Stability-mediated epistasis constrains the evolution of an influenza protein
- Fred Hutchinson Cancer Research Center, United States
- University of California, Los Angeles, United States
Figures
Figure 1

Outline of experiment designed to parallel Maynard Smith’s analogy.
The actual evolutionary trajectory involves the accumulation of mutations, and consists of a series of evolutionary intermediates. We recreate and experimentally assay each of these evolutionary intermediates. We also introduce each mutation individually into the original parent sequence, and experimentally assay these single mutants. If Maynard Smith is correct, each of the naturally occurring evolutionary intermediates should be a functional protein. However, some of the single mutants could exhibit impaired function if there is significant epistasis among mutations along the evolutionary trajectory.
Figure 2 with 2 supplements
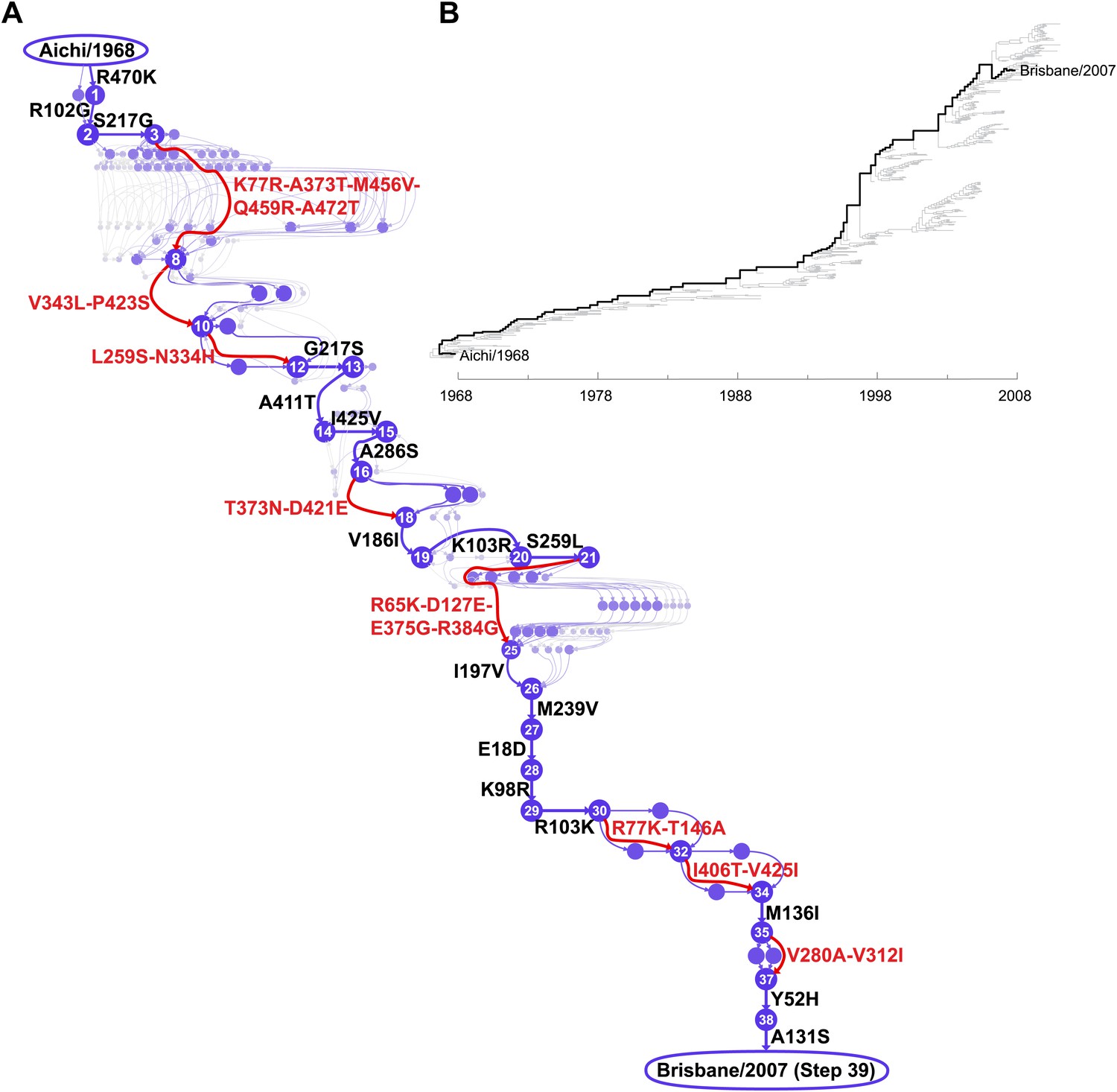
Inferred evolutionary trajectory.
(A) Evolutionary trajectory through protein sequence space from Aichi/1968 to Brisbane/2007 NP. Each circle represents a unique inferred sequence, with areas and intensities proportional to the probability that sequence was part of the evolutionary trajectory (Figure 2—figure supplement 1). Mutations for which the parent and descendent are clearly resolved are in black; mutations that occurred in an unknown order are in red. High-confidence evolutionary intermediates have numeric labels. The estimated date of occurrence of each mutation is shown in Figure 2—figure supplement 2. (B) Phylogenetic tree of the human H3N2 NPs from 1968 to 2011 that were used to infer the evolutionary trajectory. The lines of descent connecting Aichi/1968 and Brisbane/2007 to their common ancestor are in black. Data and computer code are provided in Figure 2—source code 1.
-
Figure 2—source code 1
The sequence data and source code used to generate the evolutionary trajectory and phylogenetic tree.
This code is in the form of a ZIP file with BEAST input files and Python code. A README file explains the contents.
- https://doi.org/10.7554/eLife.00631.005
Figure 2—figure supplement 1
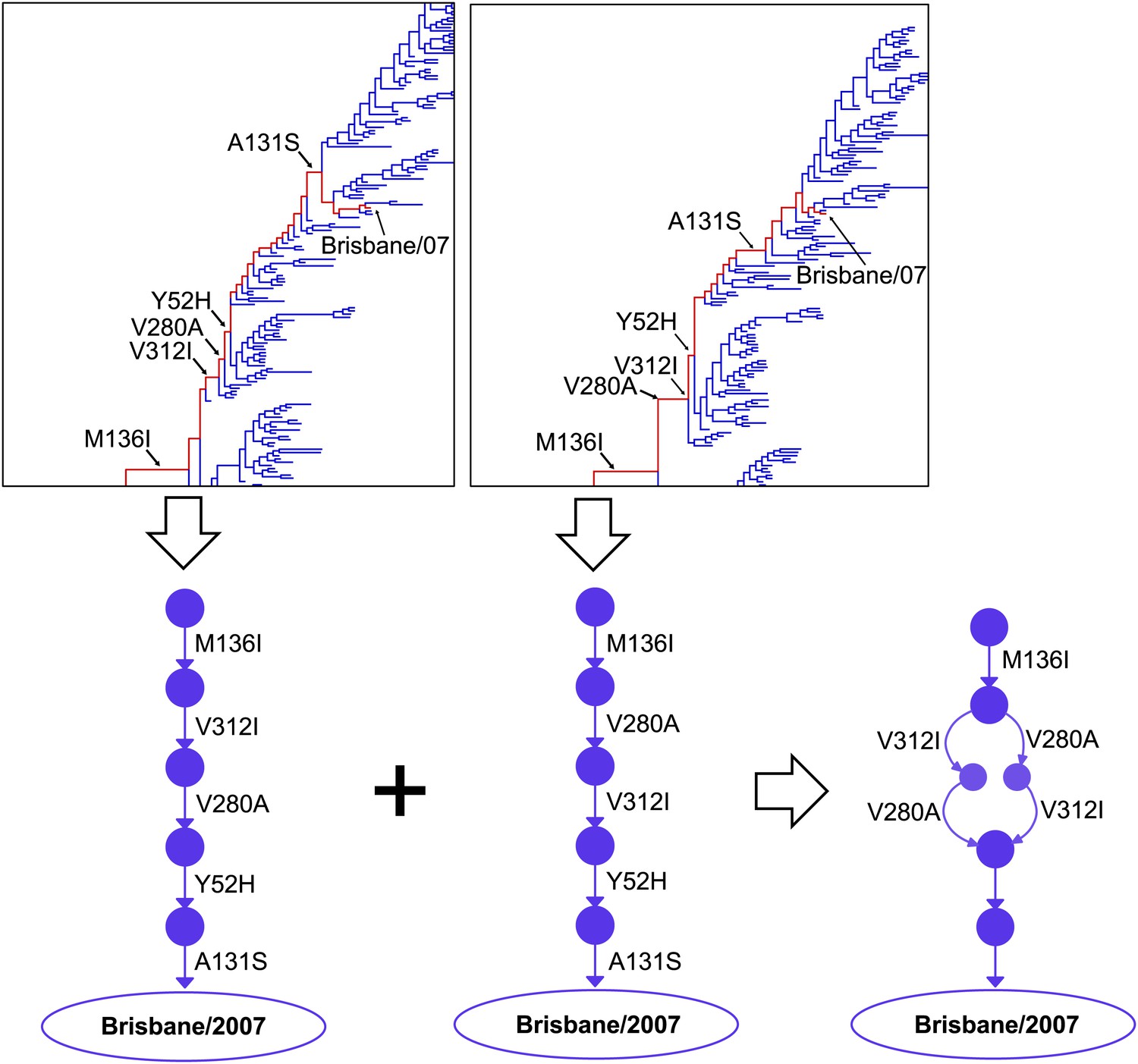
Construction of the evolutionary trajectory.
