Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorYi Arial ZengChinese Academy of Sciences, Shanghai, China
- Senior EditorJonathan CooperFred Hutch Cancer Center, Seattle, United States of America
Reviewer #1 (Public review):
In their paper, Kang et al. investigate rigidity sensing in amoeboid cells, showing that, despite their lack of proper focal adhesions, amoeboid migration of single cells is impacted by substrate rigidity. In fact, many different amoeboid cell types can durotax, meaning that they preferentially move towards the stiffer side of a rigidity gradient.
The authors observed that NMIIA is required for durotaxis and, buiding on this observation, they generated a model to explain how durotaxis could be achieved in the absence of strong adhesions. According to the model, substrate stiffness alters the diffusion rate of NMAII, with softer substrates allowing for faster diffusion. This allows for NMAII accumulation at the back, which, in turn, results in durotaxis.
The authors responded to all my comments and I have nothing to add. The evidence provided for durotaxis of non adherent (or low-adhering) cells is strong. I am particularly impressed by the fact that amoeboid cells can durotax even when not confined. I wish to congratulate the authors for the excellent work, which will fuel discussion in the field of cell adhesion and migration.
Reviewer #2 (Public review):
Summary:
The authors developed an imaging-based device, that provides both spatial confinement and stiffness gradient, to investigate if and how amoeboid cells, including T cells, neutrophils and Dictyostelium can durotax. Furthermore, the authors showed that the mechanism for the directional migration of T cells and neutrophils depends on non-muscle myosin IIA (NMIIA) polarized towards the soft-matrix-side. Finally, they developed a mathematical model of an active gel that captures the behavior of the cells described in vitro.
Strengths:
The topic is intriguing as durotaxis is essentially thought to be a direct consequence of mechanosensing at focal adhesions. To the best of my knowledge, this is the first report on amoeboid cells that are not dependent on FAs to exert durotaxis. The authors developed an imaging-based durotaxis device that provides both spatial confinement and stiffness gradient and they also utilized several techniques such as quantitative fluorescent speckle microscopy and expansion microscopy. The results of this study have well-designed control experiments and are therefore convincing.
Weaknesses:
Overall this study is well performed but there are still some minor issues I recommend the authors address:
(1) When using NMIIA/NMIIB knockdown cell lines to distinguish the role of NMIIA and NMIIB in amoeboid durotaxis, it would be better if the authors take compensatory effects into account.
(2) The expansion microscopy assay is not clearly described and some details are missed such as how the assay is performed on cells under confinement.
(3) In this study, an active gel model was employed to capture experimental observations. Previously, some active nematic models were also considered to describe cell migration, which is controlled by filament contraction. I suggest the authors provide a short discussion on the comparison between the present theory and those prior models.
(4) In the present model, actin flow contributes to cell migration while myosin distribution determines cell polarity. How does this model couple actin and myosin together?
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In their paper, Kang et al. investigate rigidity sensing in amoeboid cells, showing that, despite their lack of proper focal adhesions, amoeboid migration of single cells is impacted by substrate rigidity. In fact, many different amoeboid cell types can durotax, meaning that they preferentially move towards the stiffer side of a rigidity gradient.
The authors observed that NMIIA is required for durotaxis and, building on this observation, they generated a model to explain how durotaxis could be achieved in the absence of strong adhesions. According to the model, substrate stiffness alters the diffusion rate of NMAII, with softer substrates allowing for faster diffusion. This allows for NMAII accumulation at the back, which, in turn, results in durotaxis.
The experiments support the main message of the paper regarding durotaxis by amoeboid cells. In my opinion, a few clarifications on the mechanism proposed to explain this phenomenon could strengthen this research:
(1) According to your model, the rear end of the cell, which is in contact with softer substrates, will have slower diffusion rates of MNIIA. Does this mean that bigger cells will durotax better than smaller cells because the stiffness difference between front and rear is higher? Is it conceivable to attenuate the slope of the durotactic gradient to a degree where smaller cells lose their ability to durotact, while longer cells retain their capacity for directional movement?
We thank the reviewer for this comment. In fact, it is not always the case that bigger cells will durotax better than smaller cells. Although bigger cells will sense higher stiffness difference between the front and rear, cells placed on different regions of underlying substrates may respond differently. This is because diffusion coefficient difference is not proportional to stiffness difference in our theoretical model. Therefore, when cells are placed on a very stiff substrate, cells may not durotax. When cells are placed on a region with suitable stiffness, where cells are sensitive to stiffness gradient, bigger cells will durotax better than smaller cells. In this situation, as you mentioned, lowering the stiffness gradient will make smaller cells become adurotactic while longer cells still durotax.
