Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorFlorent GinhouxSingapore Immunology Network, Singapore, Singapore
- Senior EditorAdèle MarstonUniversity of Edinburgh, Edinburgh, United Kingdom
Reviewer #1 (Public Review):
Tsai and Seymen et al. investigate associations between RTE expression and methylation and age and inflammation, using multiple public datasets. Compared to the previous round of review, the text of the manuscript has been polished and the phrasing of several findings has been made clearer and more precise. The authors also provided ample discussion to the prior reviewer comments in their rebuttal, including new analyses. All these changes are in the correct direction, however, I believe that part of the content of the rebuttal should be incorporated in the main text, for reasons that I will outline below.
Both reviewers found the reliance on microarray expression data to detract from the study. The authors argued that their choices are supported by existing publications which performed a similar quantification of TE expression using microarray data. It could still be argued that (as far as I can tell) Reichmann et al. used a substantially larger number of probes than this study, as a consequence of starting from different arrays, however, this is a minor point which the authors do not need to address. It is still undeniable that including the validation with RNA-seq data performed in the rebuttal would strengthen the manuscript. I especially believe that many readers would want to see this analysis be prominent in the manuscript, considering that both reviewers independently converged on the issue with microarray expression data. Personally, I would have included an RNA-seq dataset next to the microarray data in the main figures, however, I understand that this would require considerable restructuring and that placing RNA-seq data besides array data might be misleading. Instead, I would ask that the authors include their rebuttal figures R1 and R2 as supplementary figures.
I would suggest introducing a new paragraph, between the section dedicated to expression data and the one dedicated to DNA methylation, mentioning the issues with microarray data (Some of which were mentioned by the reviewers and other which were mentioned by the authors in the discussion and introduction) to then introduce the validation with RNA-seq data.
Figure R3 is also a good addition and should be expanded to include the GTP and MESA study and possibly mentioned in the paragraph titled "RTE expression positively correlates with BAR gene signature scores except for SINEs."
"In this study, we did not compare MESA with GTP etc. We have analysed each dataset separately based on the available data for that dataset. Therefore, sacrificing one analysis because of the lack of information from the other does not make sense. We would do that if we were after comparing different datasets. Moreover, the datasets are not comparable because they were collected from different types of blood samples."
Indeed, the datasets are not compared directly, but the associations between age, BER and TE expression for each dataset are plotted and discussed right next to each other. It is therefore natural to wonder if the differences between datasets are due to differences in the type of blood sample or if they are a consequence of the different probe sets. Using a common set of probes would help answer that question.
Author response:
The following is the authors’ response to the original reviews.
eLife assessment
This study investigates associations between retrotransposon element expression and methylation with age and inflammation, using multiple public datasets. The study is valuable because a systematic analysis of retrotransposon element expression during human aging has been lacking. However, the data provided are incomplete due to the sole reliance on microarray expression data for the core analysis of the paper.
Both reviewers found this study to be important. We have selected the microarray datasets of human blood adopted by a comprehensive study of ageing published in a Nature
Communications manuscript (DOI: doi: 10.1038/ncomms9570). We only included the datasets specifically collected for ageing studies. Therefore, the large RNA-seq cohorts for cancer, cardiovascular, and neurological diseases were not relevant to this study and cannot be included.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
Tsai and Seymen et al. investigate associations between RTE expression and methylation and age and inflammation, using multiple public datasets. The concept of the study is in principle interesting, as a systematic analysis of RTE expression during human aging is lacking.
We thank the reviewer for the positive comment.
Unfortunately, the reliance on expression microarray data, used to perform the core analysis of the paper places much of the study on shaky ground. The findings of the study would not be sufficiently supported until the authors validate them with more suitable methods.
In our discussion section in the manuscript, we have clarified that “we are aware of the limitations imposed by using microarray in this study, particularly the low number of intergenic probes in the expression microarray data. Our study can be enriched with the advent of large RNA-seq cohorts for aging studies in the future.” However, the application of microarray for RTE expression analysis was introduced previously (DOI: 10.1371/journal.pcbi.1002486) and applied in some highly cited and important publications before (DOI: 10.1038/ncomms1180, DOI: 10.1093/jnci/djr540). In fact, in a manuscript published by Reichmann et al. (DOI: 10.1371/journal.pcbi.1002486) which was cited 76 times, the authors showed and experimentally verified that cryptic repetitive element probes present in Illumina and Affymetrix gene expression microarray platforms can accurately and sensitively monitor repetitive element expression data. Inspired by this methodological manuscript with reasonable acceptance by other researchers, we trusted that the RTE microarray probes could accurately quantify RTE expression at class and family levels.
