The genome sequence of the colonial chordate, Botryllus schlosseri
- Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, United States
- Stanford University, United States
- Howard Hughes Medical Institute, Stanford University, United States
- Università degli Studi di Milano, Italy
- University of Haifa-Oranim, Israel
- Stanford University School of Medicine, United States
Figures
Figure 1 with 1 supplement
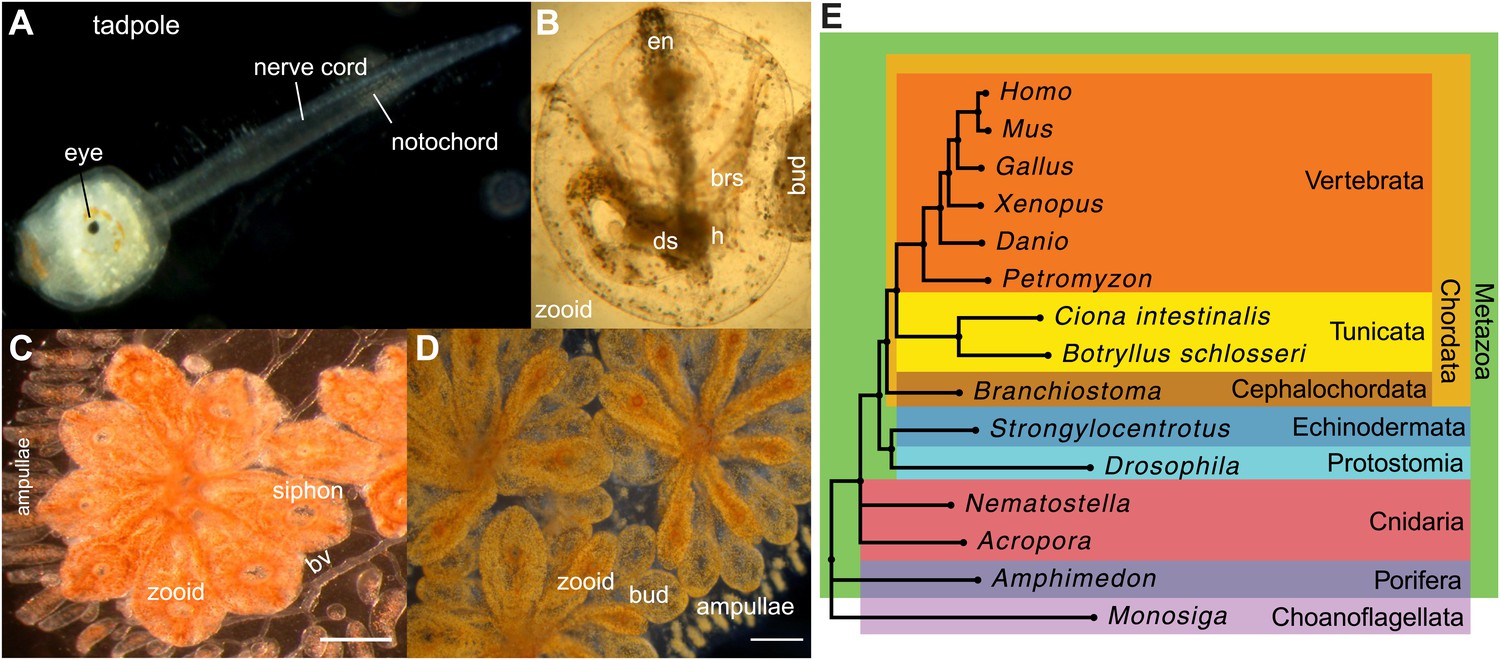
Botryllus schlosseri anatomy, life cycle, and phylogeny.
B. schlosseri reproduces both through sexual and asexual (budding) pathways, giving rise to virtually identical adult body plans. Upon settlement, the tadpole phase of the B. schlosseri lifecycle (A) will metamorphose into a founder individual (oozooid) (B), which through asexual budding, generates a colony. The colony includes three overlapping generations: an adult zooid, a primary bud, and a secondary bud, all of which are connected via a vascular network (bv) embedded within a gelatinous matrix (termed tunic). The common vasculature terminates in finger-like protrusions (termed ampullae; B–D). Bud development commences in stage A (C). Through budding, B. schlosseri generates its entire body, including digestive (ds) and respiratory (brs) systems, a simple tube-like heart (h), an endostyle (en) that harbors a stem cell niche, a primitive neural complex, and siphons used for feeding, waste, and releasing larvae (B–D). Each week, successive buds grow large (D) and complete replication of all zooids in the colony, ultimately replacing the previous generation’s zooids, which die through a massive apoptosis. (E) A phylogenomic tree produced from analysis of 521 nuclear genes (40,798 aligned amino acids) from 15 species, including B. schlosseri. Scale bar-1 mm.
Figure 1—figure supplement 1
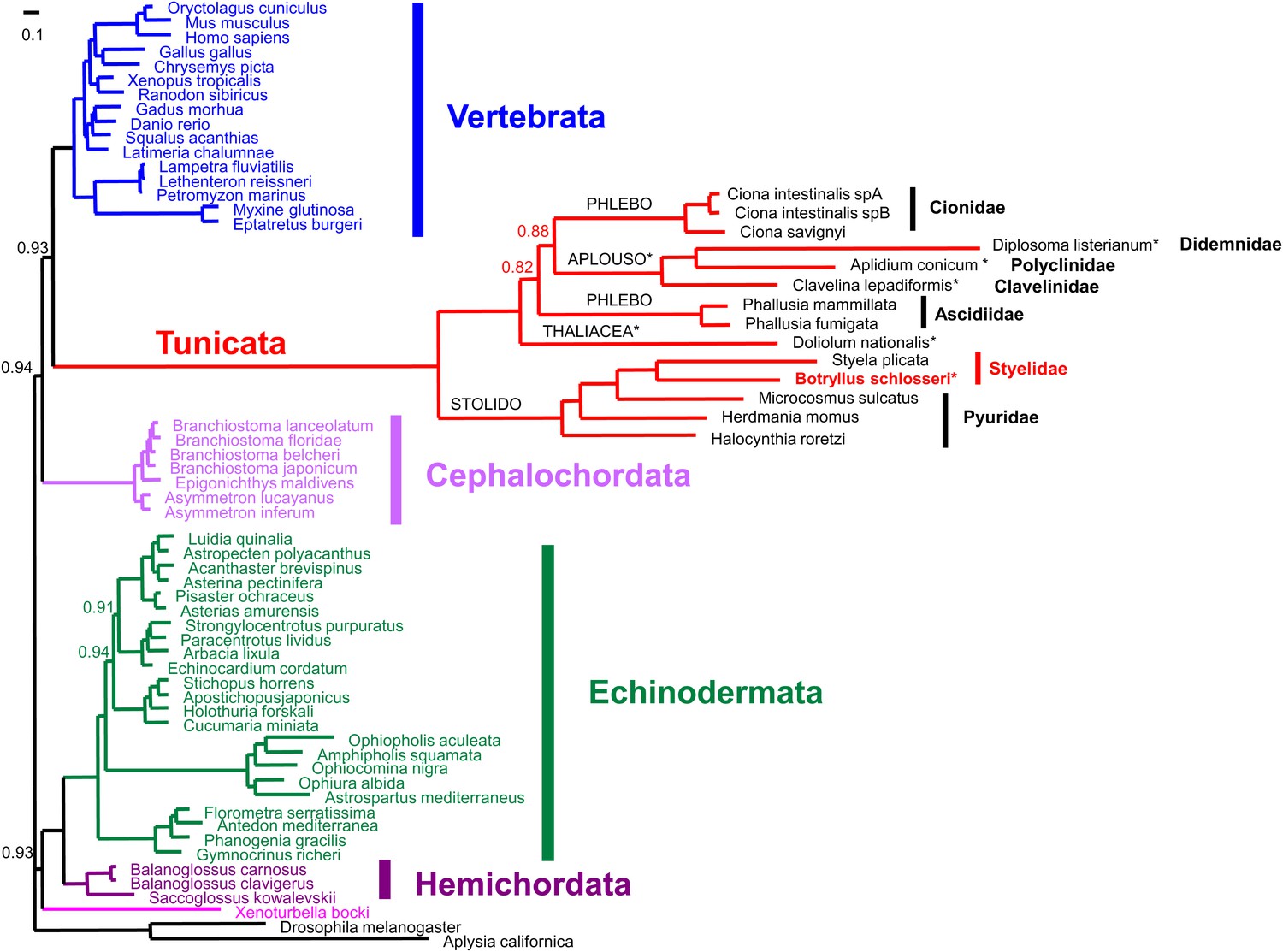
Mitogenomic analysis of tunicates and deuterostomes.
