSD of PF06882961, as well as the RMSD of ECL3 (residues A3686.57-T3787.33, Ca atoms), were supervised" was the RMSD computed without superimposing the ligand to estimate its roto-translations?
We have added details about the selections used for computing centroids throughout the methods section. For example, all the heavy atoms of F06882961 and the Ca atoms of L379-F381 were considered. RMSD values during GLP-1R activation were computed after superimposition on TM2, ECL1, and TM3 residues 170-240 (Ca atoms). This now has been specified in the text.
The authors considered the 7LCJ GLP1R-danuglipron complex as a fully active reference state instead of considering the receptor from a ternary complex with Gs. The ternary complex (7LCI) was indeed considered as a reference only in simulations of receptor-G protein recognition.
7LCJ and 7LCI are both fully active states. The main difference is that in 7LCJ, Gs coordinates were not deposited. Indeed, their RMSD computed on the TMD Ca atoms and F06882961 is 0.63 Å and 0.54 Å, respectively.
Most importantly, the ternary complex chosen by the authors is not adequate as a reference for simulating the "opening" of the AHD because it bears a miniGs, hence, missing the AHD. In that framework, such an opening is rather vague and was not properly supervised by mwSuMD. The authors must repeat metrics supervisions by using, as a reference, the 6X1A ternary complex, which bears a displaced AHD. This would likely lead to a different path of GDP release.
To the best of our knowledge, there is no evidence that a specific open conformation of the AHD is linked to GDP release. In support, we note that in GPCR ternary complexes, the AHD is usually not modelled because of its high flexibility. The only body of evidence we are aware of is that AHD must open up to allow GDP release. For this reason, we decided to supervise the distance between AHD and the Ras domain without using a reference.
In the statement: "The AHD opening was simulated starting from the best GLP-1R:Gs binding mwSuMD replica" the definition "best binding" requires clarification.
This has been amended, specifying that Replica 2 was considered the “best replica” due to the closed deviation to the cryoEM structure.
As for the case study on β2-AR-Gs recognition, I strongly suggest to eliminate it. However, I'd like to make some comments. The sentence: "the adrenergic β2 receptor (b2 AR) in an intermediate active state was downloaded from GPCRdb (https://gpcrdb.org/)" is vague as it does not indicate what intermediate active state structure was used. Since the goal of the case study was to probe the method in simulating receptor-G protein binding, it would have been better to start with a fully active state of the receptor like the 4LDO structure, employed by the authors only to extract epinephrine.
mwSuMD is designed to provide insights into structural transitions. We started from an intermediate active state of β2-AR in complex with adrenaline because resembling the most populated state stabilised by a full agonist according to NMR studies (DOI:10.1016/j.cell.2015.08.045); the fully-active β2-AR conformation is stabilized only after Gs binding. However, following the Reviewer’s suggestion, we have reduced the presented results for the β2-AR-Gs recognition.
Also in this case, it is not clear if the supervised receptor-G protein distance is between the centroid of the whole 7-helix bundle and the centroid of Gs α5. It is not clear why the TM6 RMSD concerned only the cytosolic end of the helix and did not include the kink region. With that selection, to estimate the outward displacement, RMSD should have been computed without superimposing the considered portion (once all remaining Cα-atoms of the receptors are superimposed).
As the Reviewer pointed out above, some knowledge of the system is required to set up mwSuMD. Using more generic metrics as we did in this case, like the distance between the whole TMD and Gs α5 represents a general approach applicable to other GPCRs, that should allow orthogonal metrics to evolve independently from the supervision.
As now specified in the text, the superimposition for RMSD calculation was performed on residues 40 to 140 Ca atoms, hence not considering TM6.
As for the A1R-Gi recognition, as already stated, I strongly suggest eliminating it. However, I'd like to add some comments. I would discourage the employment of an AlphaFold model for simulations deputed to model validation in general and, in particular, when highresolution structures are available. In this case, the authors would have used the 1GP2 structure of heterotrimeric Gi no matter if from the rat species.
Following the Reviewer’s suggestion, we have dramatically reduced the results presented for the A1R-Gi recognition. We considered 1GP2 for the simulations but H5 lacks the Cterminal six residues and therefore some extent of modelling was still necessary. However, we take the Reviewer’s comment on board and consider it for future work.
Also, the palmitoylation and geranylgeranylation process is quite tortuous and it is not clear why the NVT ensemble was employed in the second stage of equilibration. This is reflected also on the GLP1R case study.
We have amended the text to clarify this passage. The second NVT stage is required for stabilizing the G protein and its orientation in the simulation box. The figure below shows that a plateau of the Ca RMSD during the NVT step was reached after 700 ns for both Gi (black) and Gs (orange).
Author response image 1.