We estimated the joint posterior distribution of phylogenetic trees with mutations mapped on the branches in BEAST. Portions of two example trees with mutations are shown at the top. Tracing along the line of descent (shown in red) gives a specific sequence of mutations; we can use these to reconstruct the corresponding path through sequence space (a directed graph through the space of all possible protein sequences), as shown at the bottom of the figure. We summarize samples of individual directed graphs drawn from the posterior distribution, with the posterior probabilities of specific points and connections equaling the fraction of the samples that correspond to directed graphs containing these points and connections. We represent visually the probabilities of the points and connections by varying the areas of the circles/widths of the lines, and their intensities. At bottom right is the result of integrating just the two partial paths shown in this figure. The full evolutionary trajectory in Figure 2 represents the integration of many full paths.
Figure 2—figure supplement 2
Dates at which mutations fixed along the evolutionary trajectory in Figure 2A.
This plot shows the posterior median and 80% Bayesian credible intervals for the dates. In many cases the date intervals for two mutations overlap even though in the evolutionary trajectory one mutation can be placed before another. This is because the dates are correlated in the individual tree-mutation samples drawn from the posterior, so that the difference in times between two mutations tend to be more precisely defined than the absolute date ranges. Two mutations (red) tend to arise on branches going from the common ancestor to Aichi/1968, while the remaining mutations (blue) tend to arise on branches from the common ancestor to Brisbane/2007. The code used to generate this figure is in Figure 2—source code 1.
Figure 3 with 3 supplements

Three mutations are strongly deleterious when introduced individually into the parent Aichi/1968 NP, despite eventually becoming fixed along the evolutionary trajectory without apparent negative effect.
(A) The transcriptional activity for the high-confidence evolutionary intermediates, quantified using a GFP reporter (Figure 3—figure supplement 1). Activity is scaled so that the parent Aichi/1968 NP has an activity of one. The numeric labels of the evolutionary intermediates match those used in Figure 2A. (B) The change in activity caused by introducing each mutation individually into the parent Aichi/1968 NP. The deleterious effects on activity caused by L259S, R348G, and V280A are not caused by the genetic background of influenza polymerase proteins, as these three mutants are impaired regardless of whether the polymerase proteins are derived from Nanchang/1995, Aichi/1968, or Brisbane/2007 (Figure 3—figure supplement 3). (C) All three of the individual mutations that reduce activity also impair growth, yet there is no defect in the growth of viruses carrying the NPs of the first high-confidence evolutionary intermediates in which these individually deleterious mutations were actually fixed. Viral growth is quantified as described in Figure 3—figure supplement 2. Numerical data are in Figure 3—source data 1–3.
-
Figure 3—source data 1
Summary of transcriptional activity data (mean and standard error) for all variants from this study.
The means and standard errors are computed from at least triplicate assays, and are reported standardized so the value for Aichi/1968 NP is at 1. The data are provided in a CSV file.
- https://doi.org/10.7554/eLife.00631.009
-
Figure 3—source data 2
Summary of viral growth data (mean and standard error) for all variants for which this property was measured in this study.
The means and standard errors are computed from at least triplicate assays, and are reported standardized so the value for Aichi/1968 NP is at 1. The data are provided in a CSV file.
- https://doi.org/10.7554/eLife.00631.010
-
Figure 3—source data 3
Transcriptional activity data in the alternative polymerase genetic backgrounds shown in Figure 3—figure supplement 3.
The data are provided in a CSV file.
- https://doi.org/10.7554/eLife.00631.011
Figure 3—figure supplement 1
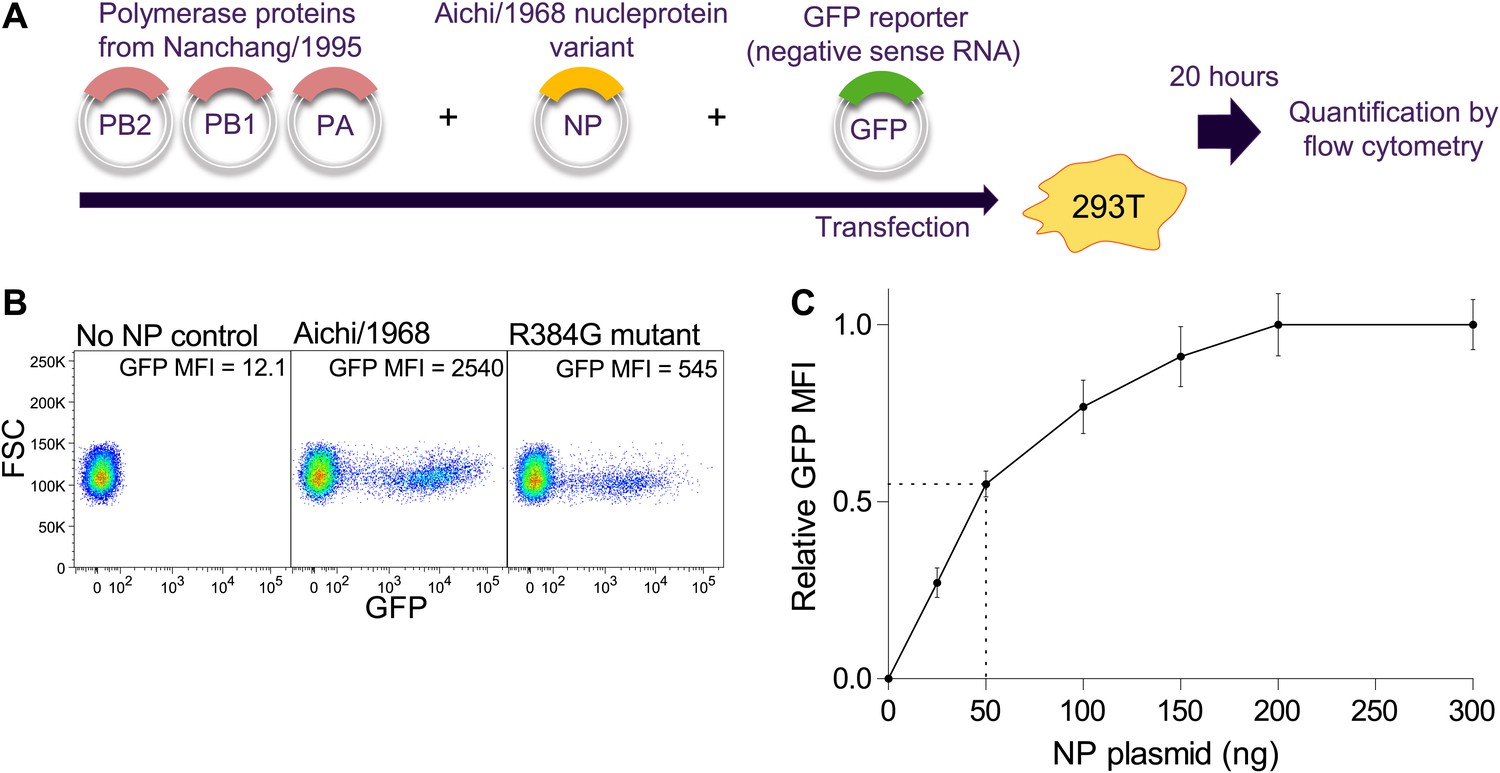
Schematic of NP transcriptional activity assay.
(A) 293T cells are co-transfected with plasmids encoding PB2, PB1, and PA from Nanchang/1995, an NP variant, and a reporter plasmid expressing a negative-sense influenza viral-RNA encoding GFP. The four influenza proteins (PB2, PB1, PA, and NP) collaborate to transcribe the reporter GFP vRNA into mRNA, which is translated into protein, causing the cells to fluoresce. Activity is quantified by flow cytometry as the mean fluorescence intensity (MFI) above the background of control cells that receive no NP, relative to the activity of three wild-type Aichi/1968 NP replicates. (B) Example flow cytometry plots. (C) Total transcriptional activity as a function of the amount of transfected NP plasmid. The PB2, PB1, PA, and GFP reporter plasmids are transfected at 200 ng each. The NP plasmid was varied from 0 to 300 ng, and the fluorescence was quantified. At low levels of NP plasmid, the signal increases with increasing plasmid concentration, but at high levels the signal is saturated. We performed our assays with 50 ng of NP plasmid, which is near the midpoint of the curve—with this amount of plasmid, the signal changes in a sensitive manner with amount of NP.