Based on the 13 mitochondrially-encoded proteins. The tree was inferred by PhyloBayes under a GTR+G+CAT model. Support values at nodes represents Bayesian Posterior Probability (PP) and are reported only when >0.5 and <0.95. Nodes with PP < 0.5 were collapsed. The tree was rooted with the non-deuterostome Drosophila and Aplysia species. The main deuterostome lineages are represented in different colours. Abbreviations for tunicate orders: Stolido: Stolidobranchia; Phlebo: Phlebobranchia; Aplouso: Aplousobranchia. Colonial tunicates are indicated by an asterisk and include Botryllus schlosseri, all Aplousobranchia ascidians, and the thaliacean Doliolum nationalis.
Figure 2 with 6 supplements

A novel short read genome sequencing and assembly method for complex, repeat-rich genomes.
(A) Genomic DNA is sheared into 6–8 kb fragments, partitioned into twelve 96-well plates, further fragmented to 600–800 bp, barcoded and sequenced separately for each well (Illumina HiSeq 2000 2x100bp), and assembled by Velvet. (B) Size distribution of contigs assembled from a representative library preparation (BL5). (C) Limiting the number of amplifiable molecules per well (barcode) to the level that almost 100% of all amplifiable molecules are present as single copies (<1000 gDNA molecules) greatly reduces the chance of having a repeated or homologous sequence within a well. Thus, sample complexity is significantly reduced, which reduces ambiguity in the reconstruction of a consensus sequence. As an example, two different predicted repeat-containing genes (g2001,1189bp; and g2002, 688bp) were assembled from two different wells (005 and 145 respectively). Although they contain highly homologous repeats (represented as a Dot Matrix plot, (D) these repetitive genes were resolved and reconstructed properly in the final assembly.
Figure 2—figure supplement 1
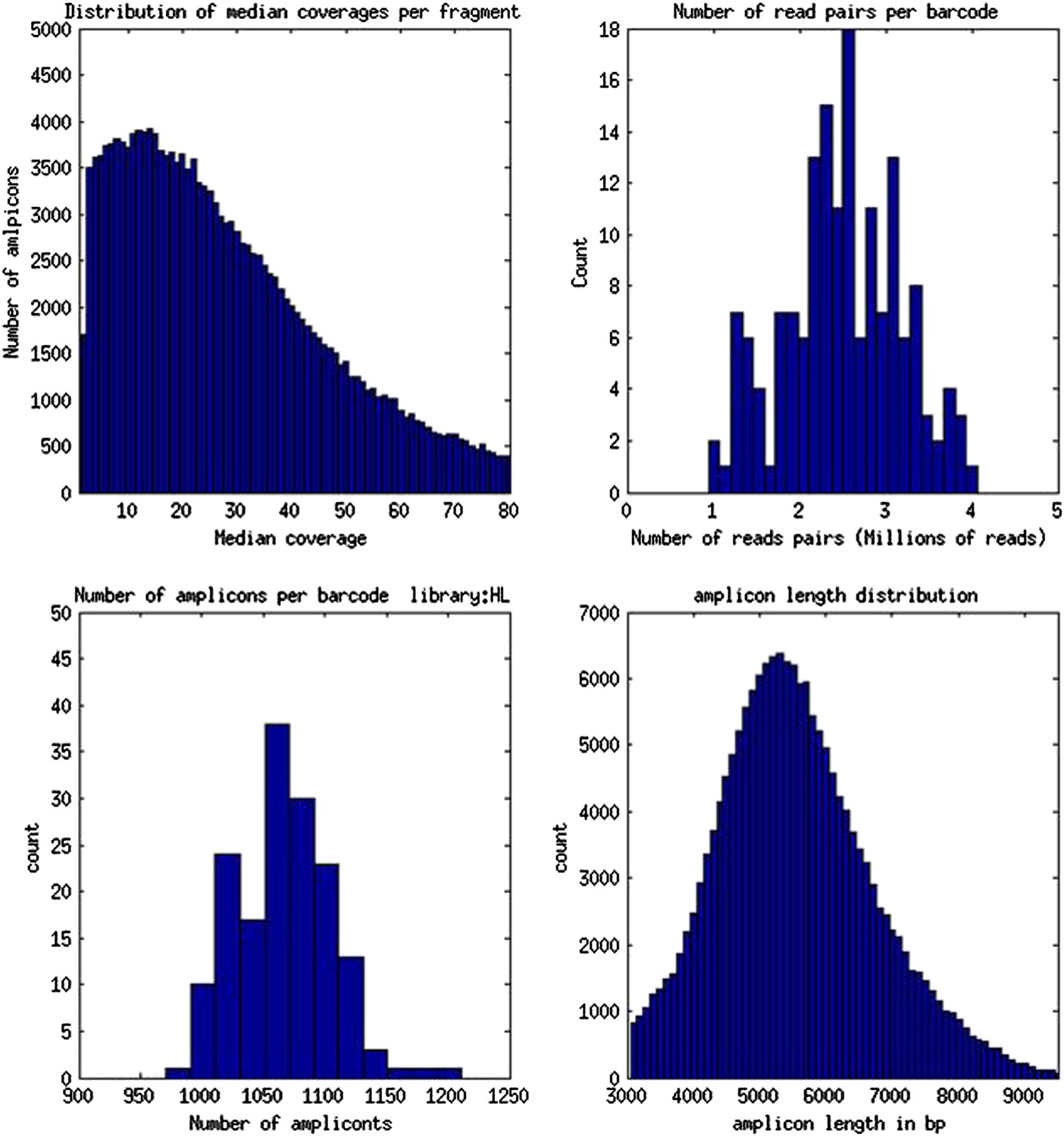
Validation of LRseq approach on human genomic DNA.
Genomic DNA from HapMap NA7019 was prepared for LRseq. These figures show LRseq assembly statistics, obtained by mapping sequenced reads to human genome reference 36. These data were also used to estimate the concentration of amplifiable molecules in B. schlosseri 356a DNA samples prepared by an identical protocol.