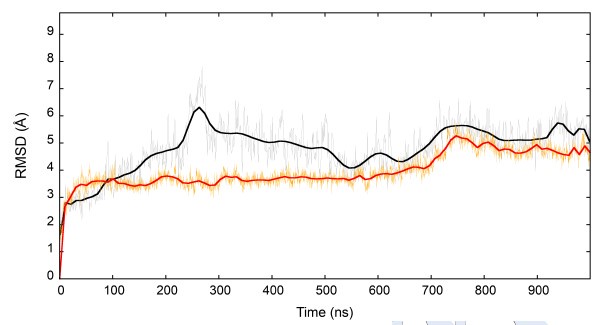
Here, it is not clear if the RMSD of α5 of Gi was computed with or without superposition.
The RMSD of α5 was computed after superimposing on A1R residues 40-140 Ca atoms (the less flexible region of the receptor). We have now amended the text to report this information.
Reviewer #3 (Recommendations For The Authors):
Points to address:
(1) Root Mean Square Deviation (RMSD) data are often reported as minimum values. It would be useful to provide the average value along the stable part of the trajectories. From the plots in Figure 2ab, it seems that the minimum values reported in the paper are very far from the average ones and thus represent special cases that are seldom reached during simulation. The authors should clarify this point;
For the revised manuscript, we moved Figure 2 to the supplementary material and added average RMSD values for the most notable replicas in Figures 4e and S8a,b. As a reference, in the text, we now report RMSDs from our previous classic MD simulations (https://doi.org/10.1038/s41467-021-27760-0) of Gs:GLP-1R cryoEM structure (Gα = 6.18 ± 2.40 Å; Gβ \= 7.22 ± 3.12 Å; Gγ = 9.30 ± 3.65 Å) which show how flexible G proteins bound to GPCRs are and give better context to the RMSD values we measured during mwSuMD simulations.
(2) The RMSD values reported in the paper always refer to single molecules or proteins. It would be useful to also report the RMSD computed over the whole complexes (ligand/GPCR or GPCR/G protein). It would provide a better metric for understanding the general distance between the results and the reference experimental structures;
We have now removed the results sections for A1R and β2 AR to focus on GLP-1R, whose RMSD is analyzed in detail in Figures 2, 3 and 4.
(3) A number of computational works investigated the GPCR/G protein interaction and these studies should be cited and discussed. Examples are the works from Mafi et al. 2023 (doi: 10.1038/s41557-023-01238-6), Fleetwood et al. 2020 (doi: 10.1021/acs.biochem.9b00842), Calderon et al. 2023 and 2024 (doi: 10.1021/acs.jcim.3c00805 and doi: 10.1021/acs.jcim.3c01574), Maria-Solano and Choi 2023 (doi: 10.7554/eLife.90773.1), Mitrovic et al. 2023 (doi: 10.1021/acs.jpcb.3c04897), and D'Amore et al. 2023 (doi: 10.1101/2023.09.14.557711). Many of these works focused on the activation of B2AR and the interaction with its G protein. In addition, Maria-Solano and Choi 2023 and D'Amore et al. 2023 also characterized the rotation of TM6 during the A1R and A2AR activation. Therefore, the claim "To the best of our knowledge, this is the first time an MD simulation captures the TM6 rotation upon receptor activation as results reported so far are largely limited to the TM6 opening and kinking55." is untimely;
We thank the Reviewer for the suggested references. We have added them to the introduction as examples of energy-biased (Calderon et al. 2023 and 2024, Maria-Solano and Choi, Mitrovic et al., D'Amore et al) or adaptive sampling (Fleetwood et al) approaches to GPCR. Since the above articles focus on β2 AR and A1R, we do not discuss them in detail because the results sections for A1R and b2 AR have been drastically reduced in the manuscript.
We note that among the suggested references, only Mafi et al report about a simulated G protein (in a pre-formed complex) and none of the work sampled TM6 rotation without input of energy. However, we have removed the claim from the text.
(4) In the discussion section, the authors claim that a distance-based approach can be employed when the structural data of the endpoints is limited. However, the results obtained from the distance-based protocol during the validation of the approach, which was done using V2R as a reference, are unsatisfying, as acknowledged by the authors themselves. For instance, the RMSD mode value reported for the AVP C alpha atoms with respect to 7DW9 is high, 0.7 nm, whereas the minimum value is 0.38 nm. In addition, some side chains are not oriented in the experimental conformation and might have a different interaction pattern with the receptor if compared with the experimental structure. Considering that in this case the endpoint is known, it is plausible that the performance of the method would degrade even further when data about the target structure is limited. In a real case scenario, the ligand binding mode is unknown and in such a case no RMSD matrix can be used. This represents the major concern of this study that is no prediction is provided, but only - rather inaccurate - reproduction of the known structural data;
The goal of the first part of the work was to compare mwSuMD to SuMD to justify its application on ligand binding using a challenging case study like vasopressin. The general validation of the parent method SuMD as a predictive tool for ligand binding mode has been extensively reported over the years (a few examples: https://doi.org/10.1021/ci400766b ; https://doi.org/10.1021/acs.jcim.5b00702 ; https://doi.org/10.1038/s41598-020-77700-z) and fell beyond the scope of this work.