Figure 3—figure supplement 2
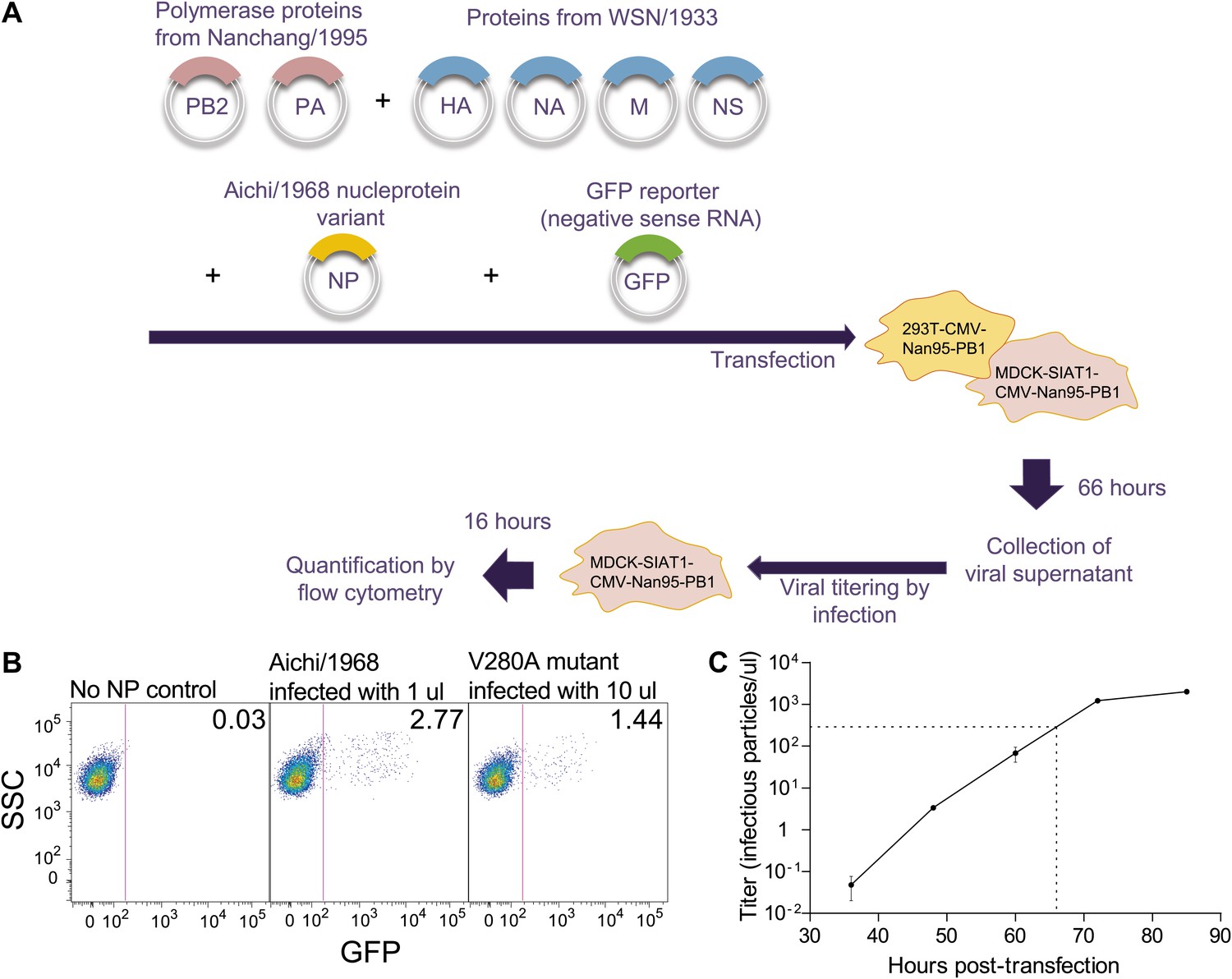
Schematic of viral growth assay.
(A) Co-cultures of 293T-CMV-Nan95-PB1 and MDCK-SIAT1-CMV-Nan95-PB1 cells were co-transfected with reverse-genetics plasmids encoding PB2/PB1/PA from Nanchang/1995, an NP variant, HA/NA/M/NS from A/WSN/1933, and a reporter plasmid expressing a negative-sense influenza viral-RNA encoding GFP with flanking sequences from PB1. A media change to low-serum media was performed after 20–24 hr, and at 66 hr post-transfection the viral supernatant was harvested. Dilutions of the supernatant were used to infect fresh MDCK-SIAT1-CMV-Nan95-PB1 cells, and 16 hr post-infection, the viral titer was determined by quantifying the fraction of GFP positive cells by flow cytometry using the Poisson formula. The titers are standardized to the average of three Aichi/1968 NP controls. (B) Example flow cytometry plots. The cells are at 105 per well, so the computed titers for these example plots are −105 × ln(1 − 0.0277) = 2809 infectious particles per μl for Aichi/1968, and −105 × ln(1 − 0.0144)/10 = 145 infectious particles per μl for V280A. (C) Viral titers in the supernatant at the indicated times post-transfection for the Aichi/1968 NP. Based on this timecourse, we chose to determine titers for all of the tested variants at 66 hr, when the virus is near but not yet at peak titers. Typically at this time point we would observe titers for the Aichi/1968 NP of around 103 infectious particles per μl; however, titers varied somewhat from day-to-day, which is why the values for each assay were standardized to three Aichi/1968 NP controls run on that same day.
Figure 3—figure supplement 3
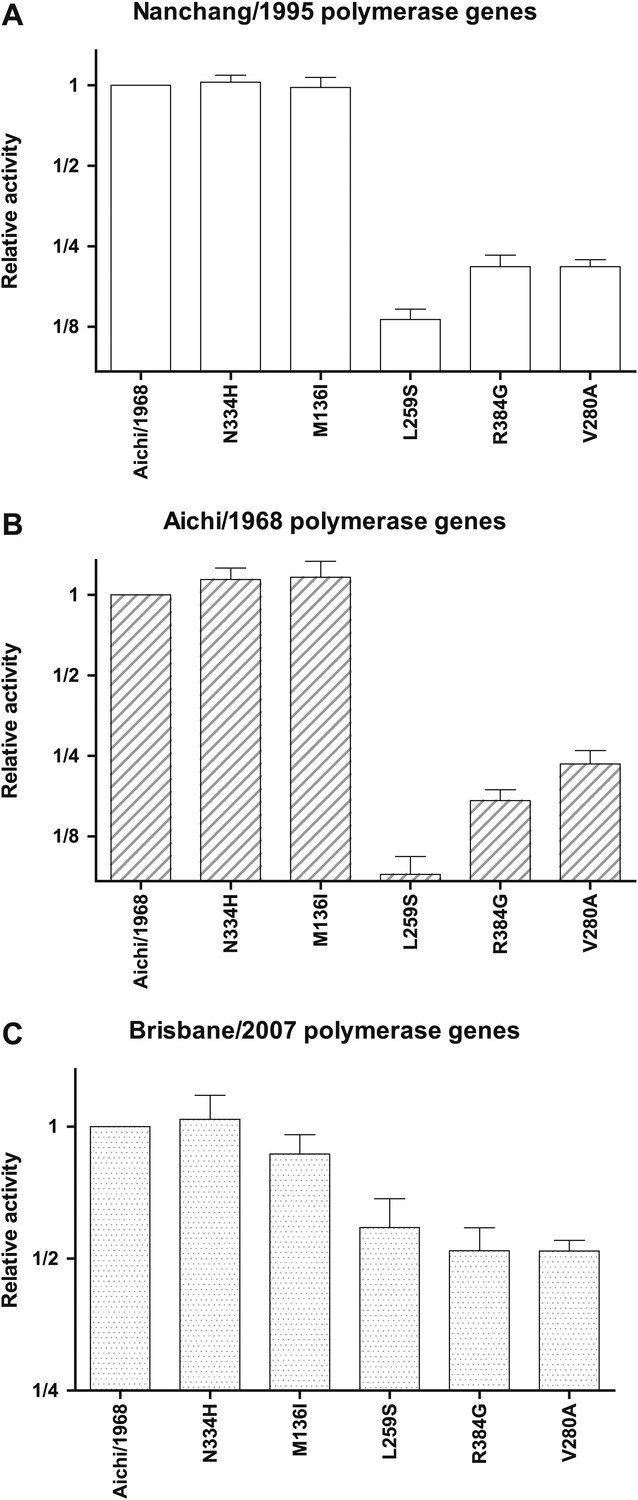
Effects of key NP mutations on transcriptional activity in different polymerase genetic background of the viral polymerase genes.
The six indicated NP variants were tested in the transcriptional activity assay with (A) the PB2, PB1, and PA genes derived from Nanchang/1995 as in all of the assays shown in Figure 3; (B) with these polymerase genes derived from Aichi/1968; or (C) with these polymerase genes derived from Brisbane/2007. In the last case (the Brisbane/2007 polymerase genes), the magnitude of the activity reduction is somewhat less, but all three mutants are still clearly impaired. In each plot, the activity is normalized relative to that observed for the parental Aichi/1968 NP in that polymerase background, and so the different panels have y-axis units that cannot be compared across panels. The NP plasmid amount is the same (50 ng) in all three backgrounds, but note that although this makes the readings at the middle of the dose-response curve for the Nanchang/1995 polymerase background (Figure 3—figure supplement 1), this may not be true for the other polymerase backgrounds. As can be seen from the plots, L259S, R384G, and V280A are impaired in all three backgrounds, suggesting that the mutational effects are intrinsic to NP and not due to interactions with polymerase genetic background. Data are in Figure 3—source data 3.
