Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorRandy StockbridgeUniversity of Michigan, Ann Arbor, United States of America
- Senior EditorKenton SwartzNational Institute of Neurological Disorders and Stroke, Bethesda, United States of America
Reviewer #1 (Public review):
Summary:
This study resolves a cryo-EM structure of the GPCR, GPR30, in the presence of bicarbonate, which the author's lab recently identified as the physiological ligand. Understanding the ligand and the mechanism of activation is of fundamental importance to the field of receptor signaling. This solid study provides important insight into the overall structure and suggests a possible bicarbonate binding site.
Strengths:
The overall structure, and proposed mechanism of G-protein coupling are solid. Based on the structure, the authors identify a binding pocket that might accommodate bicarbonate. Although assignment of the binding pocket is speculative, extensive mutagenesis of residues in this pocket identifies several that are important to G-protein signaling. The structure shows some conformational differences with a previous structure of this protein determined in the absence of bicarbonate (PMC11217264). To my knowledge, bicarbonate is the only physiological ligand that has been identified for GPR30, making this study an important contribution to the field. However, the current study provides novel and important circumstantial evidence for the bicarbonate binding site based on mutagenesis and functional assays.
Weaknesses:
Bicarbonate is a challenging ligand for structural and biochemical studies, and because of experimental limitations, this study does not elucidate the exact binding site. Higher resolution structures would be required for structural identification of bicarbonate. The functional assay monitors activation of GPR30, and thus reports on not only bicarbonate binding, but also the integrity of the allosteric network that transduces the binding signal across the membrane. However, biochemical binding assays are challenging because the binding constant is weak, in the mM range.
The authors appropriately acknowledge the limitations of these experimental approaches, and they build a solid circumstantial case for the bicarbonate binding pocket based on extensive mutagenesis and functional analysis. However, the study does fall short of establishing the bicarbonate binding site.
Reviewer #2 (Public review):
Summary:
In this manuscript, "Cryo-EM structure of the bicarbonate receptor GPR30," the authors aimed to enrich our understanding of the role of GPR30 in pH homeostasis by combining structural analysis with a receptor function assay. This work is a natural development and extension of their previous work on Nature Communications (PMID: 38413581). In the current body of work, they solved the cryo-EM structure of the human GPR30-G-protein (mini-Gsqi) complex in the presence of bicarbonate ions at 3.15 Å resolution. From the atomic model built based on this map, they observed the overall canonical architecture of class A GPCR and also identified 3 extracellular pockets created by ECLs (Pockets A-C). Based on the polarity, location, size, and charge of each pocket, the authors hypothesized that pocket A is a good candidate for the bicarbonate binding site. To identify the bicarbonate binding site, the authors performed an exhaustive mutant analysis of the hydrophilic residues in Pocket A and analyzed receptor reactivity via calcium assay. In addition, the human GPR30-G-protein complex model also enabled the authors to elucidate the G-protein coupling mechanism of this special class A GPCR, which plays a crucial role in pH homeostasis.
Strengths:
As a continuation of their recent Nature Communications publication, the authors used cryo-EM coupled with mutagenesis and functional studies to elucidate bicarbonate-GPR30 interaction. This work provided atomic-resolution structural observations for the receptor in complex with G-protein, allowing us to explore its mechanism of action, and will further facilitate drug development targeting GPR30. There were 3 extracellular pockets created by ECLs (Pockets A-C). The authors were able to filter out 2 of them and hypothesized that pocket A was a good candidate for the bicarbonate binding site based on the polarity, location, and charge of each pocket. From there, the authors identified the key residues on GPR30 for its interaction with the substrate, bicarbonate. Together with their previous work, they mapped out amino acids that are critical for receptor reactivity.
Weaknesses:
When we see a reduction of a GPCR-mediated downstream signaling, several factors could potentially contribute to this observation: 1) a reduced total expression of this receptor due to the mutation (transcription and translation issue); 2) a reduced surface expression of this receptor due to the mutation (trafficking issue); and 3) a dysfunctional receptor that doesn't signal due to the mutation. In the current revision, based on the gating strategy, the surface expression of the HA-positive WT GPR30-expressing cells is only 10.6% of the total population, while the surface expression levels of the mutants range from 1.89% (P71A) to 64.4% (D111A). Combining this information with the functional readout in Figure 3F and G, as well as their previous work, the authors concluded that mutations at P71, E115, D125, Q138, C207, D210, and H307 would decrease bicarbonate responses. Among those sites,
E115, Q138, and H307 were from their previous Nature Comm paper.