(5) In the discussion, the authors write "A complete characterization of the possible interfaces between GPCR monomers, which falls beyond the goal of the present work, should be achieved by preparing different initial unbound states characterized by divergent relative orientations between monomers to dynamically dock." It would be useful for the reader to refer to and cite here advanced computational approaches that allow a comprehensive sampling of GPCR dimerization independently from the starting conformation of the receptors. One example is coarse-grained metadynamics as shown in doi: 10.1038/s41467-023-42082-z;
The A2A</sub/D2</sub receptors dimerization has been removed from the manuscript.
(6) In many cases, it is not reported how residues missing from the experimental structures used to model the proteins were reconstructed. This information is important, considering that the authors comment on the results of their calculations on addressing these regions, such as in the case of B2AR. Furthermore, the authors did not report how their initial models were validated. The authors should also explain why they did not model the IC loops of A2AR and D2R;
In the current version of the manuscript, for V2R ECL2 and GLP-1R, we specify that we produced 10 solutions with Modeller and considered the best one in terms of the DOPE score.
The only receptor model used, β2 AR, is now presented as preliminary data focusing on Gs and avoiding any structural detail of the Gs recognition.
As reported above the A2A-D2 dimerization has been removed from the manuscript.
(7) In several cases, the authors state that residues never investigated before play an important role in the interaction between different proteins. An example is provided on page 6 for the B2AR/G protein association. Since this claim is quite significant, it would benefit from validation, at least for further calculations such as in silico mutagenesis studies. Another example is at the end of page 10 where the authors report a hidden interaction between D344 and R385 that is pivotal for Gs coupling by GLP-1R. Is there other evidence supporting this result (previously reported literature data, conservation rate of these residues, etc.)?;
We have removed the supplementary table reporting B2AR/G protein interactions to reduce speculations and added a reference that reports GLP-1 EC50 reduction upon mutation of position 344 to Ala (https://doi.org/10.1021/acscentsci.3c00063).
(8) The authors should provide a deeper discussion about the conformational rearrangement of GPCR and G protein during the coupling. In detail, the conformational changes of microswitich amino acids of GPCR (e.g., PIF, NPxxY, inactivating ionic lock) and alpha helix 5 of G proteins should be discussed in relation to the literature data and experimental structures;
We have removed the A1R and b2 AR results to focus on GLP-1R. Key structural motifs in the polar central network and TM6 kink are analyzed more in detail in Figure 3.
(9) The chronology of the conformational changes of GLP-1R is arbitrarily chosen. During the simulation, the RMSD values reported in Fig. 3 are high and do not demonstrate the full accomplishment of the simulation of the activation process of the receptor;
We agree with the Reviewer that the GLP-1R inactive to active transition was not fully accomplished, compared to other work on class A GPCRs. Unlike class A, class B GPCRs represent a challenging system to work with in silico because inactive starting conformations (e.g 6LN2) are extremely distant from the active one (e.g 7LCJ, 7LCI or 6X18), as demonstrated in Figure S6 for GLP-1R. Here we report the first attempt to model a class B GPCR activation mechanism starting from the inactive state, and even if not fully achieved, we believe it represents state-of-the-art simulations for this class of receptors.
(10) It would be helpful for the reader not familiar with the employed technique that the authors explain in one sentence in the main text the pros and cons of using multiple walkers instead of single walker SuMD;
We thank the Reviewer for the excellent suggestion. In the Discussion, we have now commented that: “more extensive sampling obtainable by seeding multiple parallel short simulations instead of a single simulation for batch”, while in the Methods we explain that “mwSuMD is designed to increase the sampling from a specific configuration by seeding user-decided parallel replicas (walkers) rather than one short simulation as per SuMD. Since one replica for each batch of walkers is always considered productive, mwSuMD gives more control than SuMD on the total wall-clock time used for a simulation. On the flip side, mwSuMD requires multiple GPUs to be the most effective, although any multi-threaded GPU can run more walkers on the same hardware keeping the sampling variety.”.
Minor points to address:
(11) Page 3: the following sentence is duplicated (also found on page 2) "GPCRs preferentially couple to very few G proteins out of 23 possible counterparts";
(12) Page 20: Figure S13 refers to the QM validation of PF06882961 torsional angle, not to the image of the receptor conformational changes, which is instead Figure S14 (please correct figure caption).
We thank the Reviewer for the accurate reading of the manuscript. These typos have been corrected.