Strengths:
This is a very important biological problem.
Weaknesses:
RNA microarray probes are obviously biased to genes, and thus quantifying transposon analysis based on them seems dubious. Based on how arrays are designed there should at least be partial (perhaps outdated evidence) that the probe sites overlap a protein-coding or non-coding RNA.
We disagree with the reviewer that quantifying transposon analysis based on microarray data is dubious. As previously shown by Reichmann et al., the quantification is reliable as long as the probes do not overlap with annotated genes and they are in the correct orientation to detect sense repetitive element transcripts. Reichman et al. identified 1,400 repetitive element probes in version 1.0, version 1.1 and version 2.0 of the Illumina Mouse WG-6 Beadchips by comparing the genomic locations of the probes with the Repeatmasked regions of the mouse genome. We applied the same criteria for Illumina Human HT-12 V3 (29431 probes) and V4 (33963) to identify the RTE-specific probes.
The authors state they only used intergenic probes, but based on supplementary files, almost half of RTE probes are not intergenic but intronic (n=106 out of 264).
All our identified RTE probes overlap with intergenic regions. However, due to their repetitive natures, some probes overlap with intronic regions, too. We have replaced "intergenic" with "non-coding" in our resubmission to show that they do not overlap with the exons of protein-coding genes. However, we do not rule out the possibility that some of our detected RTE probes might overlap non-coding RNAs. In fact, the border between coding and non-coding genomes has recently become very fuzzy with new annotations of the genome. RTE RNAs can be easily considered as non-coding RNAs if we challenge our traditional junk DNA view.
This is further complicated by the fact that not all this small subset of probes is available in all analyzed datasets. For example, 232 probes were used for the MESA dataset but only 80 for the GTP dataset. Thus, RTE expression is quantified with a set of probes which is extremely likely to be highly affected by non-RTE transcripts and that is also different across the studied datasets. Differences in the subsets of probes could very well explain the large differences between datasets in multiple of the analyses performed by the authors, such as in Figure 2a, or 3a. It is nonetheless possible that the quantification of RTE expression performed by the authors is truly interpretable as RTE expression, but this must be validated with more data from RNA-seq. Above all, microarray data should not be the main type of data used in the type of analysis performed by the authors.
In this study, we did not compare MESA with GTP etc. We have analysed each dataset separately based on the available data for that dataset. Therefore, sacrificing one analysis because of the lack of information from the other does not make sense. We would do that if we were after comparing different datasets. Moreover, the datasets are not comparable because they were collected from different types of blood samples.
Reviewer #2 (Public Review):
Summary:
Yi-Ting Tsai and colleagues conducted a systematic analysis of the correlation between the expression of retrotransposable elements (RTEs) and aging, using publicly available transcriptional and methylome microarray datasets of blood cells from large human cohorts, as well as single-cell transcriptomics. Although DNA hypomethylation was associated with chronological age across all RTE biotypes, the authors did not find a correlation between the levels of RTE expression and chronological age. However, expression levels of LINEs and LTRs positively correlated with DNA demethylation, and inflammatory and senescence gene signatures, indicative of "biological age". Gene set variation analysis showed that the inflammatory response is enriched in the samples expressing high levels of LINEs and LTRs. In summary, the study demonstrates that RTE expression correlates with "biological" rather than "chronological" aging.
Strengths:
The question the authors address is both relevant and important to the fields of aging and transposon biology.
We thank the reviewer for finding this study relevant and important.
Weaknesses:
The choice of methodology does not fully support the primary claims. Although microarrays can detect certain intergenic transposon sequences, the authors themselves acknowledge in the Discussion section that this method's resolution is limited. More critical considerations, however, should be addressed when interpreting the results. The coverage of transposon sequences by microarrays is not only very limited (232 unique probes) but also predetermined. This implies that any potential age-related overexpression of RTEs located outside of the microarray-associated regions, or of polymorphic intact transposons, may go undetected. Therefore, the authors should be more careful while generalising their conclusions.