Authors claim P71 and C207 make a structural-stability contribution, as their mutations result in a significant reduction in surface expression: P71A (1.89%) and C207A (2.71%). However, compared to 10.6% of the total population in the WT, (P71A is 17.8% of the WT, and C207A is 25.6% of the WT), this doesn't rule out the possibility that the mutated receptor is also dysfunctional: at 10 mM NaHCO3, RFU of WT is ~500, RFU of P71 and C207 are ~0.
The authors also interpret "The D125ECL1A mutant has lost its activity but is located on the surface" and only mention "D125 is unlikely to be a bicarbonate binding site, and the mutational effect could be explained due to the decreased surface expression". Again, compared to 10.6% of the total population in the WT, D125A (3.94%) is 37.2% of the WT. At 10 mM NaHCO3, the RFU of the WT is ~500, the RFU of D125 is ~0. This doesn't rule out the possibility that the mutated receptor is also dysfunctional. It is not clear why D125A didn't make it to the surface.
Other mutants that the authors didn't mention much in their text: D111A (64.4%, 607.5% of WT surface expression), E121A (50.4%, 475.5% of WT surface expression), R122 (41.0%, 386.8% of WT surface expression), N276A (38.9%, 367.0% of WT surface expression) and E218A (24.6%, 232.1% of WT surface expression) all have similar RFU as WT, although the surface expression is about 2-6 times more. On the other hand, Q215A (3.18%, 30% of WT surface expression) has similar RFU as WT, with only a third of the receptor on the surface.
Altogether, the wide range of surface expression across the different cell lines, combined with the different receptor function readouts, makes the cell functional data only partially support their structural observations.
Reviewer #3 (Public review):
Summary
GPR30 responds to bicarbonate and plays a role in regulating cellular pH and ion homeostasis. However, the molecular basis of bicarbonate recognition by GPR30 remains unresolved. This study reports the cryo-EM structure of GPR30 bound to a chimeric mini-Gq in the presence of bicarbonate, revealing mechanistic insights into its G-protein coupling. Nonetheless, the study does not identify the bicarbonate-binding site within GPR30.
Strengths
The work provides strong structural evidence clarifying how GPR30 engages and couples with Gq.
Weaknesses
Several GPR30 mutants exhibited diminished responses to bicarbonate, but their expression levels were also reduced. As a result, the mechanism by which GPR30 recognizes bicarbonate remains uncertain, leaving this aspect of the study incomplete.
Author response:
The following is the authors’ response to the original reviews.
The parts of the text that have been changed.The major changes are as follows:
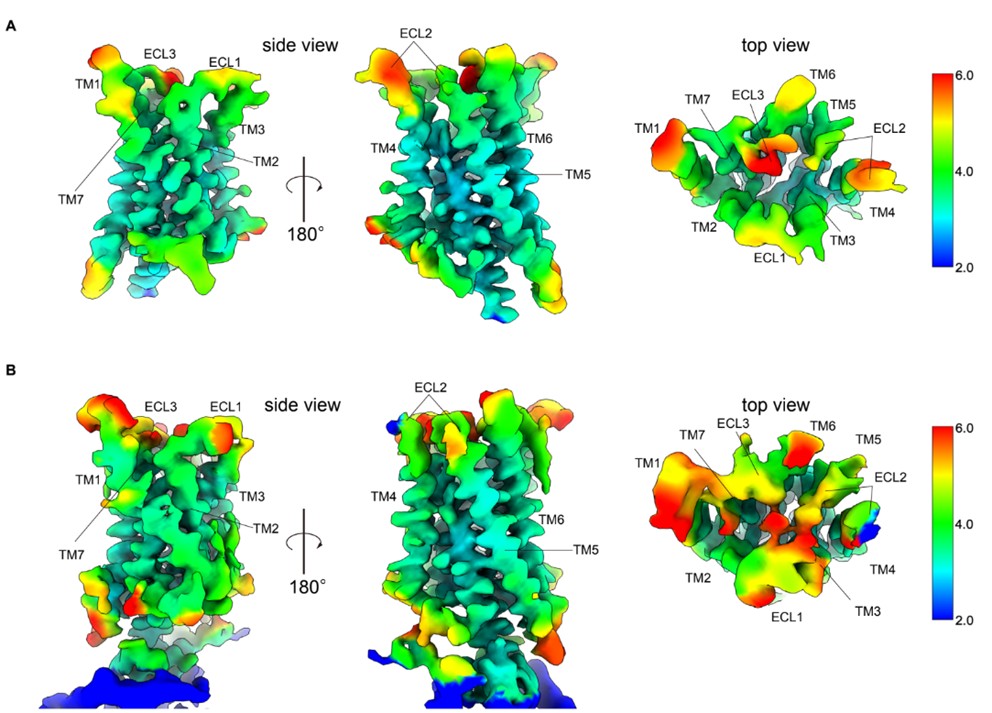
We re-analyzed the dataset and improved the local resolution of the extracellular region (Author response image 1).