Figure 4
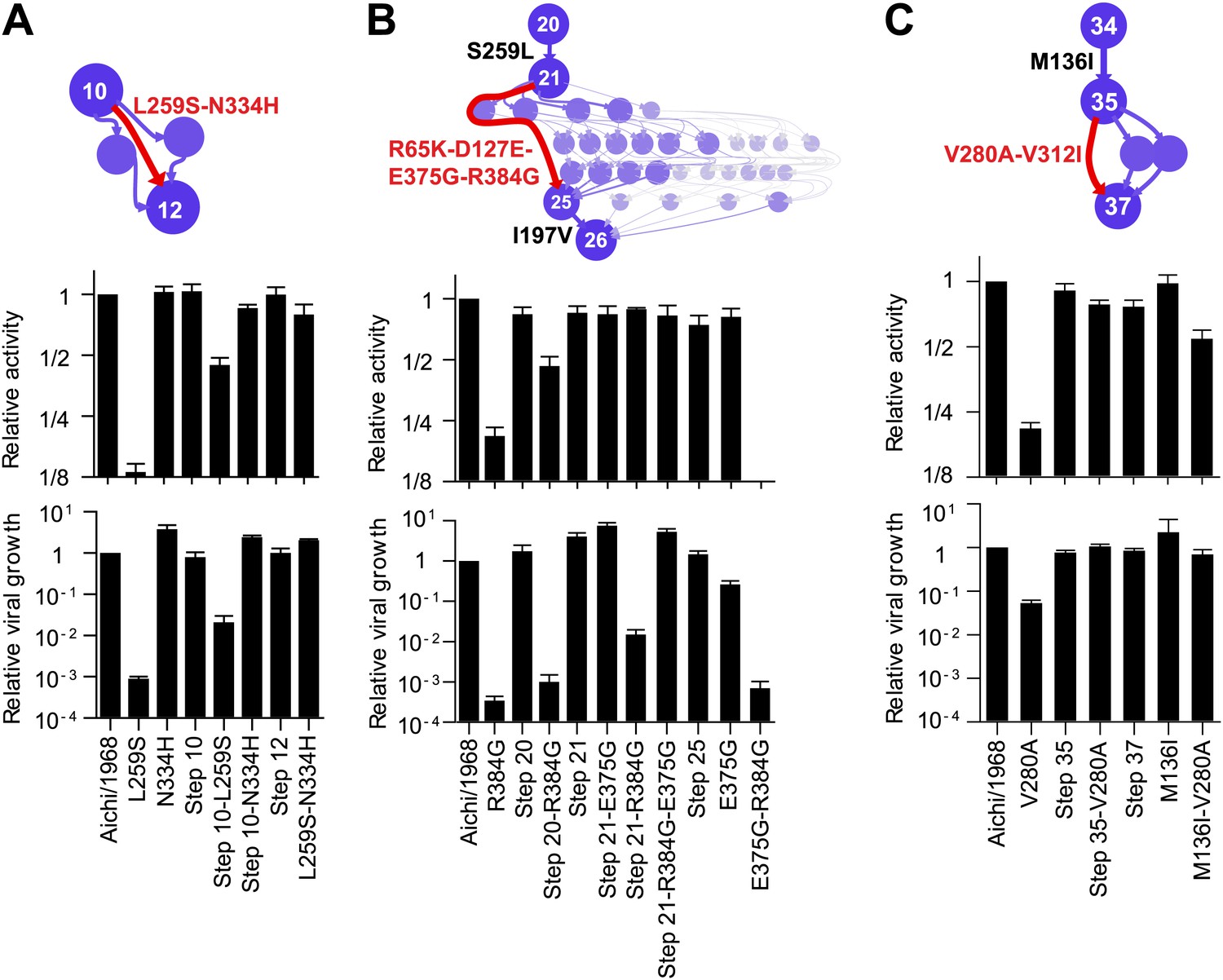
Effects of the individually deleterious mutations in the evolutionary intermediates in which they occurred.
(A) L259S impairs the transcriptional activity and viral growth of both the parent Aichi/1968 and the evolutionary intermediate Step 10, but is rescued by N334H in both backgrounds. N334H alone has little effect on activity or growth in either background. The actual evolutionary trajectory involved the fixation of L259S and N334H in an unknown order. (B) R384G impairs activity and ablates growth of Aichi/1968, but has no effect on activity and a reduced adverse effect on growth in the high-confidence evolutionary intermediate (Step 21) in which it and several other mutations occurred in an unknown order. Addition of E375G to Step 21 with R384G fully rescues viral growth, but E375G alone worsens the impact of R384G. The reversion of L259S that preceded Step 21 plays an important role in enabling R384G, as the evolutionary intermediate without this reversion (Step 20) is more negatively affected by R384G. (C) V280A is deleterious in Aichi/1968 but not in the Step 35 evolutionary intermediate in which it actually occurred. M136I, which precedes V280A, largely rescues its effect.
Figure 5 with 1 supplement
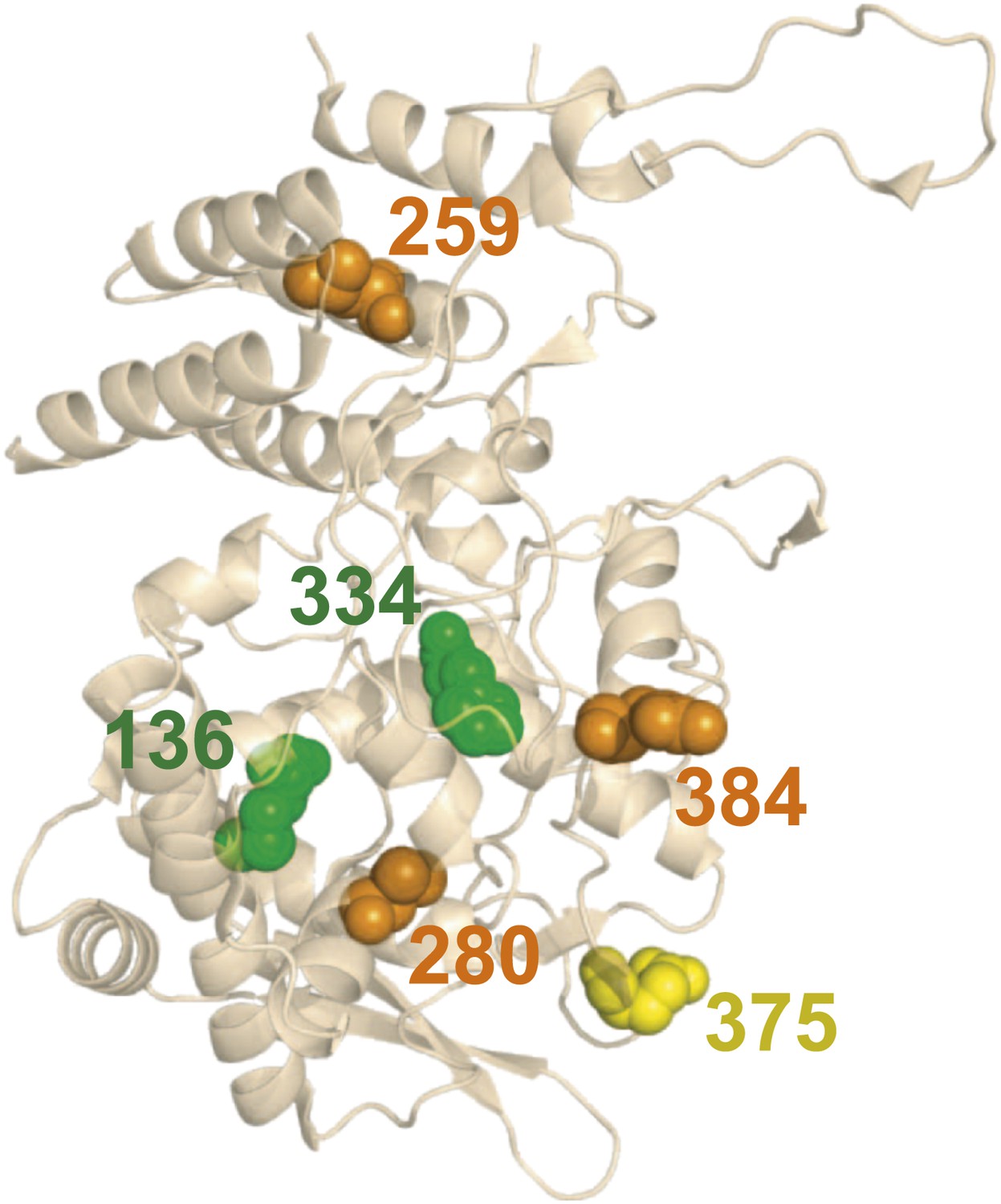
There is no obvious structural basis for the observed epistasis, as none of the epistatically interacting mutations are in contact in the solved crystal structures of NP.
Shown above is one monomer from PDB structure 2IQH; the mutations are also not in contact in any of the known oligomeric structures (Figure 5—figure supplement 1). The sites of the three individually deleterious mutations are in orange, those of the rescuing mutations are in green, and the site of E375G (which can rescue R384G depending on genetic background) is in yellow.