This is a bioinformatics study, and we have already admitted and discussed the limitations in the discussion section of this manuscript. All technologies have their own limitations, and this should not stop us from shedding light on scientific facts because of inadequate information. In the manuscript, we have discussed that all large and proper ageing studies were performed using microarray technology. Peters et al. (DOI: doi: 10.1038/ncomms9570) adopted all these datasets in their transcriptional landscape of ageing manuscript, which was used in previous studies of ageing as well. Our study essentially applies the Reichmann et al. method to the peripheral blood-related data from the Peters et al. manuscript. Since hypomethylation due to ageing is a well-established and broad epigenetic reprogramming, it is unlikely that only a fraction of RTEs is affected by this phenomenon. Therefore, the subsampling of RTEs should not affect the result so much. Indeed, this is supported in our study by the inverse correlation between DNA methylation and RTE expression for LINE and SINE classes despite having limited numbers of probes for LINE and SINE expressions.
Additionally, for some analyses, the authors pool signals from RTEs by class or family, despite the fact that these groups include subfamilies and members with very different properties and harmful potentials. For example, while sequences of older subfamilies might be passively expressed through readthrough transcription, intact members of younger groups could be autonomously reactivated and cause inflammation. The aggregation of signals by the largest group may obscure the potential reactivation of smaller subgroups. I recommend grouping by subfamily or, if not possible due to the low expression scores, by subgroup. For example, all HERV subfamilies are from the ERVL family.
We agree with the reviewer that different subfamilies of RTEs play different roles through their activation. However, we will lose our statistical power if we study RTE subfamilies with a few probes. Global epigenetic alteration and derepression of RTEs by ageing have been observed to be genome-wide. While our systematic analysis across RTE classes and families cannot capture alterations in subfamilies due to statistical power, it is still relevant to the research question we are addressing.
Next, Illumina arrays might not accurately represent the true abundance of TEs due to nonspecific hybridization of genomic transposons. Standard RNA preparations always contain traces of abundant genomic SINEs unless DNA elimination is specifically thorough. The problem of such noise should be addressed.
We have checked the RNA isolation step from MESA, GTP, and GARP manuscripts. The total RNA was isolated using the Qiagen mini kit following the manufacturer’s recommendations. The authors of these manuscripts did not mention whether they eliminated genomics DNA, but we assumed they were aware of the DNA contamination and eliminated it based on the manufacturer’s recommendations. We have looked up the literature about nonspecific hybridization of RTEs but could not find any evidence to support this observation. We would appreciate the reviewers providing more evidence about such RTE contaminations.
Lastly, scRNAseq was conducted using 10x Genomics technology. However, quantifying transposons in 10x sequencing datasets presents major challenges due to sparse signals.
Applying the scTE pipeline (https://www.nature.com/articles/s41467-021-21808-x), we have found that the statical power of quantifying RTE classes (LINE, SINE, and LTR) or RTE families (L1, L2, All, ERVK, etc.) are as good as each individual gene. However, our proposed method cannot analyse RTE subfamilies, and we did not do that.
Smart-seq single-cell technology is better suited to this particular purpose.
We agree with the reviewer that Smart-seq provides higher yield than 10x, but there is no Smartseq data available for ageing study.
Anyway, it would be more convincing if the authors demonstrated TE expression across different clusters of immune cells using standard scRNAseq UMAP plots instead of boxplots.
Since the number of RTE reads per cell is low, showing the expression of RTEs per cell in UMAP may not be the best statistical approach to show the difference between the aged and young groups. This is why we chose to analyse with Pseudobulk and displayed differential expression using boxplot rather than UMAP for each immune cell type.
I recommend validating the data by RNAseq, even on small cohorts. Given that the connection between RTE overexpression and inflammation has been previously established, the authors should consider better integrating their observations into the existing knowledge.
Please see below. We have analysed RNA-seq data suggested by Reviewer 1 in the Recommendations for the Authors section.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
I can recommend two sizeable human PMBC RNA-seq datasets that the authors could use:
Marquez et al. 2020 (phs001934.v1.p1, controlled access) and Morandini et al. 2023 (GSE193141, public access). There are likely other suitable datasets that I am not aware of. I would also recommend using identical sets of probes to quantify RTE expression across studies. If certain datasets have too few probes and would thus limit the number of probes available across all studies it might be a good idea to exclude the dataset, especially if the analysis has been supplemented by the additional RNA-seq datasets.