Figure 2—figure supplement 2
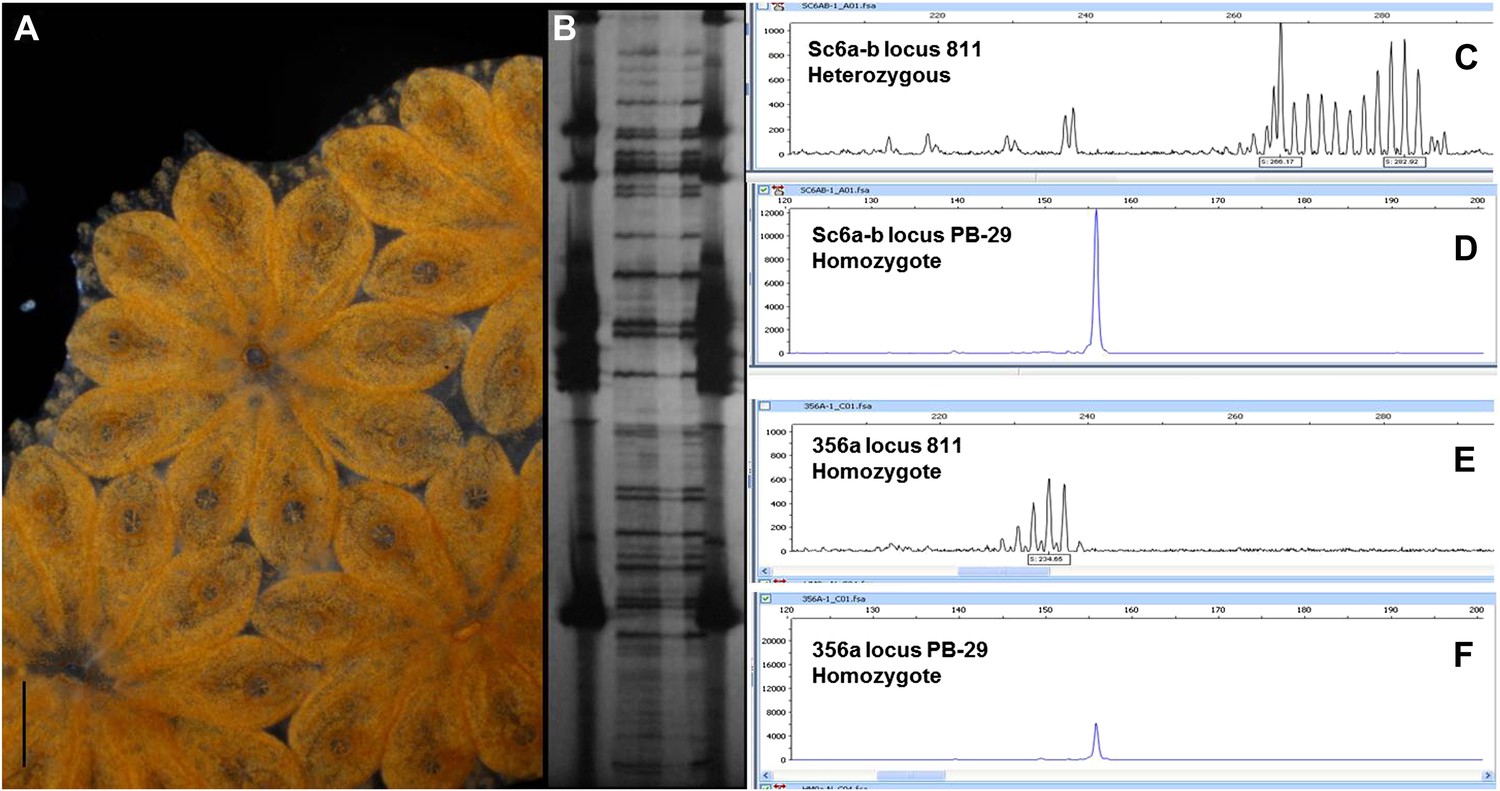
Clonality confirmation of the genome of clone Sc6a-b and clone 356a.
(A) Sc6a-b clone, a long lived (7 years old when sampled), highly regenerative colony was chosen to be sequenced. Sc6a-b subclones were starved for 48 hr prior to sampling, and 400 individuals (zooids) were sampled for sequencing. Subclones of this colony are still alive and maintained in our mariculture facility. (B) A few zooids were taken from every sample set and tested via AFLP’s genotyping analysis, confirming that all zooids belong to one genotype. (C and D). Sc6a-b microsatellite loci were homozygous (2 loci) and heterozygous (1 loci) confirming one genotype. (E and F) 356a clone was a highly regenerative long lived colony. 150 individuals were sampled and their gDNA was sequenced. Microsatellite loci were homozygous (E and F), confirming one genotype. Scale bar-1 mm
Figure 2—figure supplement 3
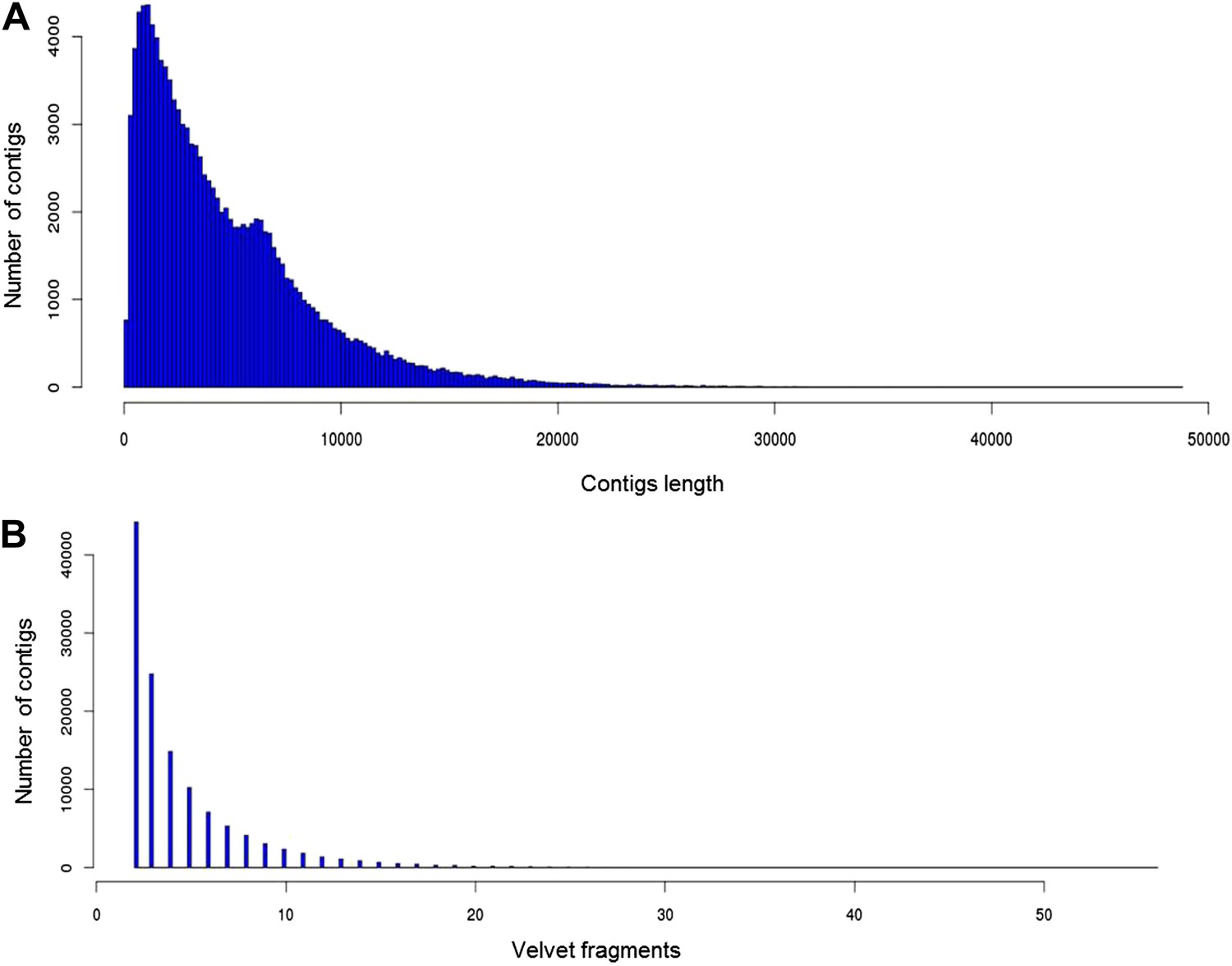
Statistics for 356a assembly.
(A) Contig length distribution. (B) Distribution of coverage of 356a assembled Celera contigs by Velvet assembled fragments.