Figure 5—figure supplement 1
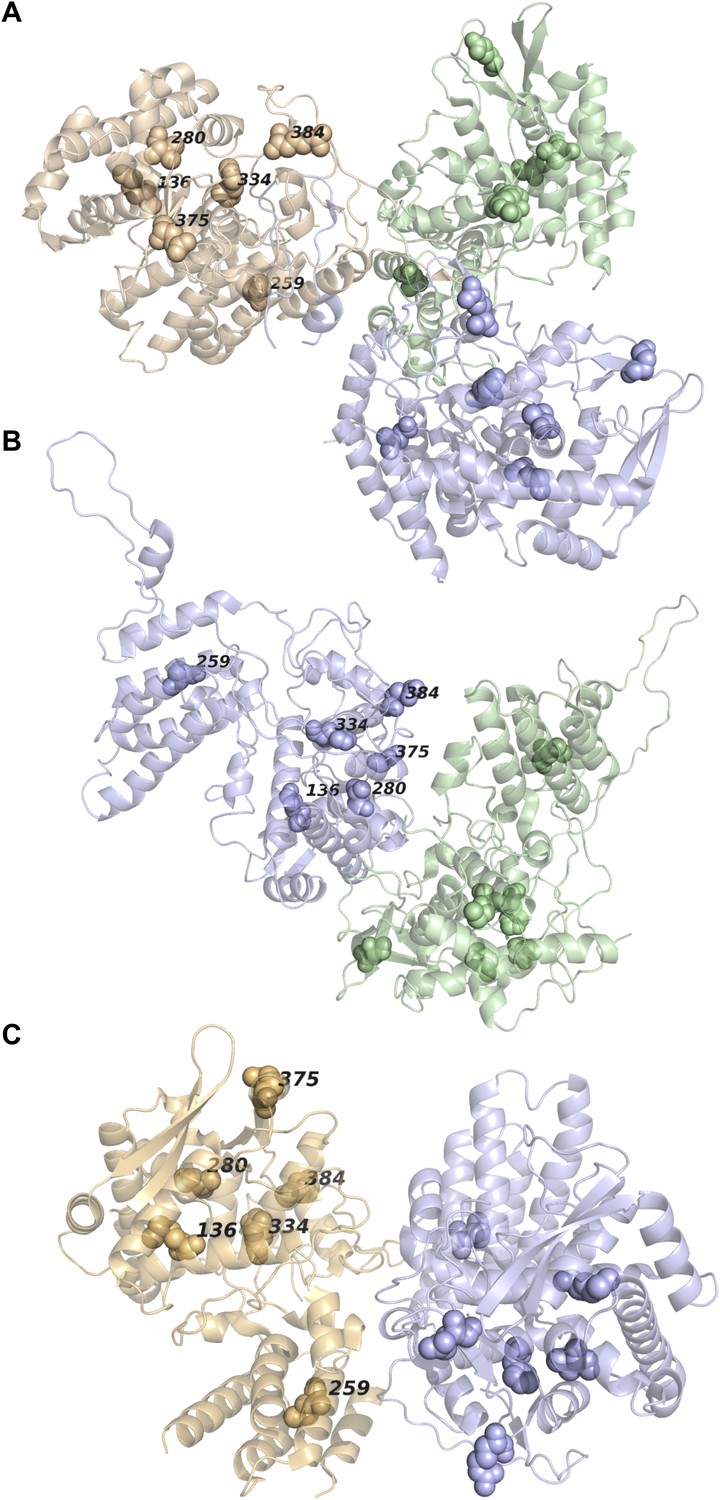
None of the epistatically interacting mutations are in contact in the known oligomeric structures of NP.
(A) Sites of the mutations (spheres) in the trimeric structure of NP from PDB 2IQH. (B) Sites of the mutations in the dimeric structure from PDB 2Q06. (C) Sites of the mutations in the dimeric structure from PDB 3MIR.
Figure 6 with 8 supplements

The epistasis correlates with mutational effects on NP stability.
(A) and (B) N334H rescues activity and mostly rescues viral growth of all three individually deleterious mutations. (C) Western blot showing that the three individually deleterious mutations all reduce NP levels in transfected cells relative to an mCherry control expressed from an IRES in the same plasmid; this effect is counteracted by N334H. (D) Quantification of NP levels from triplicate Western blots (Figure 6—figure supplement 1). (E) The deleterious mutations decrease and N334H increases the stability of NP, as measured by thermal denaturation of purified protein monitored by circular dichroism (Figure 6—figure supplements 2–6 and Figure 6—source data 1). Figure 6—figure supplement 7 shows that M136I, which precedes V280A in the natural evolution, and is modestly stabilizing (Figure 6—figure supplements 4–7), also partially rescues the levels of V280A NP in transfected cells and the activity of all three individually deleterious mutations. Together, the two stabilizing mutations N334H and M136I can rescue the activity of combinations of the individually deleterious mutations (Figure 6—figure supplement 8).
-
Figure 6—source data 1
A table of all of the melting temperatures and the changes in stability relative to Aichi/1968, in CSV format.
- https://doi.org/10.7554/eLife.00631.019
Figure 6—figure supplement 1
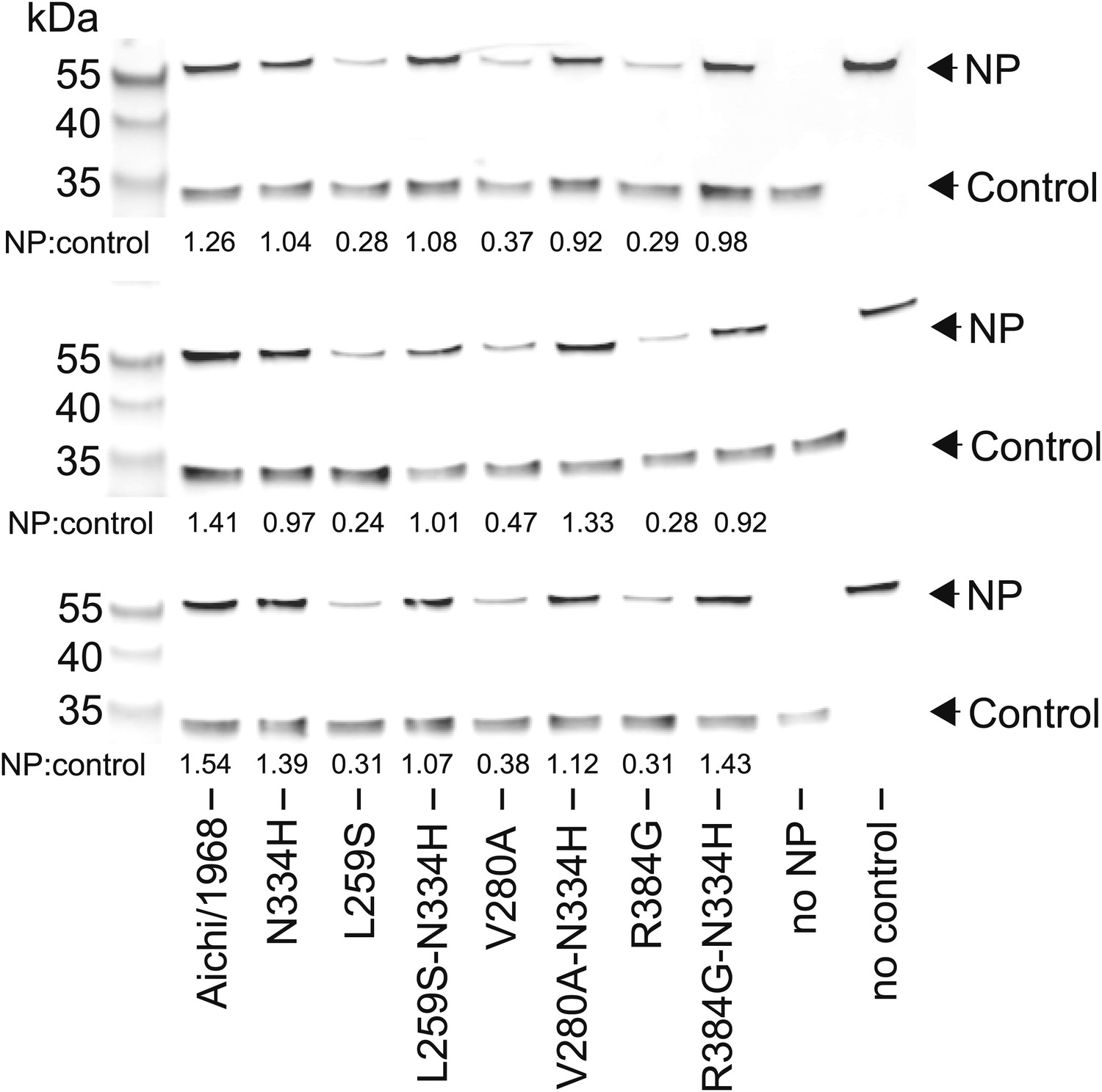
The full set of triplicate Western blots used to quantify the NP levels in transfected cells.
NP levels were quantified by transfecting 293T cells with a FLAG-tagged NP on a plasmid also containing a FLAG-tagged mCherry under an IRES element. The mCherry therefore serves as a transfection/loading control. Three triplicate blots each from independent transfections were performed to quantify NP levels. For each blot, the ratio of NP to mCherry was quantified, and is shown below the band. These ratios were then standardized relative to that for the Aichi/1968 NP, and the averages of the triplicates computed. Note that all three blots confirm that L259S, V280A, and R384G are present at reduced levels—and that N334H can rescue this defect.
