Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorCarla RothlinYale University, New Haven, United States of America
- Senior EditorCarla RothlinYale University, New Haven, United States of America
Reviewer #1 (Public Review):
Summary:
This manuscript by Aybar-Torres et al investigated the effect of common human STING1 variants on STING-mediated T cell phenotypes in mice. The authors previously made knock-in mice expressing human STING1 alleles HAQ or AQ, and here they established a new knock-in line Q293. The authors stimulated cells isolated from these mice with STING agonists and found that all three human mutant alleles resist cell death, leading to the conclusion that R293 residue is essential for STING-mediated cell death (there are several caveats with this conclusion, more below). The authors also bred HAQ and AQ alleles to the mouse Sting1-N153S SAVI mouse and observed varying levels of rescue of disease phenotypes with the AQ allele showing more complete rescue than the HAQ allele. The Q293 allele was not tested in the SAVI model. They conclude that the human common variants such as HAQ and AQ have a dominant negative effect over the gain-of-function SAVI mutants.
Strengths:
The authors and Dr. Jin's group previously made important observations of common human STING1 variants, and these knock-in mouse models are essential for understanding the physiological function of these alleles.
Weaknesses:
However, although some of the observations reported here are interesting, the data collectively does not support a unified model. The authors seem to be drawing two sets of conclusions from in vitro and in vivo experiments, and neither mechanism is clear. Several experiments need better controls, and these knock-in mice need more comprehensive functional characterization.
(1) In Figure 1, the authors are trying to show that STING agonist-induced splenocytes cell death is blocked by HAQ, AQ and Q alleles. The conclusion at line 134 should be splenocytes, not lymphocytes. Most experiments in this figure were done with mixed population that may involve cell-to-cell communication. Although TBK1-dependence is likely, a single inhibitor treatment of a mixed population is not sufficient to reach this conclusion.
(2) Q293 knock-in mouse needs to be characterized and compared to HAQ and AQ. Is this mutant expressed in tissues? Does this mutant still produce IFN and other STING activities? Does the protein expression level altered on Western blot? Is the mutant protein trafficking affected? In the authors' previous publications and some of the Western blot here, expression levels of each of these human STING1 protein in mice are drastically different. HAQ and AQ also have different effects on metabolism (pmid: 36261171), which could complicate interoperation of the T cell phenotypes.
(3) HAQ/WT and AQ/WT splenocytes are protected from STING agonist-induced cell death equally well (Figure 1G). HAQ/SAVI shows less rescue compared to AQ/SAVI. These are interesting observations, but mechanism is unclear and not clearly discussed. E.g., how does AQ protect disease pathology better than HAQ (that contains AQ)? Does Q293 allele also fully rescue SAVI?
(4) Figure 2 feels out of place. First of all, why are the authors using human explant lung tissues? PBMCs should be a better source for lymphocytes. In untreated conditions, both CD4 and B cells show ~30% dying cells, but CD8 cells show 0% dying cells. This calls for technical concerns on the CD8 T cell property or gating strategy because in the mouse experiment (Figure 1A) all primary lymphocytes show ~30% cell death at steady-state. Second, Figure 2C, these type of partial effect needs multiple human donors to confirm. Three, the reconstitution of THP1 cells seems out of place. STING-mediated cell death mechanism in myeloid and lymphoid cells are likely different. If the authors want to demonstrate cell death in myeloid cells using THP1, then these reconstituted cell lines need to be better validated. Expression, IFN signaling, etc. The parental THP1 cells is HAQ/HAQ, how does that compare to the reconstitutions? There are published studies showing THP1-STING-KO cells reconstituted with human variants do not respond to STING agonists as expected. The authors need to be scientifically rigorous on validation and caution on their interpretations.
(5) Figure 2G, H, I are confusing. AQ is more active in producing IFN signaling than HAQ and Q is the least active. How to explain this?
(6) The overall model is unclear. If HAQ, AQ and Q are loss-of-function alleles and Q is the key residue for STING-mediated cell death, then why AQ is the most active in producing IFN signaling and AQ/SAVI rescues disease most completely? If these human variants act as dominant negatives, which would be consistent with the WT/het data, then how do you explain AQ is more dominant negative than HAQ?
(7) As a general note, SAVI disease phenotypes involve multiple cell types. Lymphocyte cell death is only one of them. The authors' characterization of SAVI pathology is limited and did not analyze immunopathology of the lung.