Figure 2—figure supplement 4
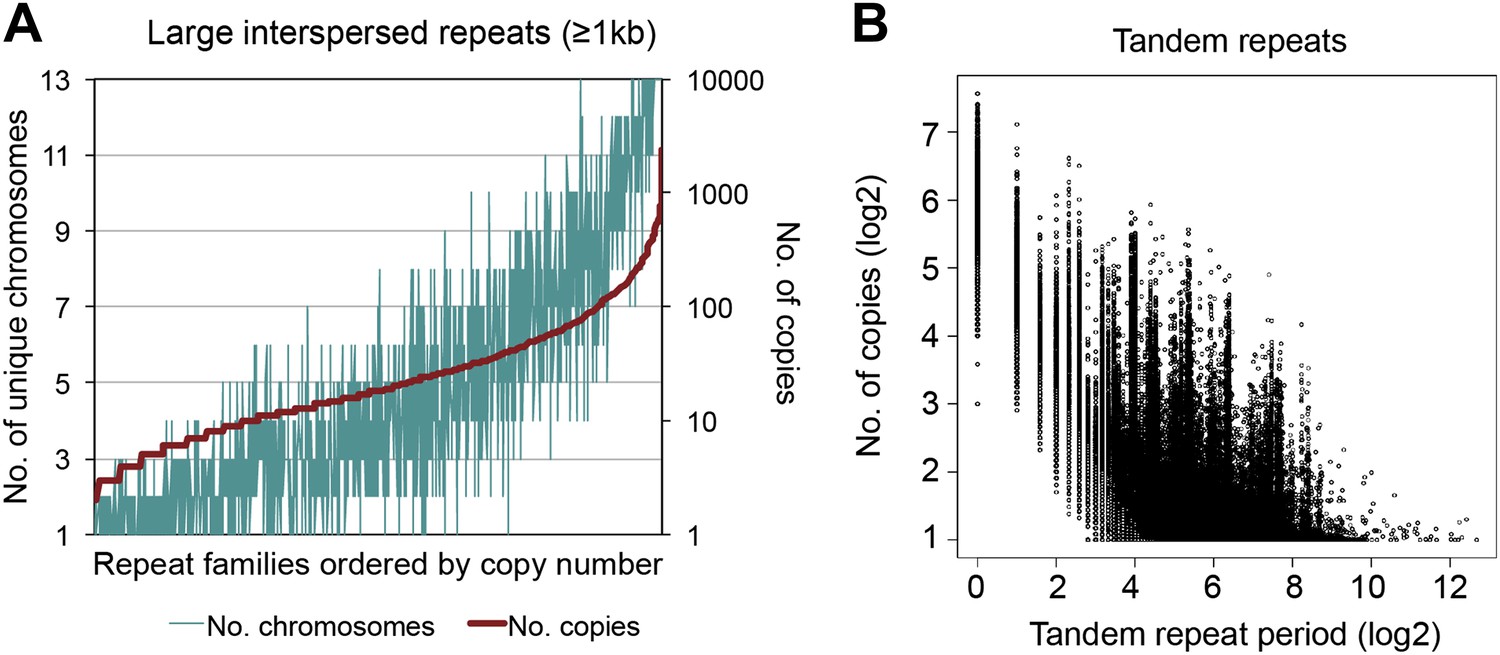
Interspersed and tandem repeats distribution in the B. schlosseri genome.
(A) RepeatScout (version 1.0.5; Price et al., 2005) was used to identify interspersed repeat elements de novo using a k-mer length of 14. All identified repeats were subsequently filtered for tandem repeat and low complexity content, using RepeatScout. Genome-wide interspersed repeats were catalogued using RepeatMasker (version open-4.0; Smit et al., 1996-2010). The distribution of large interspersed repeats families (≥1kb) ordered by copy number is presented. (B) To identify both perfect (100% sequence identity) and degenerate genomic tandem repeats, we used XSTREAM (Newman and Cooper, 2007), with a minimum repeat length of 20 bp, minimum word match of 0.8, and otherwise default parameters. 3,183,988 tandem repeats were identified, period range: 1–6525 bp, copy number range: 2.7–1096x
Figure 2—figure supplement 5

Coverage of 4 fosmids by the B. schlosseri assembly.
Fosmid sequences (red lines; gi; ac numbers are shown, number=bp), were compared with B. schlosseri contigs using blast (e-value < e−10). Best alignments between contigs >500bp (black lines) are shown. Repetitive regions are marked (blue).
Figure 2—figure supplement 6
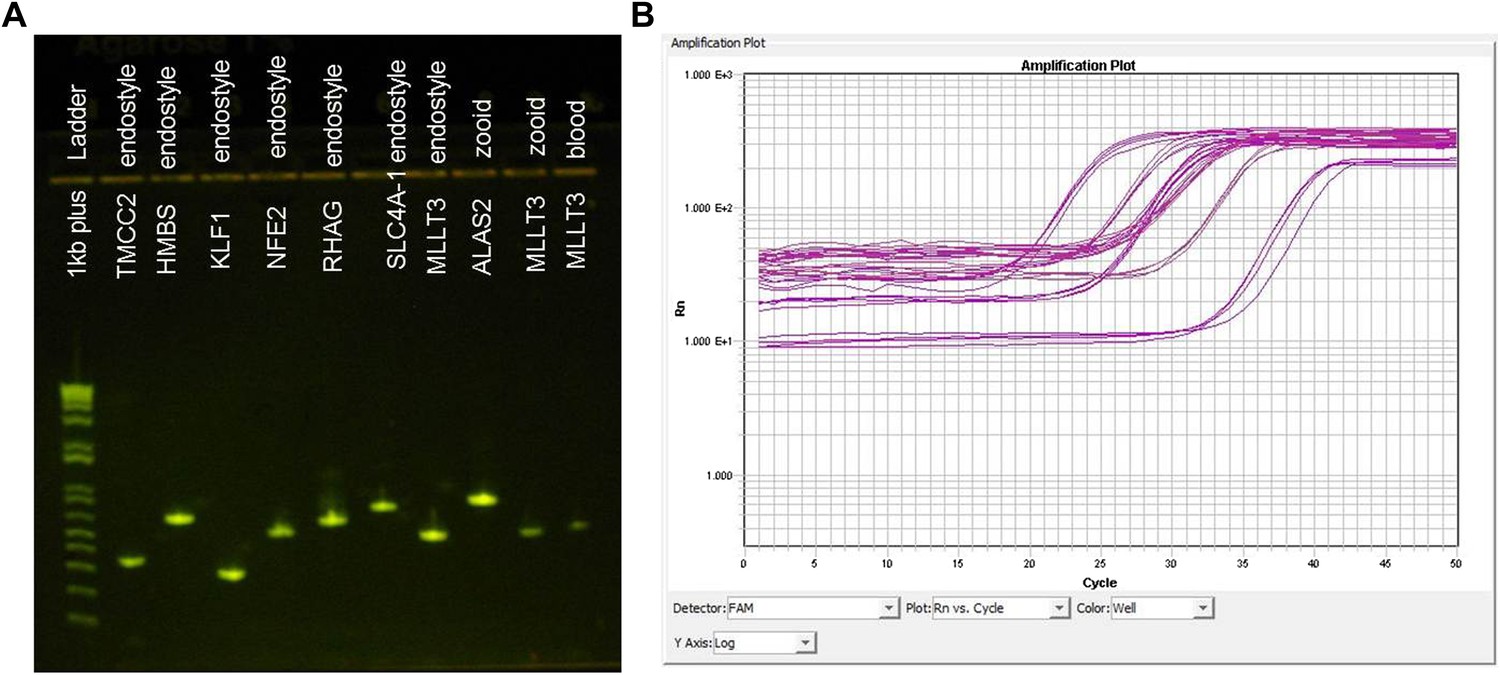
Validation of putative B. schlosseri genes.
We experimentally validated 145 B. schlosseri predicted genes. Genes were validated by observing expression in B. schlosseri cDNAs and gDNA via PCR and qPCR assays and resequencing them on Sanger. (A) cDNA PCR product of several early erythroid and HSC putative genes identified in B. schlosseri tissues (endostyle, blood or zooid). Names of the putative genes and the tissues that were tested in this experiment are indicated on the gel image. (B) qPCR expression in B. schlosseri blood of six putative immunity genes.