Figure 6—figure supplement 2
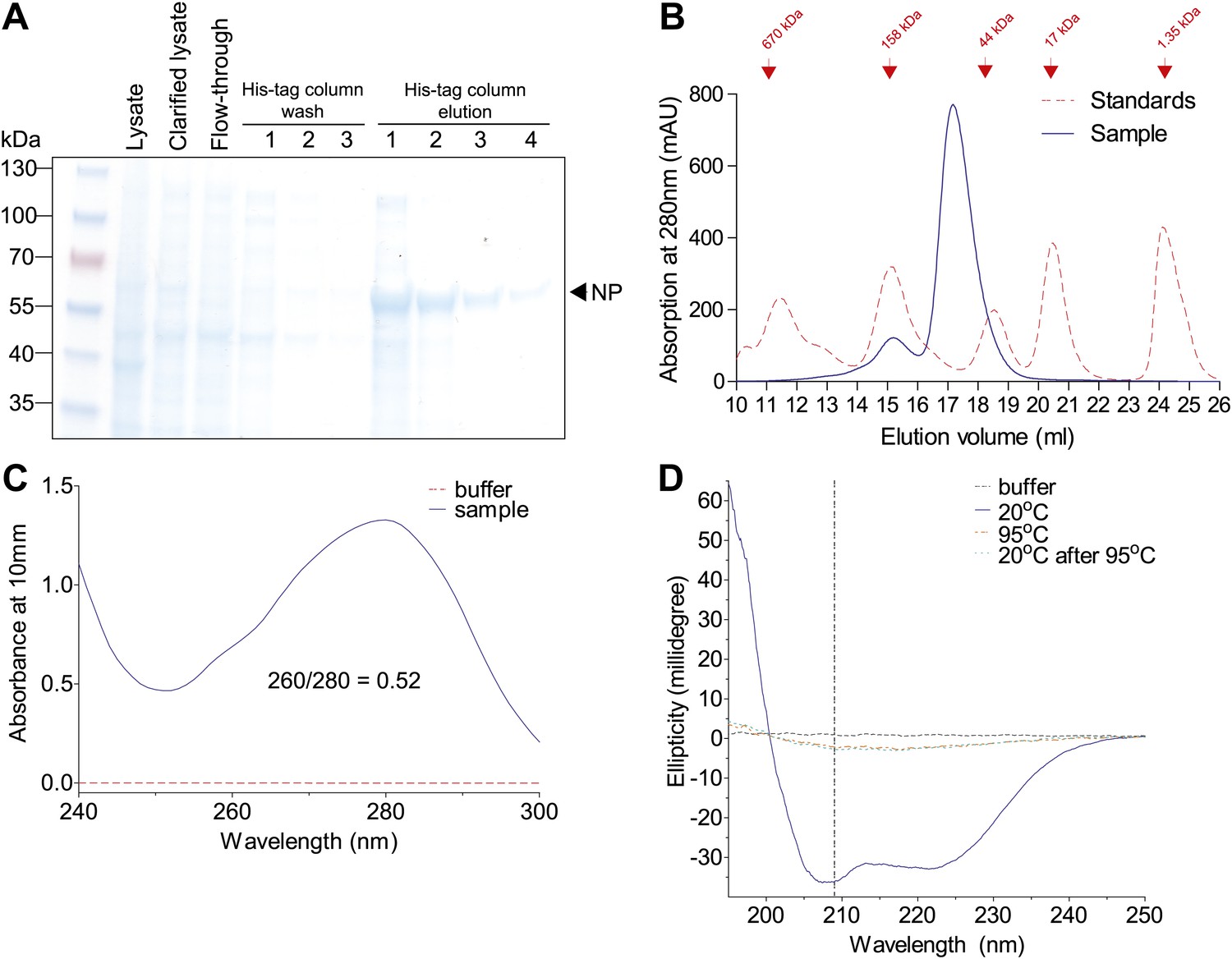
Purification of Aichi/1968 NP with deletion of residues 2–7, mutation R416A, and a C-terminal 6-His tag (expected molecular weight 56.6 kDa).
These modifications were necessary to obtain monomeric RNA-free in NP in a CD-compatible buffer. The other NP variants behaved similarly by the measures in this figure. (A) NP was at high purity after elution off a cobalt column, as judged by SDS-PAGE. (B) NP was further purified over a Superdex 200 size-exclusion column. It eluted at roughly the expected size. (C) After size-exclusion, NP had a 260/280 nm ratio much less than 1, indicating protein largely free of RNA. (D) NP at 20°C exhibits a CD spectrum characteristic of an alpha-helical protein. This spectrum disappears after heating, and does not reappear after cooling back to 20°C (indicating irreversible denaturation). We monitored unfolding at 209 nm (dashed vertical line). All CD was performed at a NP concentration of 5 μM with a cuvette path length of 0.1 cm.
Figure 6—figure supplement 3
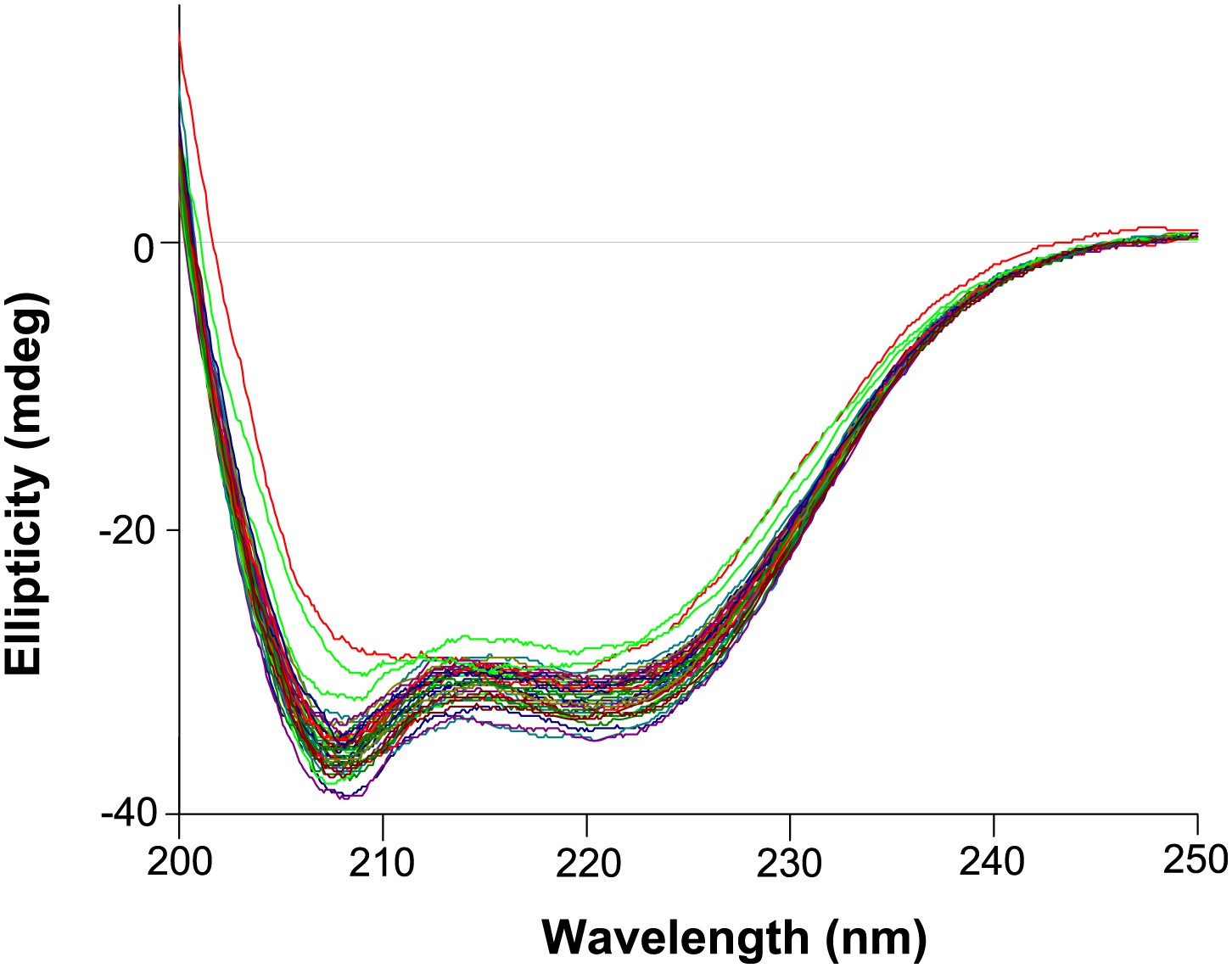
Circular dichroism wavelength scans for all variants that were tested (those with thermal melts shown in Figure 6—figure supplements 4–6).
All variants exhibit similar CD spectra characteristic of an alpha-helical protein. The magnitude of the spectra are also similar, indicating all protein variants are indeed at roughly the same concentration (which was measured at 5 μM as judged by absorbance at 280 nm).
Figure 6—figure supplement 4

The first 15 thermal denaturation curves.
NP variants were thermally denatured with unfolding monitored by ellipticity at 209 nm, at a scan rate of 2°C per minute. Melting temperatures were obtained from sigmoidal fits over the range 20–60°C.