(8) Line 281, the discussion on HIV T cell death mechanism is not relevant and over-stretching. This study did not evaluate viral infection in T cells at all. The original finding of HAQ/HAQ enrichment in HIV/AIDS was 2/11 in LTNP vs 0/11 in control, arguably not the strongest statistics.
Reviewer #2 (Public Review):
Aybar-Torres and colleagues utilize common human STING alleles to dissect the mechanism of SAVI inflammatory disease. The authors demonstrate that these common alleles alleviate SAVI pathology in mice, and perhaps more importantly use the differing functionality of these alleles to provide insight into requirements of SAVI disease induction. Their findings suggest that it is residue A230 and/or Q293 that are required for SAVI induction, while the ability to induce an interferon-dependent inflammatory response is not. This is nicely exemplified by the AQ/SAVI mice that have an intact inflammatory response to STING activation, yet minimal disease progression. As both mutants seem to be resistant STING-dependent cell death, this manuscript also alludes to the importance of STING-dependent cell death, rather than STING-dependent inflammation, in the progression of SAVI pathology. While I have some concerns, I believe this manuscript makes some important connections between STING pathology mouse models and human genetics that would contribute to the field.
Some points to consider:
(1) While the CD4+ T cell counts from HAQ/SAVI and AQ/SAVI mice suggest that these T cells are protected from STING-dependent cell death, an assay that explores this more directly would strengthen the manuscript. This is also supported by Fig 2C, but I believe a strength of this manuscript is the comparison between the two alleles. Therefore, if possible, I would recommend the isolation of T cells from these mice and direct stimulation with diABZI or other STING agonist with a cell death readout.
(2) Related to the above point - further exemplifying that the Q293 locus is essential to disease, even in human cells, would also strengthen the paper. It seems that CD4 T cell loss is a major component of human SAVI. While not completely necessary, repeating the THP1 cell death experiments from Fig 2 with a human T cell line would round out the study nicely.
(3) While I found the myeloid cell counts and BMDM data interesting, I think some more context is needed to fully loop this data into the story. Is myeloid cell expansion exemplified by SAVI patients? Do we know if myeloid cells are the major contributors to the inflammation these patients experience? Why should the SAVI community care about the Q293 locus in myeloid cells?
(4) The functional assays in Figure 4 are exciting and really connect the alleles to disease progression. To strengthen the manuscript and connect all the data, I would recommend additional readouts from these mice that address the inflammatory phenotype shown in vitro in Figure 5. For example, measuring cytokines from these mice via ELISA or perhaps even Western blots looking for NFkB or STING activation would be supportive of the story. This would also allow for some tissue specificity. I believe looking for evidence of inflammation and STING activation in the lungs of these mice, for example, would further connect the data to human SAVI pathology.
Author response:
We deeply appreciate the editors’ and reviewers’ invaluable time and effort. We would also like to extend our gratitude to eLife for its unwavering commitment to a transparent review and publication model. Below, we present our point-by-point responses to the comments.
Besides the WT allele, equivalent to the mouse TMEM173 gene, the human TMEM173 gene has two common alleles: the HAQ and AQ alleles carried by billions of people. The main conclusions and interpretation, summarized in the Title and Abstract, are (i) Different from the WT TMEM173 allele, the HAQ or AQ alleles are resistant to STING activation-induced cell death; (ii) STING residue 293 is critical for cell death; (iii) HAQ, AQ alleles are dominant to the SAVI allele; iv) One copy of the AQ allele rescues the SAVI disease in mice. We propose that STING research and STING-targeting immunotherapy should consider human TMEM173 heterogeneity. These interpretations and conclusions were based on Data and Logic. We welcome alternative, logical interpretations from our peers and potential collaborations to advance the human TMEM173 research.
Reviewer #1 (Public Review):
Responses to Comment 1: We greatly appreciate Reviewer 1's insights. We will change the “lymphocytes” to “splenocytes” (line 134) as suggested. We respectfully disagree with Reviewer 1’s comments on TBK1 (lines 129 – 134). First, we used two different TBK1 inhibitors: BX795 and GSK8612. Second, because BX795 also inhibits PDK1, we used a PDK1 inhibitor GSK2334470; Third, both BX795 and GSK8612 completely inhibited diABZI-induced splenocyte cell death (Figure 1B). The logical conclusion is “TBK1 activation is required for STING-mediated mouse spleen cell death ex vivo”. (line 118).