Figure 3 with 4 supplements
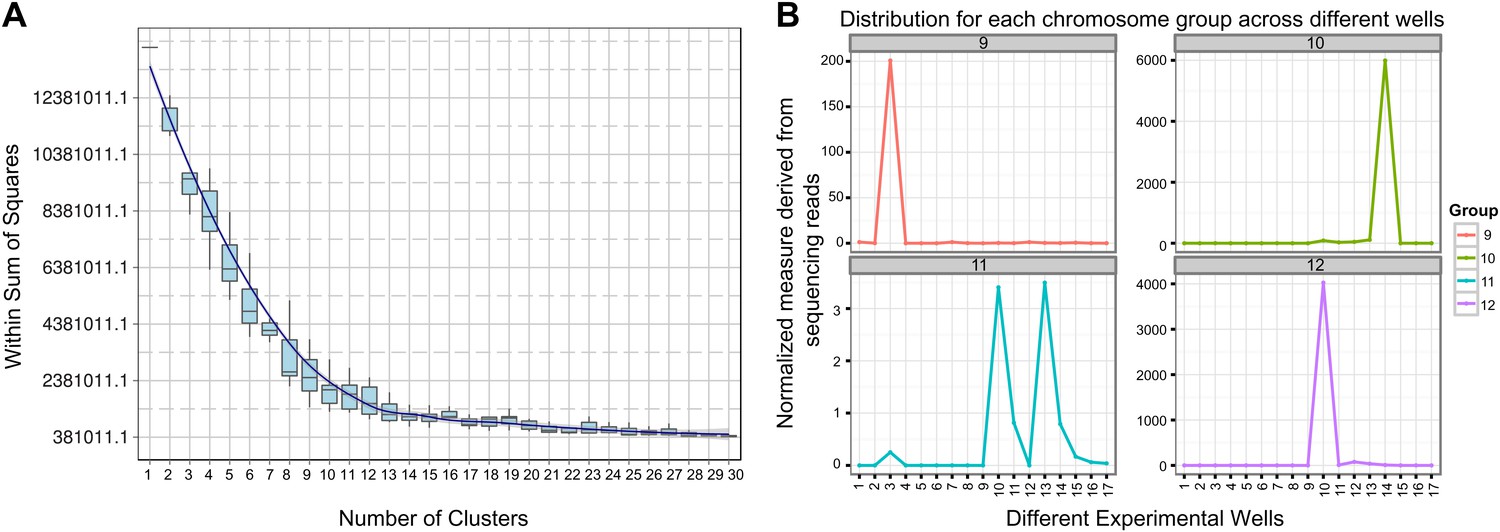
Clustering and assignment of B.schlosseri chromosomes.
(A) We isolated and sequenced 21 metaphase chromosome mixtures using a microfluidic device. Each chromosome mixtures was amplified, barcoded and sequenced separately (IlluminaHiSeq). Genomic contigs larger than 7 kb were aligned to the chromosome reads using BWA. Subsequently, assignment of scaffolds to chromosome cluster was performed using iterative K-means clustering on the correlation matrix between each scaffold. In addition, to find the number of clusters/chromosomes we performed k-means clustering iteratively across different cluster numbers. This plot demonstrates that increasing beyond 13 clusters does little to reduce the error; therefore 13 chromosomes were successfully resolved. (B) To estimate the configuration after the clustering step, 17 out of the 21 wells were deduced to contain information that is used in the clustering process. The average number of normalized reads counts from each metaphase chromosome mixture (well) that align to each scaffold in a cluster group was calculated and plotted. Each peak represented can be inferred to denote the presence of a specific chromosome in the well. Examples of four representative wells are presented, metaphase chromosome mixtures contained between 1–4 chromosomes (see also Figure 3—figure supplement 1).
Figure 3—figure supplement 1
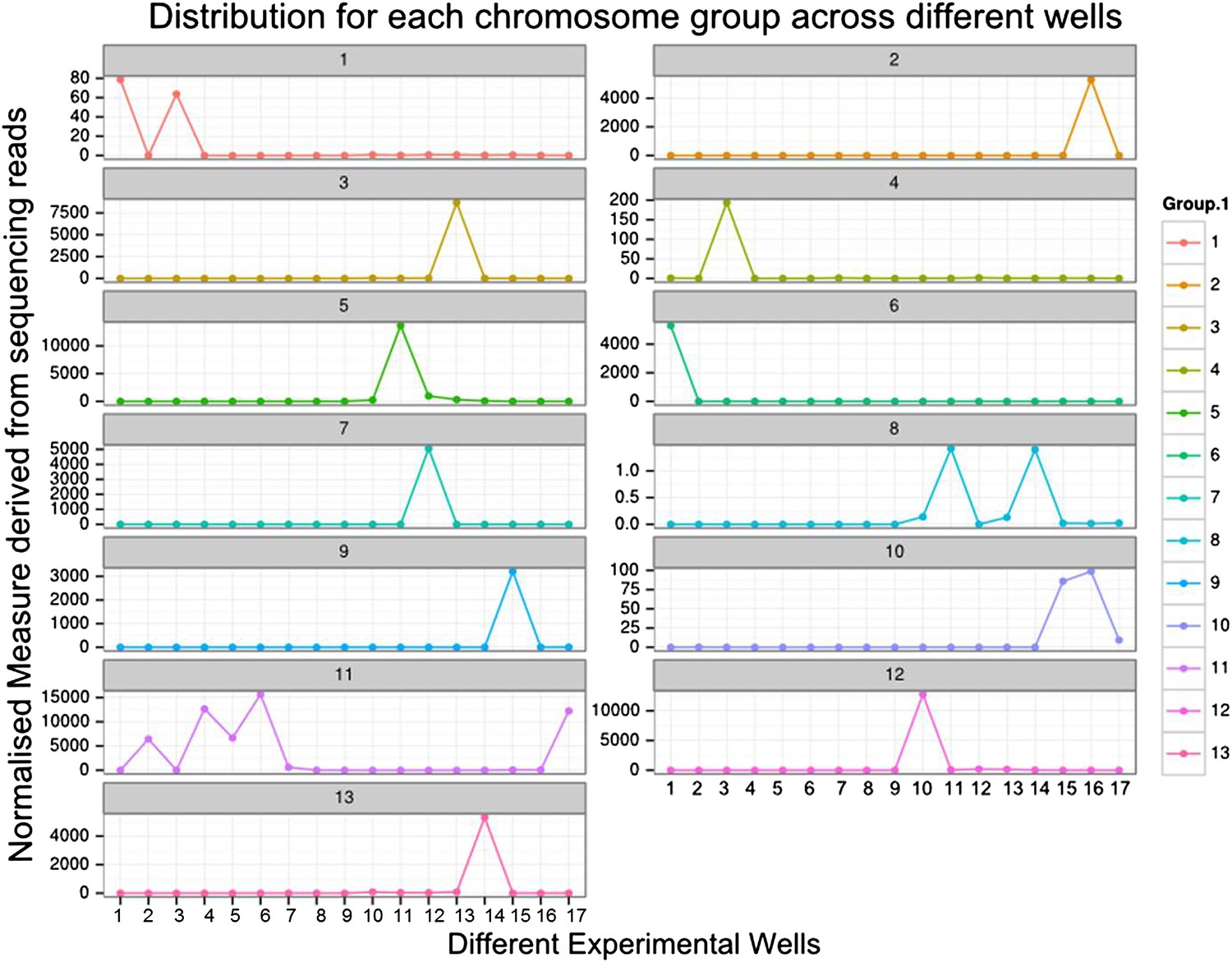
Distribution of B. schlosseri chromosome groups across different wells.
We isolated and sequenced metaphase diluted chromosome mixtures using a microfluidic device. Each chromosome mixture was amplified, barcoded and sequenced separately (IlluminaHiSeq). The average number of normalized reads counts from each diluted chromosome mixture (well) that align to each scaffold in a cluster group was calculated and plotted. Each peak represents the presence of a specific chromosome in the well. In the 17 wells presented above, chromosome mixtures contained between 1–4 chromosomes.