We tried to further address this question by our durotaxis assay but there was a challenge: the amoeboid cells we use, including CD4+ Naïve T cells, neutrophils, dHL-60 cells and Dictysotelium, frequently protrude, retract and alter contact area with the substrate which make it difficult for us to distinguish between bigger and smaller cells in a particular cell type. Previously reported durotactic cell lines, such as MDA-MB-231 and HT1080 cells, are bigger than the amoeboid cells we use but they are mesenchymal cells and adopt distinct mechanisms which always involve stable focal adhesions. Due to this, although we are eager to answer this question by experiments and that the stiffness gradient is tunable in our system, we have not found an appropriate approach and experimental setup.
(2) Where did you place the threshold for soft, middle, and stiff regions (Figure 6)? Is it possible that you only have a linear rigidity gradient in the center of your gel and the more you approach the borders, the flatter the gradient gets? In this case, cells would migrate randomly on uniform substrates. Did you perform AFM over the whole length of the gel or just in the central part?
We thank the reviewer for this comment. We have performed AFM over the whole length of our gradient gel (Fig. S1A). We divide the gel into three equal parts (stiff: 1-4 mm; middle: 4-7 mm; soft: 7-10 mm) and the stiffness gradient is almost linear within each part as shown in Fig. S1A.
(3) In which region (soft, middle, stiff) did you perform all the cell tracking of the previous figures?
We thank the reviewer for this question. We performed the cell tracking in the soft region of the gradient gel.
(4) What is the level of confinement experienced by the cells? Is it possible that cells on the soft side of the gels experience less confinement due to a "spring effect" whereby the coverslips descending onto the cells might exert diminished pressure because the soft hydrogels act as buffers, akin to springs? If this were the case, cells could migrate following a confinement gradient.
We thank the reviewer for this comment. Although the possibility that our thin hydrogel layers act as buffers cannot be completely excluded, we have performed the durotaxis assay without upper gradient gel providing confinement (Author response image 1A). In this case, CD4+ Naïve T cells, neutrophils, dHL-60 cells and Dictysotelium can still durotax (Author response image 1B-E), indicating stiffness gradient itself is sufficient to direct amoeboid cell migration.
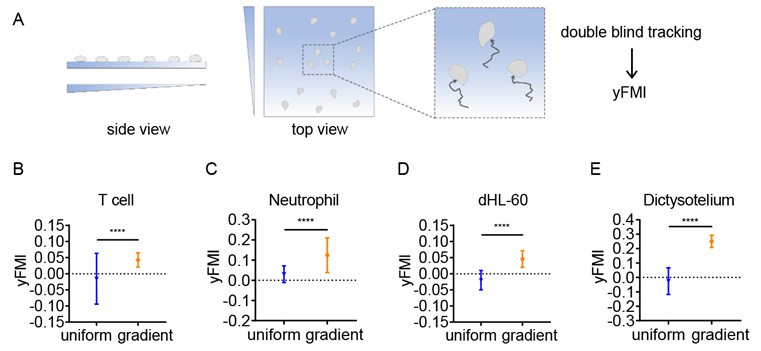
Author response image 1.
Illustration of the durotaxis system without confinement (A) and y-FMI of CD4+ Naïve T cells (B), neutrophils (C), dHL-60 cells (D) and Dictysotelium (E) cultured on uniform substrate or gradient substrate (n ≥ 30 tracks were analyzed for each experiment, N = 3 independent experiments for each condition, replicates are biological). All error bars are SEM. ****, P < 0.0001, by Student’s t-test.
Reviewer #2 (Public Review):
Summary:
The authors developed an imaging-based device that provides both spatialconfinement and stiffness gradient to investigate if and how amoeboid cells, including T cells, neutrophils, and Dictyostelium, can durotax. Furthermore, the authors showed that the mechanism for the directional migration of T cells and neutrophils depends on non-muscle myosin IIA (NMIIA) polarized towards the soft-matrix-side. Finally, they developed a mathematical model of an active gel that captures the behavior of the cells described in vitro.
Strengths:
The topic is intriguing as durotaxis is essentially thought to be a direct consequence of mechanosensing at focal adhesions. To the best of my knowledge, this is the first report on amoeboid cells that do not depend on FAs to exert durotaxis. The authors developed an imaging-based durotaxis device that provides both spatial confinement and stiffness gradient and they also utilized several techniques such as quantitative fluorescent speckle microscopy and expansion microscopy. The results of this study have well-designed control experiments and are therefore convincing.
Weaknesses:
Overall this study is well performed but there are still some minor issues I recommend the authors address:
(1) When using NMIIA/NMIIB knockdown cell lines to distinguish the role of NMIIA and NMIIB in amoeboid durotaxis, it would be better if the authors took compensatory effects into account.