We re-modeled based on the improved density and canceled the bicarbonate model based on comments from all reviewers.
We performed calcium assay using cell lines stably expressing the mutants, whose surface expression levels were analyzed by fluorescence-activated cell sorting (FACS)
(Figure 3F, G and Figure 3–figure supplement 1-3).
Thus, we significantly revised our discussion of the extracellular binding pocket and the result of the mutational study. In the revised manuscript, we speculate that H307 is a candidate for the bicarbonate binding site.
Author response image 1.
Figure Comparison of local resolution between re-analyzed and previous maps.A Side and top view of the re-analyzed receptor-focused map of GPR30 colored by local resolution. B Side and top view of the previous receptor-focused map of GPR30 colored by local resolution
Reviewer #1 (Public Review):
Summary:
This study resolves a cryo-EM structure of the GPCR, GPR30, which was recently identified as a bicarbonate receptor by the authors' lab. Understanding the ligand and the mechanism of activation is of fundamental importance to the field of receptor signaling. However, the main claim of the paper, the identification of the bicarbonate binding site, is only partly supported by the structural and functional data, leaving the study incomplete.
Strengths:
The overall structure, and proposed mechanism of G-protein coupling seem solid. The authors perform fairly extensive unbiased mutagenesis to identify a host of positions that are important to G-protein signaling. To my knowledge, bicarbonate is the only physiological ligand that has been identified for GPR30, making this study a particularly important contribution to the field.
Weaknesses:
Without higher resolution structures and/or additional experimental assessment of the binding pocket, the assignment of the bicarbonate remains highly speculative. The local resolution is especially poor in the ECL loop region where the ligand is proposed to bind (4.3 - 4 .8 Å range). Of course, sometimes it is difficult to achieve high structural resolution, but in these cases, the assignment of ligands should be backed up by even more rigorous experimental validation.The functional assay monitors activation of GPR30, and thus reports on not only bicarbonate binding, but also the integrity of the allosteric network that transduces the binding signal across the membrane. Thus, disruption of bicarbonate signaling by mutagenesis of the putative coordinating residues does not necessarily mean that bicarbonate binding has been disrupted. Moreover, the mutagenesis was apparently done prior to structure determination, meaning that residues proposed to directly surround bicarbonate binding, such as E218, were not experimentally validated. Targeted mutagenesis based on the structure would strengthen the story.
Moreover, the proposed bicarbonate binding site is surprising in a chemical sense, as it is located within an acidic pocket. The authors cite several other structural studies to support the surprising observation of anionic bicarbonate surrounded by glutamate residues in an acidic pocket (references 31-34). However, it should be noted that in general, these other structures also possess a metal ion (sodium or calcium) and/or a basic sidechain (arginine or lysine) in the coordination sphere, forming a tight ion pair. Thus, the assigned bicarbonate binding site in GPR30 remains an anomaly in terms of the chemical properties of the proposed binding site.
Thank you for your insightful comments. Based on the weaknesses you pointed out, we reconstructed the receptor based on the improved density and removed the bicarbonate model. We performed calcium assays using cell lines stably expressing the variant based on the structure.