Figure 3—figure supplement 2
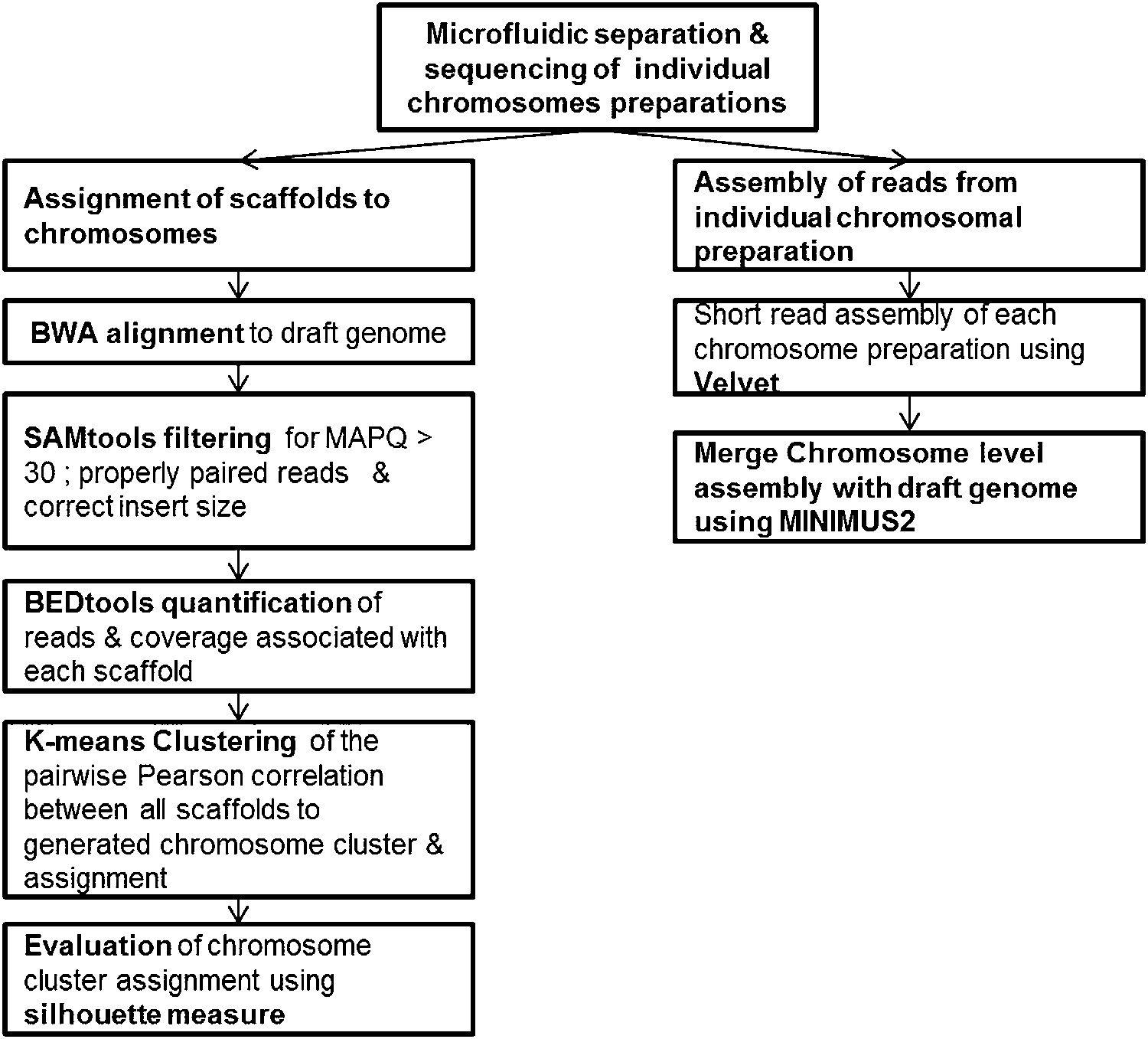
Pipeline for the assignment of chromosome scaffolds and the 356a–chromosomes hybrid assembly process.
https://doi.org/10.7554/eLife.00569.014
Figure 3—figure supplement 3
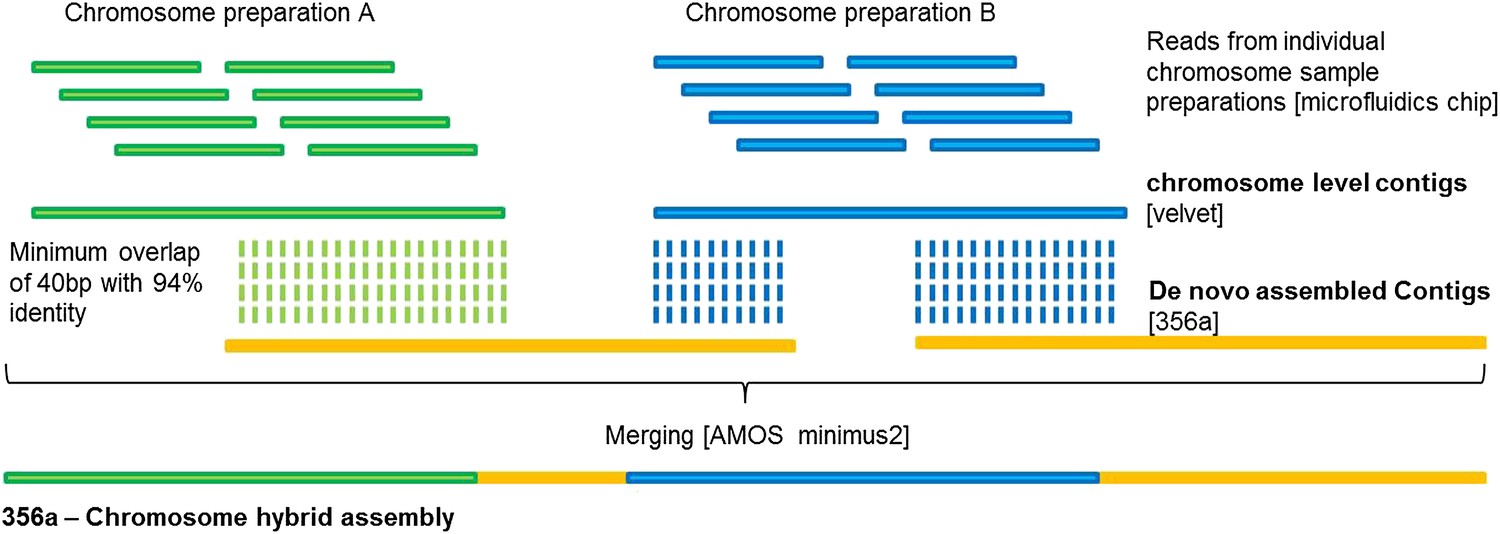
356a-Chromosome hybrid assembly of B. schlosseri.
Reads from each of the individual chromosome sample preparations were subsequently assembled using Velvet. The resulting chromosome level contigs were then merged with the 356a assembly to create a 356a-chromosome hybrid assembly.