Until recently, there was no publicly-available, non-cancerous, large cohort of RNA-seq data for ageing studies. We tried to gain access to the two RNA-seq datasets suggested by reviewer 2: Marquez et al. 2020 (phs001934.v1.p1, controlled access) and Morandini et al. 2023 (GSE193141, public access).
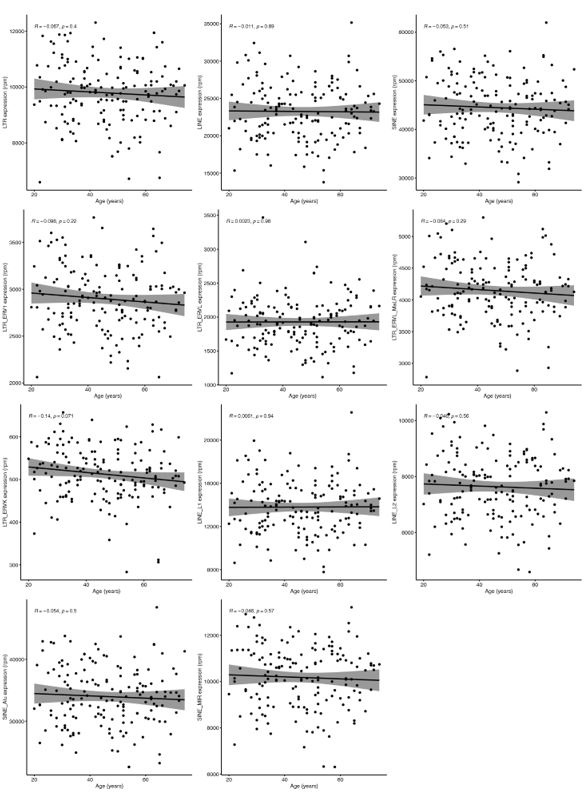
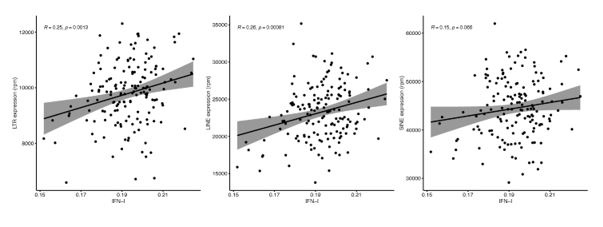
Unfortunately, Marquez et al. 2020 data is not accessible because the authors only provide the data for projects related to cardiovascular diseases. However, we did analyse Morandini et al. 2023 data, and we can confirm that no association was observed between any class and family of RTEs with chronological ageing (Author response image 1), which is the second strong piece of evidence supporting the statement in the manuscript. However, as expected, we found a positive correlation between RTE expression and IFN-I signature score (Author response image 2).
Author response image 1.
Linear analysis of RTE expression and chronological age.
Author response image 2.
Linear analysis of RTE expression and IFN gene signature expression.
The authors use "biological age" and inflammation as interchangeable concepts, including in the title. Please correct this wording.
We have now added a new terminology to the manuscript called “biological age-related (BAR)”, which has been clearly addressed this distinction. We don’t think it is needed to change the title.
The authors find correlations between RTE expression and age-associated gene signatures but not chronological age itself. This is puzzling because, as the wording suggests, the expression of these inflammatory pathways is age-associated. If RTE expression correlates with inflammation which itself correlates with age, one might expect RTE expression to also correlate with age. Do the authors see a correlation between various inflammatory gene signatures and chronological age, in the analyzed datasets? If yes, then how would you explain that discrepancy? Moreover, in this case, I would recommend using a linear model, rather than correlation, to separate the effects of chronological age and RTE expression on inflammation (Inflammation et al ~ Age + RTE expression), or equivalent designs.
As described above, we have now introduced the BAR terminology, which resolves this confusion. We did not find a correlation between RTE expression and chronological age. However, we did identify the correlation between BAR gene signatures and RTE expression.