This manuscript uncovers a significant aspect of the interplay between the common human TMEM173 alleles and the rare SAVI mutation (lines 23-26). Our discovery that the common human TMEM173 alleles are resistant to STING activation-induced cell death is a substantial finding. It further strengthens the argument that the HAQ and AQ alleles are functionally distinct from the WT allele 1-3. We wish to underscore the crucial message of this study-that 'STING research and STING-targeting immunotherapy should consider TMEM173 heterogeneity in humans' (line 37), which has been largely overlooked in current STING clinical trials 4.
Regarding STING-Cell death, as we stated in the Introduction (lines 62-79). (i) STING-mediated cell death is cell type-dependent 5-7 and type I IFNs-independent 5,7,8. (ii) The in vivo biological significance of STING-mediated cell death is not clear 7,8. (iii) The mechanisms of STING-Cell death remain controversial. Multiple cell death pathways, i.e., apoptosis, necroptosis, pyroptosis, ferroptosis, and PANoptosis, are proposed 7,9,10. SAVI patients (WT/SAVI) and mouse models had CD4 T cellpenia 8,11. SAVI/HAQ, SAVI/AQ restored T cells in mice. Thus, the manuscript provides some answers to the biological significance of STING-cell death. Next, splenocytes from Q293/Q293 mice are resistant to STING cell death. The logical conclusion is that the amino acid 293 is critical for STING cell death. How aa293 mediates this function needs future investigation. Similarly, how TBK1 mediates STING cell death, independent of type I IFNs and NFκB induction, needs future investigation.
Responses to Comment 2: These are all very interesting questions that we will address in future studies. This manuscript, titled “The common TMEM173 HAQ, AQ alleles rescue CD4 T cellpenia, restore T-regs, and prevent SAVI (N153S) inflammatory disease in mice” does not focus on Q293 mice. We have been researching the common human TMEM173 alleles since 2011 from the discovery12 , mouse model1,3, human clinical trial2, and human genetics studies 3. This manuscript is another step towards understanding these common human TMEM173 alleles with the new discovery that HAQ, AQ are resistant to STING cell death.
Responses to Comment 3: We aim to address these worthy questions in future studies. In this manuscript, Figure 6 shows AQ/SAVI had more T-regs than HAQ/SAVI (lines 246 – 256). In our previous publication on HAQ, AQ knockin mice, we showed that AQ T-regs have more IL-10 and mitochondria activity than HAQ T-regs 3. We propose that increased IL-10+
Tregs in AQ mice may contribute to an improved phenotype in AQ/SAVI compared to
HAQ/SAVI. However, we are not excluding other contributions (e.g. metabolic difference) by the AQ allele. We will explore these possibilities in future research.
Responses to Comment 4: Figure 2 is necessary because it reveals the difference between mouse and human STING cell death. Figure 2A-2B showed that STING activation killed human CD4 T cells, but not human CD8 T cells or B cells. This observation is different from Figure 1A, where STING activation killed mouse CD4, CD8 T cells, and CD19 B cells, revealing the species-specific STING cell death responses. Regarding human CD8 T cells, as we stated in the Discussion (lines 318-320), human CD8 T cells (PBMC) are not as susceptible as the CD4 T cells to STING-induced cell death 8. We used lung lymphocytes that showed similar observations (Figure 2A). For Figure 2C, we used 2 WT/HAQ and 3 WT/WT individuals (lines 738-739). We generate HAQ, AQ THP-1 cells in STING-KO THP-1 cells (Invivogen,, cat no. thpd-kostg) (lines 740-741).
A recent study found that STING agonist SHR1032 induces cell death in STING-KO THP-1 cells expressing WT(R232) human STING 10 (line 182) independent of type I IFNs. SHR1032 suppressed THP1-STING-WT(R232) cell growth at GI50: 23 nM while in the parental THP1STING-HAQ cells, the GI50 of SHR1032 was >103 nM 10. Cytarabine was used as an internal control where SHR1032 killed more robustly than cytarabine in the THP1-STING-WT(R232) cells but much less efficiently than cytarabine in the THP-1-STING-HAQ cells 10.
This manuscript rigorously uses mouse splenocytes, human lung lymphocytes, THP-1 reconstituted with HAQ, AQ, and HAQ/SAVI, AQ/SAVI mice, to demonstrate that the common human HAQ, AQ alleles are resistant to STING cell death in vitro and in vivo.