Figure 3—figure supplement 4
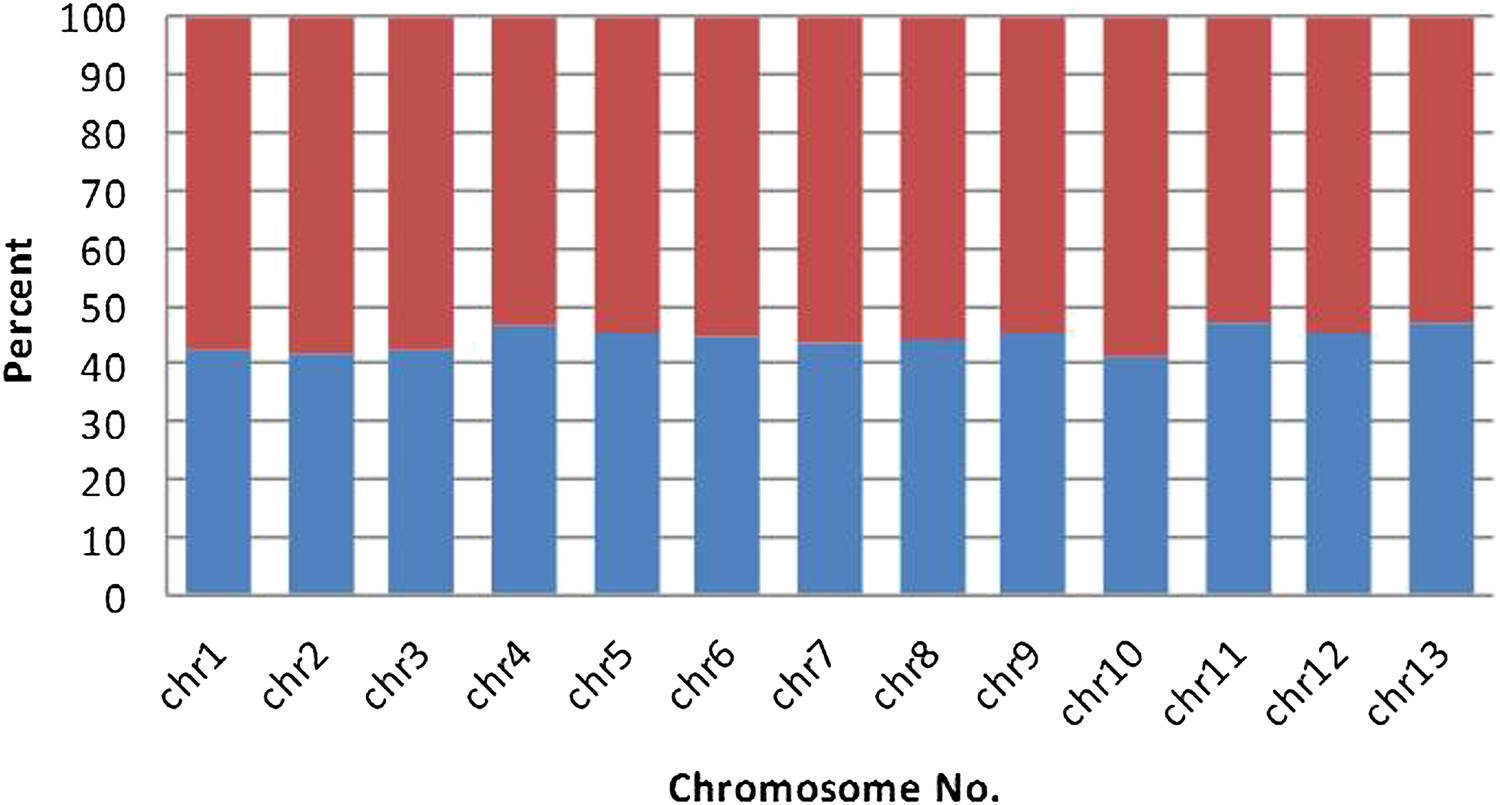
The fraction of B. schlosseri predicted intron-less genes (blue) and genes with introns (red) in the different chromosomes.
https://doi.org/10.7554/eLife.00569.016
Figure 4
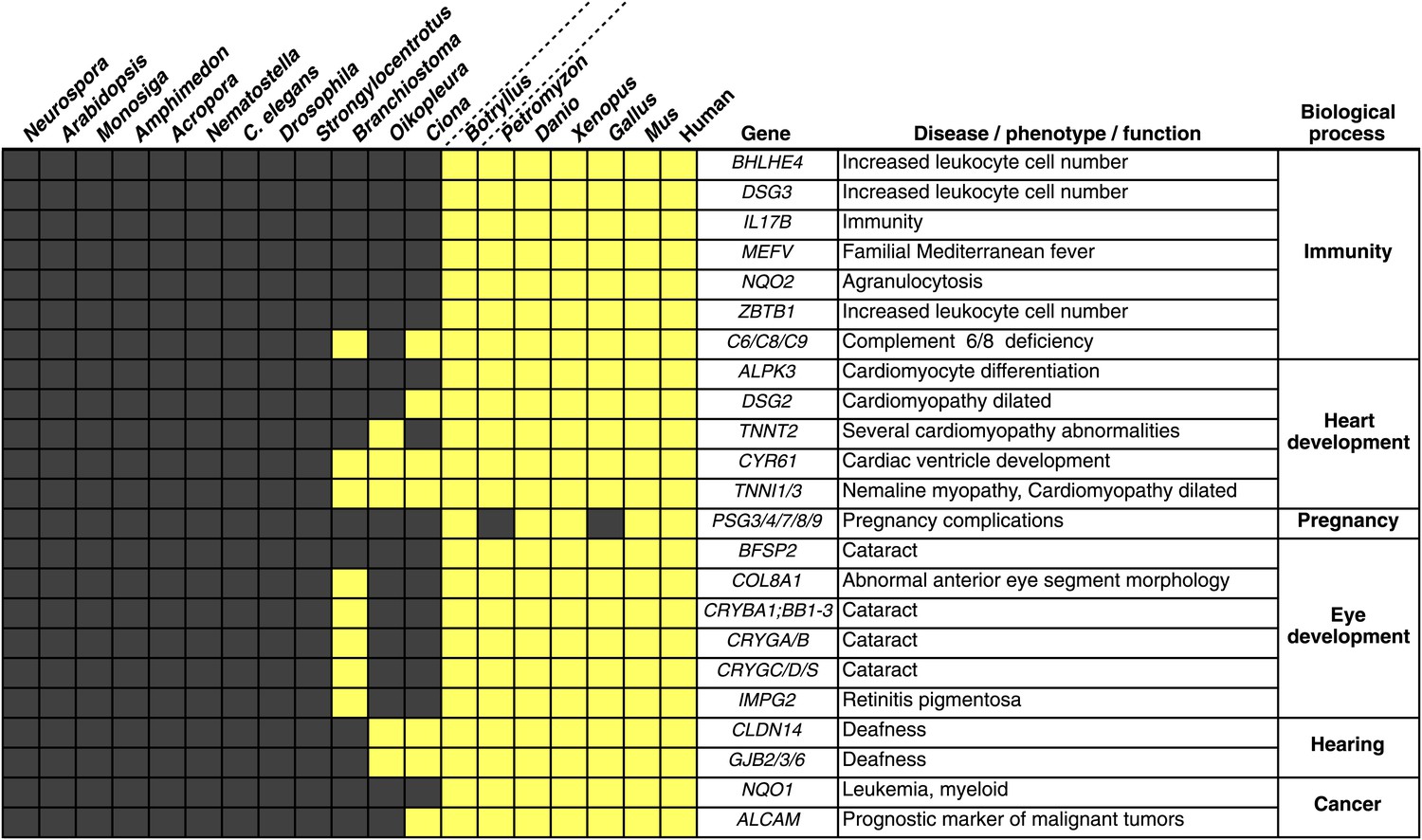
Innovations underlying the emergence and early diversification of vertebrates.
Protein-encoding genes in B. schlosseri were compared to a diverse sampling of 18 well-annotated genomes from other species, and for each genome, the presence or absence of homology to human or mouse proteins was assessed (all vs all blastp e-value threshold of 1e−10; Figure 4—source data 1A). Our data indicate that homologs of ∼660 human/mouse genes were present in the common ancestor of tunicates and vertebrates, but not non-chordate species Figure 4—source data 1B). Among them are genes associated with the development, function, and pathology of vertebrate features, including heart, eye, hearing, immunity, pregnancy and cancer (Figure 4—source data 1C). Gray box = no homology; Yellow box = homology.
-
Figure 4—source data 1
Vertebrates evolution.
(A) Innovations that underline the emergence and early diversification of vertebrates. We compared protein-encoding genes in B. schlosseri to a diverse sampling of 18 well-annotated genomes from other species. All protein sequences were compared by blastp against all other protein sequences. Based on this data set a list was generated of genes known from human and mouse and their existence (1) or absence (0) in the tested species (e-value < e−10). (B) The 660 putative genes present in protochordates, human and mouse, but absent in non-chordate species. This list was generated from Figure 4—source data 1A. Per every species, or species group we filtered for genes that were present in the tested species/species group and in human or mouse, but were absent in non-chordate species. (C) Innovations that underline the emergence and early diversification of vertebrates. This table is based on data gathered in Figure 4—source data 1B and is focused on the genes that are present in B. schlosseri and vertebrates (either alone or in combination with other protochordate species) but are absent in non-chordate species. A ToppGene analysis is presented of these sets of genes which summarized their molecular functions, biological processes, human and mouse phenotypes, and pathways they are involved in, gene families, drugs and human diseases.