Reviewer #2(Public Review):
Summary:
In this manuscript, "Cryo-EM structure of the bicarbonate receptor GPR30," the authors aimed to enrich our understanding of the role of GPR30 in pH homeostasis by combining structural analysis with a receptor function assay. This work is a natural development and extension of their previous work (PMID: 38413581). In the current body of work, they solved the first cryo-EM structure of the human GPR30-G-protein (mini-Gsqi) complex in the presence of bicarbonate ions at 3.21 Å resolution. From the atomic model built based on this map, they observed the overall canonical architecture of class A GPCR and also identified 4 extracellular pockets created by extracellular loops (ECLs) (Pockets A-D). Based on the polarity, location, and charge of each pocket, the authors hypothesized that pocket D is a good candidate for the bicarbonate binding site. To verify their structural observation, on top of the 10 mutations they generated in the previous work, the authors introduced another 11 mutations to map out the essential residues for the bicarbonate response on hGPR30. In addition, the human GPR30-G-protein complex model also allowed the authors to untangle the G-protein coupling mechanism of this special class A GPCR that plays an important role in pH homeostasis.
Strengths:
As a continuation of their recent Nature Communication publication (PMID: 38413581), this study was carefully designed, and the authors used mutagenesis and functional studies to confirm their structural observations. This work provided high-resolution structural observations for the receptor in complex with G-protein, allowing us to explore its mechanism of action, and will further facilitate drug development targeting GPR30. There were 4 extracellular pockets created by ECLs (Pockets A-D). The authors were able to filter out 3 of them and identified that pocket D was a good candidate for the bicarbonate binding site based on the polarity, location, and charge of each pocket. From there, the authors identified the key residues on GPR30 for its interaction with the substrate, bicarbonate. Together with their previous work, they carefully mapped out nine amino acids that are critical for receptor reactivity.
Weaknesses:
It is unclear how novel the aspects presented in the new paper are compared to the most recent Nature Communications publication (PMID: 38413581). Some areas of the manuscript appear to be mixed with the previous publication. The work is still impactful to the field. The new and novel aspects of this manuscript could be better highlighted.
I also have some concerns about the TGFα shedding assay the authors used to verify their structural observation. I understand that this assay was also used in the authors' previous work published in Nature Communications. However, there are still several things in the current data that raised concerns:
Thank you for your insightful comments. Based on the weaknesses you pointed out, we highlighted the new and novel aspects of this manuscript could be better highlighted.l. We performed calcium assays using cell lines stably expressing the variant based on the structure.
(1) The authors confirmed the "similar expression levels of HA-tagged hGPR30" mutants by WB in Supplemental Figure 1A and B. However, compared to the hGPR30-HA (~6.5 when normalized to the housekeeping gene, Na-K-ATPase), several mutants of the key amino acids had much lower surface expression: S134A, D210A, C207A had ~50% reduction, D125A had ~30% reduction, and Q215A and P71A had ~20% reduction. This weakens the receptor reactivity measured by the TGFα shedding assay.
Since the calcium assay data is included in the main figure, the TGFα shedding assay and WB expression quantification data are Figure 3. –– supplement figure 1-4, but we included an explanation of the expression levels in the figure caption.
(2) In the previous work, the authors demonstrated that hGPR30 signals through the Gq signaling pathway and can trigger calcium mobilization. Given that calcium mobilization is a more direct measurement for the downstream signaling of hGPR30 than the TGFα shedding assay, pairing the mutagenesis study with the calcium assay will be a better functional validation to confirm the disruption of bicarbonate signaling.
According to the suggestion, we performed calcium assay using cell lines stably expressing the mutants (Figure 3F, G and Figure 3–figure supplement 1-3).
(3) It was quite confusing for Figure 4B that all statistical analyses were done by comparing to the mock group. It would be clearer to compare the activity of the mutants to the wild-type cell line.
Thank you for your comment. As you mentioned, the comparisons are made between wild-type GPR30 and mutants in the revised manuscript (Figure 3G, Figure 3.—figure supplement 4B)
Additional concerns about the structural data include
(1) E218 was in close contact with bicarbonate in Figure 4D. However, there is no functional validation for this observation. Including the mutagenesis study of this site in the cell-based functional assay will strengthen this structural observation.
We cancelled the bicarbonate model, and we performed mutation analysis targeting all residues facing the binding pocket using cell lines that stably express variants including E218A.