We agree with reviewer 1 that STING-mediated cell death mechanisms in myeloid and lymphoid cells may be different and likely contribute to the different mechanisms proposed in STING cell death research 7,9,10. Our study focuses on the in vivo mechanism of T cellpenia.
Responses to Comment 5: We stated in the Introduction that “AQ responds to CDNs and produce type I IFNs in vivo and in vitro 3,13,14 ”(line 94, 95). We reported that the AQ knock in mice responded to STING activation 3. We previously showed that there was a negative natural selection on the AQ allele in individuals outside of Africa 3. 28% of Africans are WT/AQ but only 0.6% East Asians are WT/AQ 3. Future research on the AQ allele will address this interesting question that may shed new mechanistic light on STING action.
Responses to Comment 6: The comment here is similar to comment 3. In this manuscript, Figure 6 shows AQ/SAVI had more T-regs than HAQ/SAVI (lines 246 – 256). In our previous publication on HAQ, AQ knockin mice, we showed that AQ T-regs have more IL-10 and mitochondria activity than HAQ T-regs 3. We propose that increased IL-10+ Tregs in AQ mice may contribute to an improved phenotype in AQ/SAVI compared to HAQ/SAVI. However, we are not excluding other contributions (e.g. metabolic difference) by the AQ allele.
Responses to Comment 7: Both radioresistant parenchymal and/or stromal cells and hematopoietic cells influence SAVI pathology in mice 15,16. Nevertheless, the lack of CD 4 T cells, including the anti-inflammatory T-regs, likely contributes to the inflammation in SAVI mice and patients. We characterized lung function, lung inflammation (Figure 4), lung neutrophils, and inflammatory monocyte infiltration (Figure S4).
Responses to Comment 8: Several publications have linked STING to HIV pathogenesis 17-22 (line 271). The manuscript studies STING activation-induced cell death. It is not stretching to ask, for example, does preventing STING cell death, without affecting type I IFNs production, restore CD4 T cell counts and improve care for AIDS patients?
Reviewer #2 (Public Review):
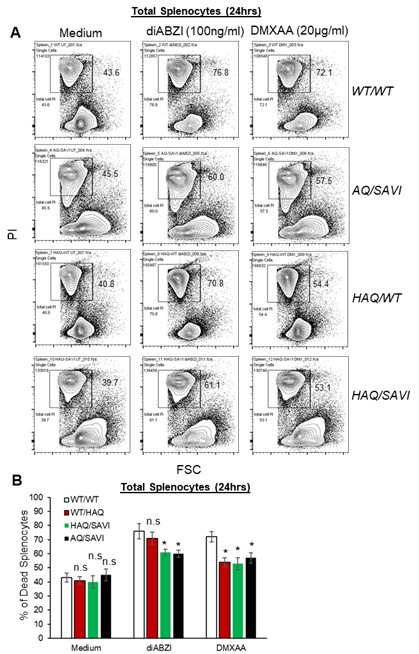
Response to Comment 1: Please see the Figure below for cell death by diABZI, DMXAA in Splenocytes from WT/WT, WT/HAQ, HAQ/SAVI, AQ/SAVI mice. The HAQ/SAVI and AQ/SAVI splenocytes showed similar partial resistance to STING activationinduced cell death.
Responses to Comment 2: We examined HAQ, AQ mouse splenocytes, HAQ human lung lymphocytes, THP-1 reconstituted with HAQ, AQ, and HAQ/SAVI, AQ/SAVI mice, to demonstrate that the common human HAQ, AQ alleles are resistant to STING cell death in vitro and in vivo. Additional human T cell line work does not add too much.
Responses to Comment 3: This is possibly a misunderstanding. We use BMDM for the purpose of comparing STING signaling (TBK1, IRF3, NFκB, STING activation) by WT/SAVI, HAQ/SAVI, AQ/SAVI. Ideally, we would like to compare STING signaling in CD4 T cells from WT/SAVI to HAQ/SAVI, AQ/SAVI mice. However, WT/SAVI has no CD4 T cells. Here, we are making the assumption that the basic STING signaling (TBK1, IRF3, NFκB, STING activation) is conserved between T cells and macrophages.