- https://doi.org/10.7554/eLife.00569.018
Figure 5
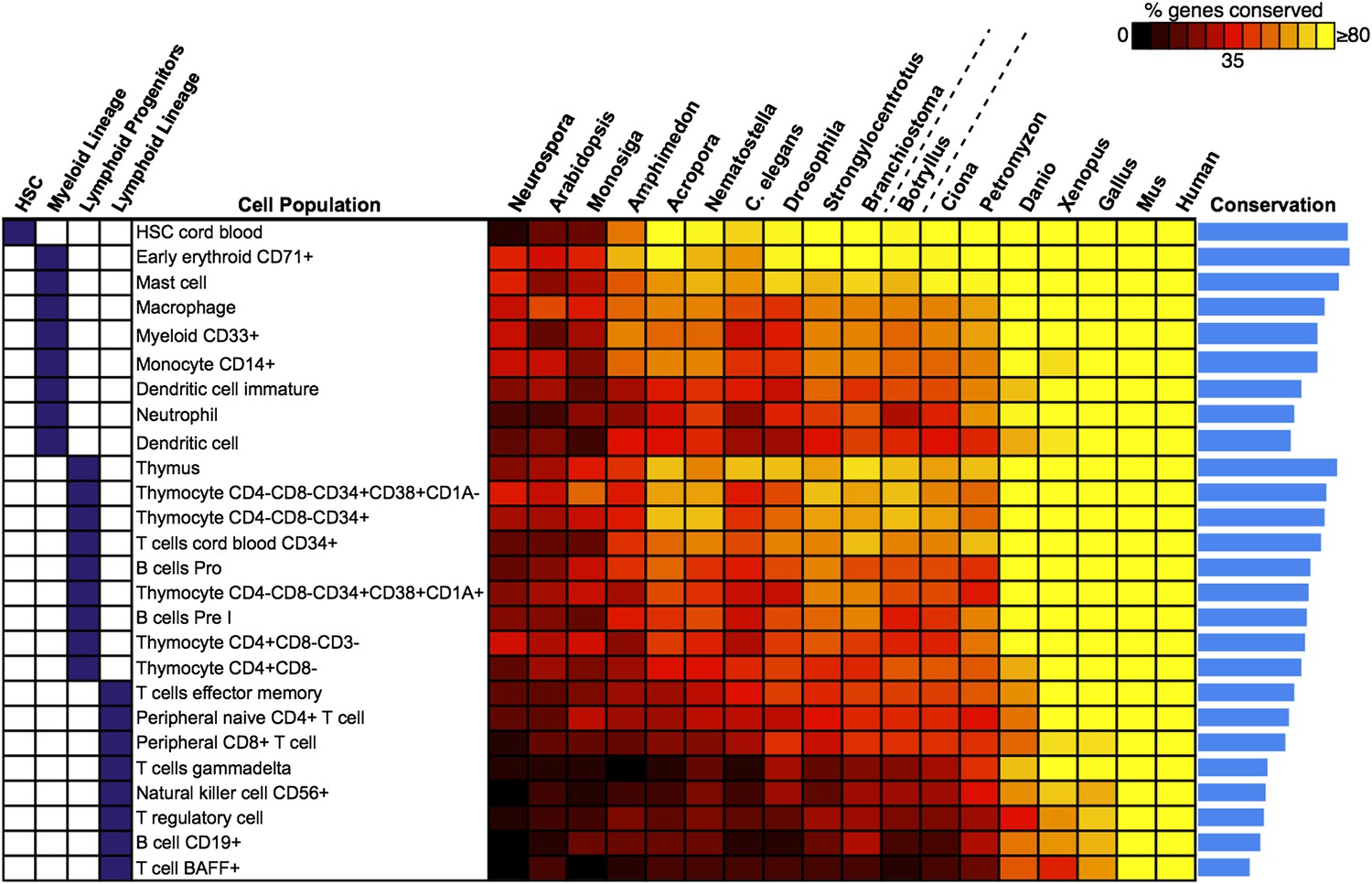
Analysis of blood and immune cell type-specific genes across evolution reveals evidence for hematopoietic precursors in B. schlosseri.
We analyzed gene expression microarray data from 26 different human blood cell populations, organized into four cell lineages (HSC; Lymphoid Progenitors; Myeloid and Lymphoid Lineage), and identified a set of twenty signature genes with highly enriched expression profiles for each population (Supplementary file 3). For each blood-related gene set, we identified homologous gene sequences in B. schlosseri and 17 other species; the fraction of genes (out of 20) found for each species is displayed as a heat map. Within each major lineage, cell populations are sorted in decreasing order by a conservation index, calculated as the average number of genes found across the 18 species (indicated by a blue bar graph).
Videos
Video 1
B. schlosseri blood circulation.
(A) Time-lapse acquisition of blood flow in the blood vessels (bv) and ampullae of a chimeric B. schlosseri colony, generated from a fusion between a mother and its offspring (fused). (B) Ampullae contract, buds develop, and a colony gets ready to replace the old generation. (C) Old generation zooids are getting resorbed (res. z) and replaced by the new generation (buds). (D) A heart beating and pumping blood in the primary bud of a different colony. (E) Blood flow through a common blood vessel between two allogeneic/compatible colonies, creating a natural chimera.
Additional files
-
Supplementary file 1
Species classification and Accession Number (AC) of the complete mtDNA sequences used in the tunicate phylogenetic reconstructions.
- https://doi.org/10.7554/eLife.00569.021
-
Supplementary file 2
B. schlosseri genome statistics.
(A) Statistics of Velvet assembly of wells per every 356a genomic library preparation. (B) Detailed statistics of Velvet assembly of wells per every 356a genomic library preparation. Name: barcode number; #reads: number of filtered reads obtained, NumCtgs: number of assembled contigs, Min: minimum contig length obtained, Max: maximum contig length obtained, Middle: length of the middle contig; Sum: sum of assembled contigs (bp), N50: length of N50 (bp), NumAbove8k: number of assembled contigs above 8k, NumAbove20k: number of assembled contigs above 20k. Kmer: Kmer used by Velvet assembly, Ecov: estimated coverage, CovCutoff: minimum coverage used. (C) Total gDNA and RNA sequence data obtained. Genomic DNA was extracted from tissue from two long-lived colonies (356a and Sc6a-b) raised in our mariculture facility. Sequence reads from colony Sc6a-b were obtained using Roche 454 Titanium and Illumina GAIIx. Sequence reads from colony 356a and 21 individual chromosomes isolated from a wild colony were obtained by Illumina HiSeq 2000. RNA-seq reads from several tissues (endostyle, vasculature, gonads and digestive system) were obtained by Illumina GAIIx and Illumina MiSeq. RNA-seq reads from 19 different lab reared colonies were obtained by Illumina HiSeq 2000. (D) B. schlosseri genome assemblies and chromosome scaffolds statistics. (E) Interspersed repeats in B. schlosseri. Analysis of interspersed repeat content in B. schlosseri, compared to C. intestinalis (version ci2, downloaded from the UCSC genome browser). RepeatScout (version 1.0.5; Price et al., 2005) was used to identify interspersed repeat elements de novo using a k-mer length of 14. All identified repeats were subsequently filtered for tandem repeat and low complexity content, using RepeatScout. Genome-wide interspersed repeats were catalogued using RepeatMasker (version open-4.0) (Smit et al., 1996–2010). (F) Alignment statistic of all of the B. schlosseri non mitochondrial genes (66), expressed sequence tags (98,611 EST’s) and fosmids (11) available on NCBI and, Sc6a-b assembly (518,856 contigs) with the B. schlosseri 356a chromosome hybrid draft genome assembly. (G) Genome assemblies statistics of several wild type species. (H) B. schlosseri predicted gene structure statistics. (I) Barcoded adapters list for 192 wells.
- https://doi.org/10.7554/eLife.00569.022
-
Supplementary file 3
Potential precursors of human hematopoietic populations in B. schlosseri.
We analyzed gene expression microarray data from 26 different human blood cell populations, along with a large diversity of non-blood human tissue samples, and identified a set of twenty signature genes with highly enriched expression profiles for each hematopoietic population. The 20 signature genes for each blood-related gene set, and the identified orthologous gene sequences in B. schlosseri are presented. Status = 1 predicted gene is present in B. schlosseri genome; Status = 0 predicted gene is absent from B. schlosseri genome.
- https://doi.org/10.7554/eLife.00569.023
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The genome sequence of the colonial chordate, Botryllus schlosseri
eLife 2:e00569.
https://doi.org/10.7554/eLife.00569




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}