(2) For the flow chart of the cryo-EM data processing in Supplemental data 2, the authors started with 10,148,422 particles after template picking, then had 441,348 Particles left after 2D classification/heterogenous refinement, and finally ended with 148,600 particles for the local refinement for the final map. There seems to be a lot of heterogeneity in this purified sample. GPCRs usually have flexible and dynamic loop regions, which explains the poor resolution of the ECLs in this case. Thus, a solid cell-based functional validation is a must to assign the bicarbonate binding pocket to support their hypothesis.
We re-analyzed the dataset and improved the local resolution of the extracellular region (Author response image 1) and cancelled the bicarbonate model. Yet, as suggested by the reviewer, solid cell-based functional validation is efficient to analyze the receptor function response to bicarbonate. Thus, we performed mutation analysis targeting all residues facing the binding pocket using cell lines stably expressing the mutants, whose surface expression levels were analyzed by FACS (Figure 3F, G and Figure 3.––figure supplement 1-3).
Reviewer #3 (Public Review):
Summary:
GPR30 responds to bicarbonate and regulates cellular responses to pH and ion homeostasis. However, it remains unclear how GPR30 recognizes bicarbonate ions. This paper presents the cryo-EM structure of GPR30 bound to a chimeric mini-Gq in the presence of bicarbonate. The structure together with functional studies aims to provide mechanistic insights into bicarbonate recognition and G protein coupling.
Strengths:
The authors performed comprehensive mutagenesis studies to map the possible binding site of bicarbonate.
Weaknesses:
Owing to the poor resolution of the structure, some structural findings may be overclaimed.
Based on EM maps shown in Figure 1a and Figure Supplement 2, densities for side chains in the receptor particularly in ECLs (around 4 Å) are poorly defined. At this resolution, it is unlikely to observe a disulfide bond (C130ECL1-C207ECl2) and bicarbonate ions. Moreover, the disulfide between ECL1 and ECL2 has not been observed in other GPCRs and the published structure of GPR30 (PMID: 38744981). The density of this disulfide bond could be noise.
The authors observed a weak density in pocket D, which is accounted for by the bicarbonate ions. This ion is mainly coordinated by Q215 and Q138. However, the Q215A mutation only reduced but not completely abolished bicarbonate response, and the author did not present the data of Q138A mutation. Therefore, Q215 and Q138 could not be bicarbonate binding sites. While H307A completely abolished bicarbonate response, the authors proposed that this residue plays a structural role. Nevertheless, based on the structure, H307 is exposed and may be involved in binding bicarbonate. The assignment of bicarbonate in the structure is not supported by the data.
Thank you for your insightful comments. Based on the weaknesses you pointed out, we reconstructed the receptor based on the improved density and removed the bicarbonate model. We performed calcium assays using cell lines stably expressing the variant based on the structure.
Reviewer #1 (Recommendations For The Authors):
(1) The experimental validation of the bicarbonate binding could be strengthened by developing an assay that directly monitors bicarbonate binding (rather than GPCR signaling)
We agree that a direct binding assay for bicarbonate would be highly attractive (i.e. Filter binding assay using 14C-HCO₃⁻). However, the weak affinity of bicarbonate ions (in the mM range) would make reliable radioisotope-based detection impossible due to minimal specific receptor occupancy and high non-specific background and thus it is highly challenging and there are limitations to what can be done in this structural paper.
and determining a structure at comparable resolution in the absence of bicarbonate. In addition, all residues that are proposed to be located adjacent to the bicarbonate should be mutated and functionally validated.
We re-modeled the receptor based on the improved density and canceled the bicarbonate model. We performed calcium assay using cell lines stably expressing the mutants (Figure 3F, G and Figure 3.–figure supplement 1-3).
(2) What are the maps contoured in Figure 4D? The legend should describe this. Is 218 within the map region shown, or is there no density for its sidechain?
We removed the corresponding figure and cancelled the bicarbonate model.
(3) The contour level of the maps in Figure 1 - Figure Supplement 2 should also be indicated. Are these all contoured at the same level?
Thank you for your comment. We re-analyzed the same data set and obtained new density maps and models. We reworked Figure 1 and Figure 1. figure supplement 2; the contour level of the map for Figure 1 and composite map for the Figure 1. figure supplement 2 is the same, 7.65.
(4) Regarding the cited structures of bicarbonate-binding proteins, for three of the four cited structures, the bicarbonate is actually coordinated by positive ligands, with the Asp/Glu playing a more peripheral role:
Capper et al: Overall basic cavity with tight bidentate coordination by Arg. The Glu is 5-6 Å away.