Figure 6—figure supplement 5
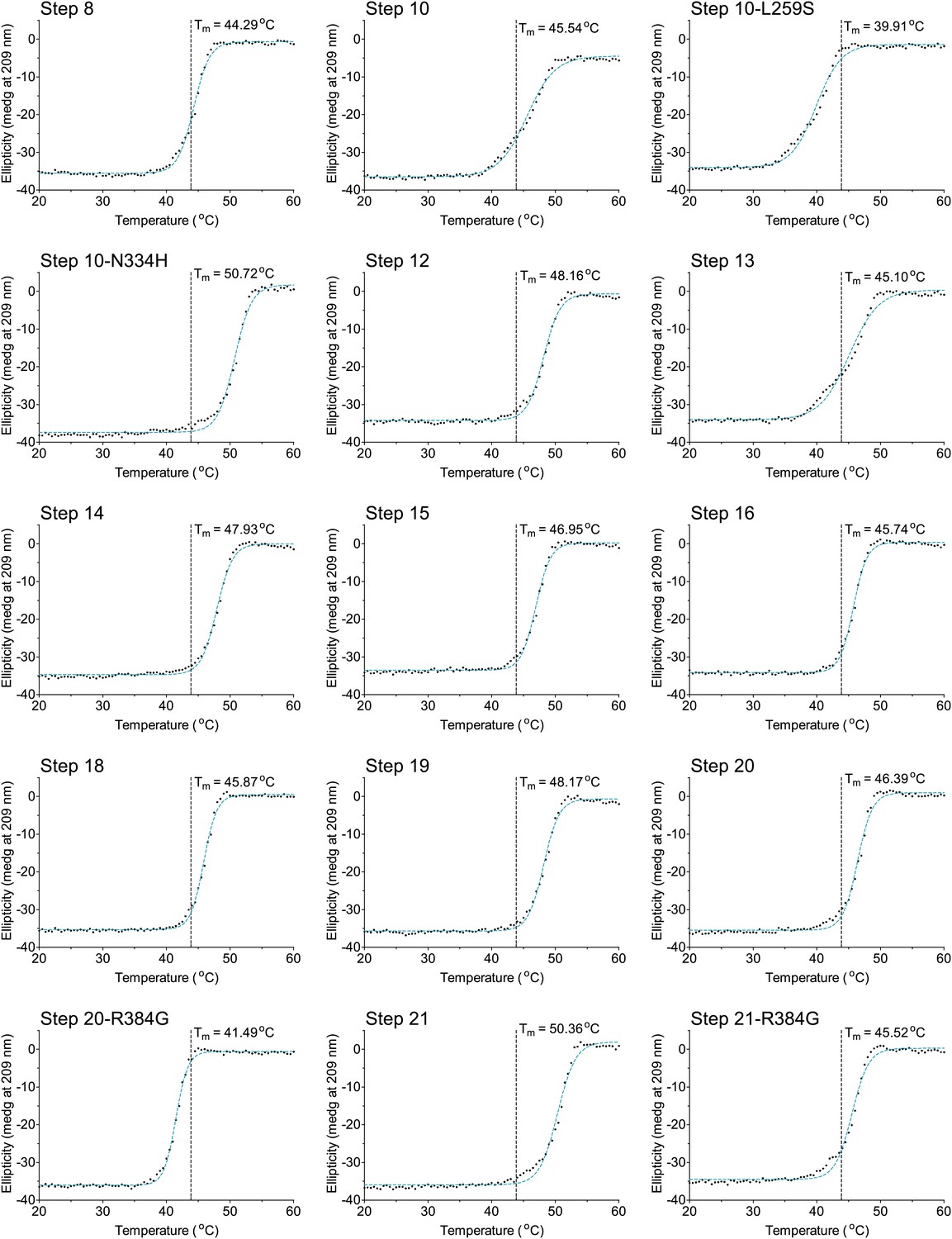
The second 15 thermal denaturation curves.
NP variants were thermally denatured with unfolding monitored by ellipticity at 209 nm, at a scan rate of 2°C per minute. Melting temperatures were obtained from sigmoidal fits over the range 20–60°C.
Figure 6—figure supplement 6
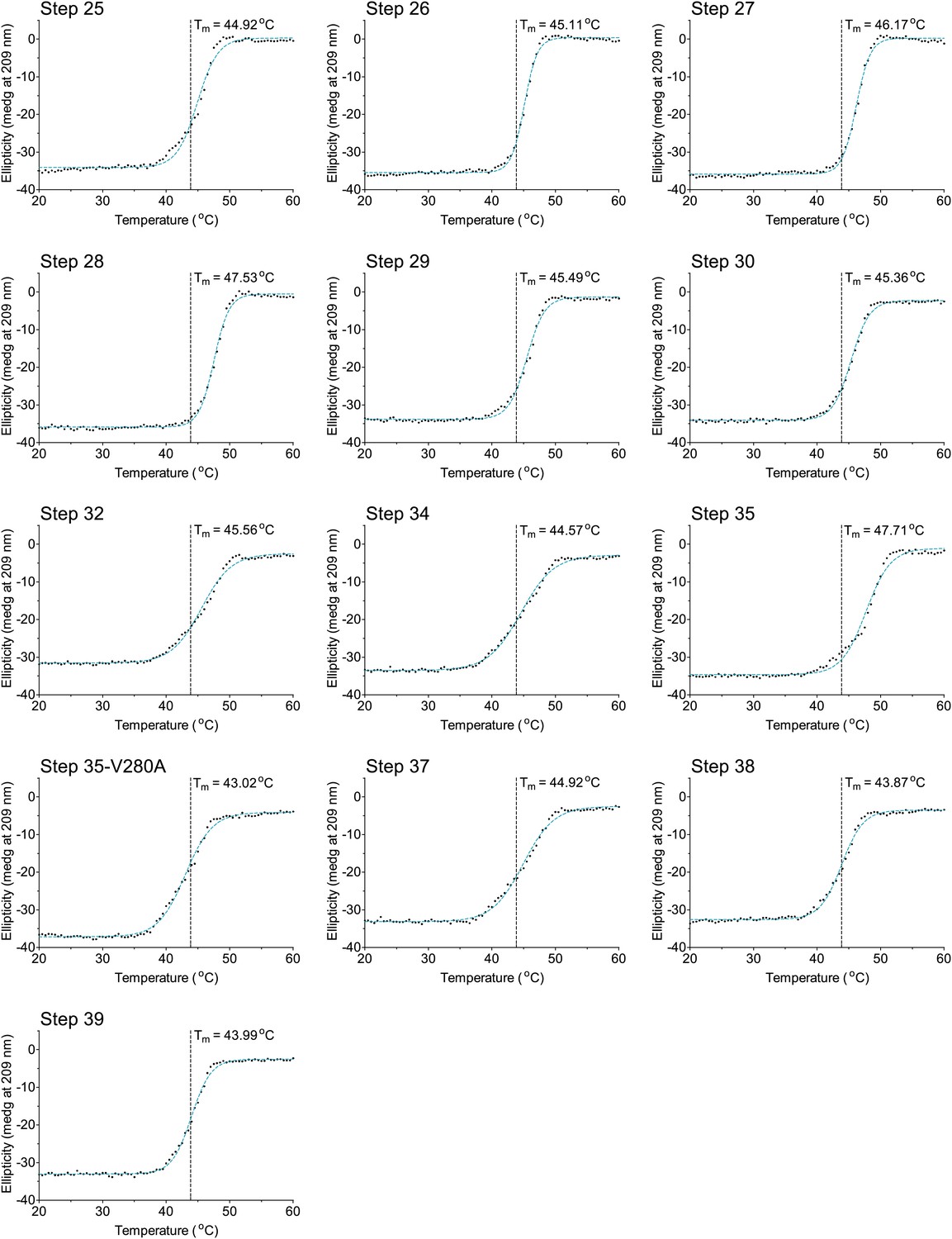
The last 13 thermal denaturation curves.
NP variants were thermally denatured with unfolding monitored by ellipticity at 209 nm, at a scan rate of 2°C per minute. Melting temperatures were obtained from sigmoidal fits over the range 20–60°C.
Figure 6—figure supplement 7
M136I partially rescues the activity of the three individually deleterious mutations, and mostly rescues protein levels for V280A.
M136I preceded V280A in the natural evolution of NP. Note that Figure 6—figure supplements 4–6 shows that M136I is modestly stabilizing. (A) In the activity assay, M136I alone has no effect, but it can partially rescue the activity of all three individually deleterious mutations. (B) Western blots showing the levels of NP protein relative to the mCherry control. M136I is present at wild-type levels, whereas levels of V280A are reduced. Adding M136I to V280A largely rescues the NP levels. Three triplicate Western blots from independent transfections are shown. The numbers below the bands represent the ratio of the NP band intensity to the mCherry band intensity. (C) Quantification of the triplicate Western blots.
Figure 6—figure supplement 8
Activities for NPs with combinations of the stabilizing and destabilizing mutations.
Various combinations of the three epistatically constrained destabilizing mutations (L259S, V280A, R384G) and the stabilizing mutations (N334H, M136I) were introduced into the Aichi/1968 NP and the total transcriptional activity was quantified. The final NP variant at the end of the trajectory contained V280A, R384G, N334H, and M136I—but not L259S, as this mutation reverts before the occurrence of R384G. An NP with just those four mutations exhibits wild-type levels of activity.
Figure 7 with 1 supplement

Most of the epistasis that we identified in NP’s evolution can be explained by counterbalancing stabilizing and destabilizing mutations.
(A) The relationship between NP stability and transcriptional activity for all variants for which both properties were measured. As long as the stability is greater than a threshold around 42°C, changes in stability are neutral with respect to activity. Below this threshold, activity declines sharply with decreasing stability. (B) The relationship between viral growth and NP stability exhibits a similar behavior. The exception is that growth of variants with R384G is fully rescued only by combining a stabilizing mutation with E375G (Figure 4B), an effect that we hypothesize is related to the electrostatic charge on one of NP’s surfaces (Figure 7—figure supplement 1). (C) The dynamics of protein stability during NP evolution. Shown are the measured stabilities for evolutionary intermediates from the trajectory in Figure 2A. The lines along the y-axis at the far left show the stabilities of the five indicated individual point-mutants of the Aichi/1968 NP. Although the destabilizing mutations L259S, R384G, and V280A are deleterious to the Aichi/1968 parent, during evolution they are counterbalanced by stabilizing mutations. In the top panels, selected points are labeled with the NP variant name; the full data plotted in this figure are in Figure 7—source data 1.