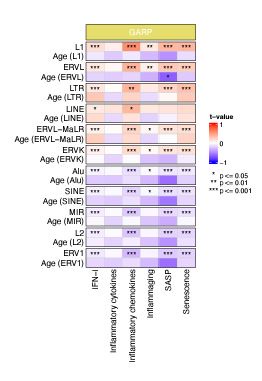
To separate the effects of chronological age and RTE expression on BAR gene signature scores, we performed a generalized linear model (GLM) analysis using BAR gene signature scores as response variables and RTE expression and chronological age as predictors (BAR gene signature scores ~ RTE expression + chronological age). Significant association was observed between BAR gene signature scores and RTE expression in the GARP cohort (Author response image 3). However, when chronological age is considered as predictor, we did not identify a correlation between chronological age and BAR gene signatures, indicating that BAR events are not corelated with chronological age (Author response image 3).
Author response image 3.
Generalized linear models (GLM) analysis (BAR gene signature scores ~ RTE expression + chronological age). For each RTE family, we separately performed GLM. Age (RTE family) indicates the chronological age when used in the design formula for that specific RTE family.
Some of the gene sets used by the authors have considerable overlap with others and are also not particularly comprehensive. I can recommend this very comprehensive gene set: https://www.gsea-msigdb.org/gsea/msigdb/human/geneset/SAUL_SEN_MAYO.
We did not choose to use large gene lists such as the suggested SEN_MAYO list, as we found Singscore struggles to generate reliable scores with sufficient variance when the number of genes increase to more than twenty. Although there is some overlap between inflammation-related genes and cellular senescence genes (e.g., IL6, IL1A, IL1B), it is important to note that each gene list focuses on different aspects of biological aging and should not be dismissed as redundant.
Minor comments:
Overall, several sentences in the manuscript feel somewhat unnatural. I would recommend further proofreading. I will mention some examples:
Thank you for your feedback. We have fixed all these issues in the new submission.
• One line 34, "like the retroviruses" should be "like retroviruses. There are several other places in the text where "the" is not required.
Fixed.
• On line 86, "to generate the RTE expression". "the" is again not necessary and I would replace "generate" with "quantify".
Fixed.
• On line 86, "we mapped the probe locations to RepeatMasker". RepeatMasker is not a genome. Do you mean you mapped the probe location to a genome annotated by RepeatMasker? The same applies to line 99.
Fixed. We changed the sentence to: “To quantify RTE expression, we mapped the microarray probe locations to RTE locations in RepeatMasker to extract the list of noncoding (intergenic or intronic) probes that cover the RTE regions.”
• Figure 1 contains a typo in the aims section: "evetns" instead of "events".
Fixed.
• On line 495 "filtered out" seems to imply your removed intergenic probes. I assume you mean that you specifically selected intergenic probes.
Fixed.
• Figure 1 nicely summarizes your datasets. Could you add a Figure 1b panel showing how you used RNA arrays to quantify RTE expression? This should include the number of probes for each RTE family, so I suggest merging this with Figure S1.
We disagree with the reviewer to merge Figure 1 and Figure S1 because they are addressing two different concepts.
Reviewer #2 (Recommendations For The Authors):
In Figure 2c, it is unclear what colour scale has been used for age.
Thank you for the comment. We have added a legend for age in this figure.
There are no figure legends for Supplementary Figures 1 to 5 and all figures after Supplementary Figure 8.
A new version with legends has been submitted.
For different datasets used, the choice of "healthy" patients should be more clear and explicit.
Are asymptomatic patients with autoimmune inflammatory disorders considered as "healthy"? If not only healthy patients' blood is analysed (such as PBMS from primary osteoarthrosis), how inflammatory signatures enrichment discovered in this study may be associated not just with "biological age" but with the disease itself?
In our analysis, we did not exclusively study "healthy" individuals, as none of our datasets were initially collected from strictly healthy populations. While the microarray datasets were not specifically collected from people with particular diseases, they were also not screened for asymptomatic conditions. To demonstrate the same pattern in healthier cohorts, we added scRNA-seq analysis of confirmed healthy individuals to our study. However, the focus of this study is not on healthy aging. Instead, it is on biological ageing that includes both healthy and non-healthy ageing.
We included the GARP (primary osteoarthritis) dataset as it is a cohort of age-related diseases (ARD). While we cannot definitively attribute inflammatory signatures enrichment to biological aging or disease, the observation of such enrichment in a cohort of ARD is worth considering. To make this clearer, we have replaced the term “healthy” with “non-cancerous” for microarray analysis throughout the paper.