Koropatkin et al: Two structures. The first, solved at pH 5, is proposed to have carbonic acid bound. The second, solved at pH 8, shows carbonate in a complex with calcium, with the calcium coordinated by carboxylates.
Wang et al: The bicarbonate is coordinated by a lysine and a sodium ion. The sodium is coordinated by carboxylates.
The authors should more thoughtfully discuss the unusual properties of this binding site with regard to the previous literature. Is it possible that bicarbonate binds in complex with a metal ion? Could this possibility be experimentally tested?
We cancelled the bicarbonate model.
(5) As a structure of GPR30 has been recently published by another group (PMID: 38744981), it would be valuable to discuss structural similarities and differences and discuss how bicarbonate activation and activation by the chloroquine ligand identified by the other group might both be accommodated by this structure.
Thank you for your valuable comment. We compared the structure presented by another group and added our discussion, as “During the revision of this manuscript, the structures of apo-GPR30-Gq (PDB 8XOG) and the exogenous ligand Lys05-bound GPR30-Gq (PDB 8XOF) were reported [42]. We compared our structure of GPR30 in the presence of bicarbonate with these structures. In the extracellular region, the position of TM5 in GPR30 in the presence of bicarbonate is similar to that in apo-GPR30. In contrast, the position of TM6 is shifted outward relative to that of apo-GPR30, resembling the conformation observed in Lys05-bound GPR30 (Figure 6A, B). Additionally, the position of ECL1 is also shifted outward compared to that of apo-GPR30 (Figure 6B). In the GPR30 structure in the presence of bicarbonate, ECL2 was modeled, suggesting differences in structural flexibility. These findings indicate that the structure of GPR30 in the presence of bicarbonate is different from both the apo structure and the Lys05-bound structure, demonstrating that the structure and the flexibility of the extracellular domain of GPR30 change depending on the type of ligand. Furthermore, focusing on the interaction with Gq, the αN helix of Gq is not rotated in the structure bound to Lys05, in contrast to the characteristic bending of the αN helix in our structure (Figure 6C, D). Although it is necessary to consider variations in experimental conditions, such as salt concentration, the differences in the Gq binding modes suggest that the downstream signals may change in a ligand-dependent manner.” (lines 249-266).
Reviewer #2 (Recommendations For The Authors):
(1) It is highly recommended that the authors carefully go through the "insights into bicarbonate binding" section. The results of the new findings in this paper were blended in with the results from the previous work: the importance of E115, Q138, and H307 in the receptor-bicarbonate interaction was shown in the Nature Communication paper but the authors didn't make it clear, which added a little confusion.
We emphasized this fact in the main text (lines 130-132).
(2) It would be nice for the authors to add some content about the physiological concentration of HCO3 or refer more to their previous work about the rationale for selecting the bicarbonate dose in their functional assay.
Thank you for your comment. The physiological concentration of bicarbonate is 22-26 mM in the extracellular fluid, including interstitial fluid and blood, and 10-12 mM in the intracellular fluid. The bicarbonate concentration alters in various physiological and pathological conditions – metabolic acidosis in chronic kidney disease causes a drop to 2-3 mM, and metabolic alkalosis induced by severe vomiting increases HCO3- concentrations more than 30 mM. Thus, our present and previous works clearly show that GPR30 is activated by physiological concentrations of bicarbonate, whether it is localized intracellularly or on the membrane, and that GPR30 can be deactivated or reactivated in various pathophysiological conditions. We added this in the discussion section (lines 267-278).
(3) In Figure 3A, in the legend, the authors mentioned: "black dashed lines indicate hydrogen bonds". No hydrogen bond was noted in the figure.
We totally corrected Figure 3.
(4) Figure 3B, it would be helpful for the authors to denote the meaning of the blue-white-red color coding in the legend.
We removed the figure.
(5) Supplemental Figure 3: since AF3 was released on May 3rd, it would be awesome in the revision version if the authors would update this to the AF3 model.
The AF2 model has been replaced with the AF3. (Figure 2–figure supplement 2A-C). The AF2 and AF3 models are almost identical, and they form incorrect disulfide bonds. This confirms the usefulness of the experimental structural determination in this study.
(6) Supplemental Figure 4: it wasn't clear to me if the expression experiments were repeated multiple times or if there was any statistical analysis for the expression level was done in this study.