-
Figure 7—source data 1
The activity, viral growth, and stability data shown in Figure 7.
The data are provided in CSV format.
- https://doi.org/10.7554/eLife.00631.029
Figure 7—figure supplement 1
We hypothesize that E375G helps counteract R348G by maintaining the electrostatic charge on one of NP’s surfaces.
As shown in Figure 4B, the activity of the destabilizing mutation R384G is fully rescued by a stabilizing mutation, such as N334H in Aichi/1968 or reversion of L259S in Step 21. However, full rescue of viral growth also requires E375G. E375G is destabilizing, and alone does not rescue growth or activity of R384G. We hypothesize that rescue of growth of R384G requires two events. The first is counteraction of NP destabilization. The second is some effect specific to E375G that is apparent in growth but not transcriptional activity. E375G (which causes loss of a negative charge) counterbalances the loss of a positive charge due to R384G. The image shows an electrostatic surface (PyMol, PDB 2IQH). R384 and E375 are on the same surface, contributing positive and negative charges. Mutating both to neutral glycine maintains the net charge. Perhaps the charge of this surface is important for interaction with other protein partners late in the life cycle (after RNA transcription), explaining why E375G is important for growth but not activity.
Figure 8 with 3 supplements
The three epistatically constrained destabilizing mutations occur at sites significantly enriched in human CTL epitopes.
Distributions of the numbers of experimentally characterized epitopes per residue for all sites or sites that experienced mutations along the evolutionary trajectory are in blue and red, respectively. Sites 259, 280, and 384 are in significantly more epitopes than three random positions from all sites (p=0.001) or the mutated sites (p=0.004); however, three random positions from the mutated sites are not in significantly more epitopes than three random positions from all sites (p=0.157). Epitopes with experimentally characterized CTL responses were mined from the Immune Epitope Database (Figure 8—source code 1 and Figure 8—figure supplement 1). The primary citation and summary information for epitopes involving sites 259, 280, and 384 are in Figure 8—source data 1. Similar results are obtained if CTL epitopes are instead predicted computationally (Figure 8—figure supplements 1, 2). A dN/dS analysis is inconclusive about whether the three sites are under positive or negative selection, probably due to lack of sequence data (Figure 8—figure supplement 3 and Figure 8—source code 2).
-
Figure 8—source data 1
Literature-characterized NP human CTL epitopes that include residues 259, 280, or 384 and contain sequences conserved or nearly conserved in Aichi/1968.
The epitopes are listed in a TXT file.
- https://doi.org/10.7554/eLife.00631.032
-
Figure 8—source code 1
The input data files and the custom Python scripts used to identify human CTL epitopes in NP.
The code and data are provided as a ZIP file, which contains a README text file that describes the contents in more detail.
- https://doi.org/10.7554/eLife.00631.033
-
Figure 8—source code 2
The data and source code used for the dN/dS analysis.
The code and data are provided in a ZIP file; a README file explains the contents.
- https://doi.org/10.7554/eLife.00631.034
Figure 8—figure supplement 1
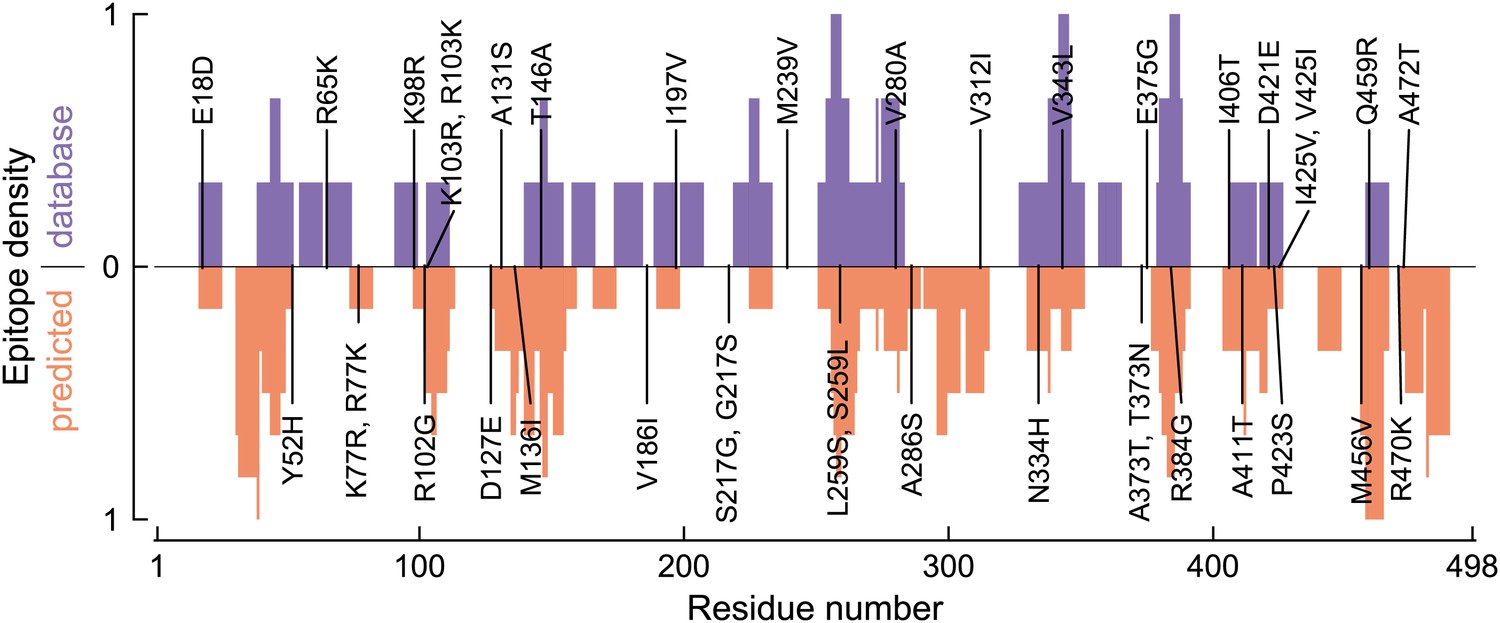
Distribution of human CTL epitopes along the NP primary sequence.
This plot shows the number of CTL epitopes for each residue in NP. Sites of specific mutations along the evolutionary trajectory are indicated. The top blue bars show the number of experimentally characterized CTL epitopes as mined from the Immune Epitope Database. The bottom red bars show the number of CTL epitopes predicted by NetCTLpan1.1, a computational epitope prediction method, applied to all HLA supertypes. In both cases, the epitope density is scaled to have a minimum of 0 and a maximum of 1.
Figure 8—figure supplement 2
The three epistatically constrained mutations are at sites significantly enriched in CTL epitopes as predicted by a computational epitope prediction program NetCTLpan1.1.
The blue curve shows the distribution of epitopes per site for all sites in NP, while the red curve shows the distribution of epitopes per site for the sites in NP that substituted along the evolutionary trajectory. The sites of the three epistatically constrained mutations (280, 259, and 384) are indicated. These three sites contain significantly more epitopes than would be expected from three randomly chosen positions from all sites (p=0.008) or from the mutated sites (p=0.028). A random selection of three positions from all mutated sites contains significantly more epitopes than a random selection of three positions from all sites (p=0.011).
Figure 8—figure supplement 3
It is largely inconclusive whether the sites of the three epistatically constrained mutations are under positive or negative selection as quantified by dN/dS values.
Human H3N2 influenza NP sequences from 1968 to 2011 were analyzed for positive selection using the dN/dS analysis implemented in FUBAR (Figure 8—source code 2). The plots show the posterior probability that sites are under (A) negative or (B) positive selection. At reasonable levels of significance, we can conclude that site 384 is not under negative selection and is probably under positive selection. For sites 259 and 280, there is not strong evidence for either positive or negative selection. We note that the lack of decisive results may be due largely to insufficient sequence data—although we used hundreds of NP sequences, they are closely related with a trunk-like phylogeny as shown in Figure 4B, which will reduce the power of the test. In addition, we note that mutations which are beneficial to viral CTL escape may still fix slowly (reducing their sites’ dN/dS values) even if they are under positive selection due to the types of epistatic constraint described in this paper.
Author response image 1

Additional files
-
Supplementary file 1
The full vRNAs in the reverse-genetics plasmids used in this study.
A text file giving the vRNAs inserted between the RNA polymerase I promoter and terminator in the reverse-genetics plasmids pHWAichi68-NP, pHWNan95-PB2, pHWNan95-PB1, pHWNan95-PA, pHWAichi68-PB2, pHWAichi68-PB1, pHWAichi68-PA, pHWBR07-PB2, pHWBR07-PB1, pHWBR07-PA, pHW184-HA, pHW186-NA, pHW187-M, pHW188-NS, and pHH-PB1flank-eGFP.
- https://doi.org/10.7554/eLife.00631.038
-
Supplementary file 2
The protein-coding sequences of all NP variants used in this study.
A text file giving the coding sequences for the NP variants used in this study.
- https://doi.org/10.7554/eLife.00631.039
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Stability-mediated epistasis constrains the evolution of an influenza protein
eLife 2:e00631.
https://doi.org/10.7554/eLife.00631




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}