We thank the reviewer for this suggestion. We have investigated the compensation of myosin in NMIIA and NMIIB KD HL-60 cells using Western blot and added this result in our updated manuscript (Fig. S4B, C). The results showed that the level of NMIIB protein in NMIIA KD cells doubled while there was no compensatory upregulation of NMIIA in NMIIB KD cells. This is consistent with our conclusion that NMIIA rather than NMIIB is responsible for amoeboid durotaxis since in NMIIA KD cells, compensatory upregulation of NMIIB did not rescue the durotaxis-deficient phenotype.
(2) The expansion microscopy assay is not clearly described and some details are missed such as how the assay is performed on cells under confinement.
We thank the reviewer for this comment. We have updated details of the expansion microscopy assay in our revised manuscript in line 481-485 including how the assay is performed on cells under confinement:
Briefly, CD4+ Naïve T cells were seeded on a gradient PA gel with another upper gel providing confinement. 4% PFA was used to fix cells for 15 min at room temperature. After fixation, the upper gradient PA gel is carefully removed and the bottom gradient PA gel with seeded cells were immersed in an anchoring solution containing 1% acrylamide and 0.7% formaldehyde (Sigma, F8775) for 5 h at 37 °C.
(3) In this study, an active gel model was employed to capture experimental observations. Previously, some active nematic models were also considered to describe cell migration, which is controlled by filament contraction. I suggest the authors provide a short discussion on the comparison between the present theory and those prior models.
We thank the reviewer for this suggestion. Active nematic models have been employed to recapitulate many phenomena during cell migration (Nat Commun., 2018, doi: 10.1038/s41467-018-05666-8.). The active nematic model describes the motion of cells using the orientation field, Q, and the velocity field, u. The director field n with (n = −n) is employed to represent the nematic state, which has head-tail symmetry. However, in our experiments, actin filaments are obviously polarized, which polymerize and flow towards the direction of cell migration. Therefore, we choose active gel model which describes polarized actin field during cell migration. In the discussion part, we have provided the comparison between active gel model and motor-clutch model. We have also supplemented a short discussion between the present model and active nematic model in the main text of line 345-347:
The active nematic model employs active extensile or contractile agents to push or pull the fluid along their elongation axis to simulate cells flowing (61).
(4) In the present model, actin flow contributes to cell migration while myosin distribution determines cell polarity. How does this model couple actin and myosin together?
We thank the reviewer for this question. In our model, the polarization field P(r,t) is employed to couple actin and myosin together. It is obvious that actin accumulate at the front while myosin diffuses in the opposite direction. Therefore, we propose that actin and myosin flow towards the opposite direction, which is captured in the convection term of actin (∇[c(v+wP)]) and myosin (∇[m(-wP)]) density field.
Reviewing Editor (Recommendations For The Authors):
We suggest that you cite the publication about confinement force microscopy from the Betz lab (https://doi.org/10.1101/2023.08.22.554088).
We thank the editor for this suggestion. We have cited this publication in line 89 in our updated manuscript.
Reviewer #1 (Recommendations For The Authors):
Minor points and text corrections:
- In line 288 you state that NMIIA basal diffusion rate is larger on softer substrates, while in line 315 you say that NMIIA is more diffusive on stiff. The two sentences seem to contradict each other.
We thank the reviewer for pointing out this mistake. In our active gel model, the basal diffusion rate of NMIIA is larger on stiffer substrate. We have corrected this mistake in line 288 (line 283 in the updated manuscript) in our revised manuscript.
- How were the non-muscle myosin images (Figure 3F) collected?
We thank the reviewer for this question. The non-muscle myosin images in Fig. 3F are single planes collected by epifluorescence-confocal microscopy. We have updated the related method in our revised manuscript in line 477-478:
After mounting medium is solidified, single plane images were captured using a 63×1.4 NA objective lens on Andor Dragonfly epi-fluorescence confocal imaging system.
- Is there a quantification of NMAII accumulation at the back?
We thank the reviewer for this question. We have a quantification of NMIIA distribution in Fig. 3G. We measured the fluorescence intensity of NMIIA and NMIIB in the soft and stiff region of cells and found that the soft/stiff fluorescence ratio of NMIIB is about 0.95 and the ratio of NMIIA is about 1.82, indicating NMIIA tend to be localized at back while NMIIB is evenly distributed in the soft and stiff region of cells.
- At which frequency were images acquired for Fluorescent Speckle Microscopy? Overall, I think it would help to state the length and frequency of videos in the legends.
We thank the reviewer for this comment. We have updated the length (10 min for movie 6-10 and 80 sec for movie11) and frequency (15 sec intervals for movie 6-10 and 2 sec intervals for movie11) of Fluorescent Speckle Microscopy videos in our revised manuscript.
Reviewer #2 (Recommendations For The Authors):
The cell contour of Figure S5C is not very clear.
We thank the reviewer for this comment. We have marked the outline of the cell in Fig. S5C in our updated manuscript.