We performed the expression experiment by western blotting once and did not perform statistical analyses. We performed repeated FACS analyses of HEK cells stably expressing N-terminally HA-tagged wild-type or mutant GPR30s to analyze their membrane and whole-cell expressions during revision (Figure 3.–figure supplement 1-3). Using these stable cells, we performed calcium assays using cell lines stably expressing the mutants (Figure 3F, G and Figure 3–figure supplement 1-3).
(7) Supplemental Figure 4: Also, is there a reason for the authors to compare the expression level of hGPR30 to the housekeeping gene NA-K-ATPase rather than the total loaded protein? Traditionally housekeeping genes have been used as loading controls to semiquantitatively compare the expression of target proteins in western blots. However, numerous recent studies show that housekeeping proteins can be altered due to experimental conditions, biological variability across tissues, or pathologies. A consensus has developed for using total protein as the internal control for loading. An editorial from the Journal of Biological Chemistry reporting on "Principles and Guidelines for Reporting Preclinical Research" from the workshop held in June 2014 by the NIH Director's Office, Nature Publishing Group, and Science stated, "It is typically better to normalize Western blots using total protein loading as the denominator".
Thank you for your instructive comment. We evaluated western blotting with the same amount of total protein loaded 20 µg for whole-cell lysate and 1.5 µg for cell surface protein (Figure 3.–figure supplement 3C-F).
Reviewer #3 (Recommendations For The Authors):
The claim about this disulfide should be removed unless the authors can provide mass spec evidence.
Thank you for your crucial comments. Firstly, C130 is a residue of TM3, not ECL1, so our misprint has been corrected to C1303.25. C207ECL2, located at position 45.50, is the most conserved residue in ECL2, and it forms a disulfide bond with cysteine at position 3.25 (PMID: 35113559). The paper was additionally cited regarding the preservation of the bond of C1303.25-C207ECL2 (line 103). Indeed, disruption of this disulfide bond by the C207ECL2 A mutation resulted in a marked reduction in receptor activity. In addition, the data set was re-analyzed to improve the local resolution of the extracellular region, and it was shown that the density of ECL2 is not noise (Figure 2. ––figure supplement 2). We are confident about the presence of the disulfide bond, based on the structural analysis data and the conservation.
The highly flexible extracellular region is greatly affected by experimental conditions and ligands, so we speculate that the ECL2 and the disulfide bond was not observed in other reported structures of GPR30. Then, we have added the following content to the discussion, as “In the GPR30 in the presence of bicarbonate, ECL2 was modelled, suggesting differences in structural flexibility.” (lines 256-257).
The authors should remove the assignment of bicarbonate in the structure, and tone down the binding site of bicarbonate.
We cancelled the bicarbonate model.
Minor:
(1) The potency of bicarbonate for GPR30 is in the mM range. Although the concentration of bicarbonate in the serum can reach mM range, how about its concentration in the tissues? Given its low potency, it may be not appropriate to claim GPR30 is a bicarbonate receptor at this point, but the authors can claim that GPR30 can be activated by or responds to bicarbonate.
The physiological concentration of bicarbonate is 22-26 mM in the extracellular fluid, including interstitial fluid and blood, and 10-12 mM in the intracellular fluid. Therefore, GPR30 is activated by physiological concentrations of bicarbonate in the tissues. Also, the bicarbonate concentration alters in various physiological and pathological conditions – metabolic acidosis in chronic kidney disease causes a drop to 2-3 mM, and metabolic alkalosis induced by severe vomiting increases HCO3- concentrations more than 30 mM. Thus, our work clearly shows that GPR30 is activated by physiological concentrations of bicarbonate, whether it is localized intracellularly or on the membrane, and that GPR30 can be deactivated or reactivated in various pathophysiological conditions. According to the reasons above, we claim GPR30 is a bicarbonate receptor (lines 267-278).
(2) The description that there is no consensus on a drug that targets GPR30 is not accurate, since lys05 has been reported as an agonist of GPR30 and their structure is published (PMID: 38744981). The published structures of GPR30 should be introduced in the paper.
We added the discussion about the structural comparison with the Lys05-bound structure (Figure 6, lines 249-266)
(3) BW numbers in Figure 4A should be shown.
We added BW numbers in the figures of the mutational studies.