Responses to Comment 4: Reviewer 2 suggests looking for evidence of inflammation and STING activation in the lungs of HAQ/SAVI, AQ/SAVI. We would like to elaborate further. First, anti-inflammatory treatments, e.g. steroids, DMARDs, IVIG, Etanercept, rituximab, Nifedipine, amlodipine, et al., all failed in SAVI patients 11. Second, Figure S4 examined lung neutrophils and inflammatory monocyte infiltration. Interestingly, while AQ/SAVI mice had a better lung function than HAQ/SAVI mice (Figure 4D, 4E vs 4H, 4I), HAQ/SAVI and AQ/SAVI lungs had comparable neutrophils and inflammatory monocyte infiltration. Last, SAVI is classified as type I interferonopathy 11, but the lung diseases of SAVI are mainly independent of type I IFNs 23-26. The AQ allele suppresses SAVI in vivo. Understanding the mechanisms by which AQ rescues SAVI can generate curative care for SAVI patients.
Author response image 1.
(A-B). Flow cytometry of HAQ/SAVI, AQ/SAVI, WT/WT or WT/HAQ splenocytes treated with diABZI (100ng/ml) or DMXAA (20µg/ml) for 24hrs. Cell death was determined by PI staining. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values are determined by one-way ANOVA Tukey’s multiple comparison test. * p<0.05. n.s: not significant.
References.
(1) Patel, S. et al. The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele. J Immunol 198, 776-787 (2017).
(2) Sebastian, M. et al. Obesity and STING1 genotype associate with 23-valent pneumococcal vaccination efficacy. JCI Insight 5 (2020).
(3) Mansouri, S. et al. MPYS Modulates Fatty Acid Metabolism and Immune Tolerance at Homeostasis Independent of Type I IFNs. J Immunol 209, 2114-2132 (2022).
(4) Sivick, K. E. et al. Comment on "The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele". J Immunol 198, 4183-4185 (2017).
(5) Gulen, M. F. et al. Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427 (2017).
(6) Kabelitz, D. et al. Signal strength of STING activation determines cytokine plasticity and cell death in human monocytes. Sci Rep 12, 17827 (2022).
(7) Murthy, A. M. V., Robinson, N. & Kumar, S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ 27, 2989-3003 (2020).
(8) Kuhl, N. et al. STING agonism turns human T cells into interferon-producing cells but impedes their functionality. EMBO Rep 24, e55536 (2023).
(9) Li, C., Liu, J., Hou, W., Kang, R. & Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol 9, 698679 (2021).
(10) Song, C. et al. SHR1032, a novel STING agonist, stimulates anti-tumor immunity and directly induces AML apoptosis. Sci Rep 12, 8579 (2022).
(11) Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371, 507-518 (2014).
(12) Jin, L. et al. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun 12, 263-269 (2011).
(13) Yi, G. et al. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One 8, e77846 (2013).
(14) Patel, S. et al. Response to Comment on "The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele". J Immunol 198, 4185-4188 (2017).
(15) Gao, K. M. et al. Endothelial cell expression of a STING gain-of-function mutation initiates pulmonary lymphocytic infiltration. Cell Rep 43, 114114 (2024).
(16) Gao, K. M., Motwani, M., Tedder, T., Marshak-Rothstein, A. & Fitzgerald, K. A. Radioresistant cells initiate lymphocyte-dependent lung inflammation and IFNgammadependent mortality in STING gain-of-function mice. Proc Natl Acad Sci U S A 119, e2202327119 (2022).
(17) Monroe, K. M. et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428-432 (2014).
(18) Doitsh, G. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509-514 (2014).
(19) Jakobsen, M. R., Olagnier, D. & Hiscott, J. Innate immune sensing of HIV-1 infection. Curr Opin HIV AIDS 10, 96-102 (2015).
(20) Silvin, A. & Manel, N. Innate immune sensing of HIV infection. Curr Opin Immunol 32, 54-60 (2015).
(21) Altfeld, M. & Gale, M., Jr. Innate immunity against HIV-1 infection. Nat Immunol 16, 554-562 (2015).
(22) Krapp, C., Jonsson, K. & Jakobsen, M. R. STING dependent sensing - Does HIV actually care? Cytokine Growth Factor Rev 40, 68-76 (2018).
(23) Luksch, H. et al. STING-associated lung disease in mice relies on T cells but not type I interferon. J Allergy Clin Immunol 144, 254-266 e258 (2019).
(24) Stinson, W. A. et al. The IFN-gamma receptor promotes immune dysregulation and disease in STING gain-of-function mice. JCI Insight 7 (2022).
(25) Warner, J. D. et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med 214, 3279-3292 (2017).
(26) Fremond, M. L. et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract 9, 803-818 e811 (2021).



