Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation
- Howard Hughes Medical Institute, University of Washington, United States
- Janelia Farm Research Campus, Howard Hughes Medical Institute, United States
Figures
Figure 1

Purification and electron microscopy of the AKAP18γ–PKAholo complex.
(A) SDS-PAGE and Coomassie staining of purified individual complex components. (B) Size-exclusion chromatography (SEC) trace for purification of the assembled AKAP18γ–PKAholo complex. Fractions at the leading edge of the peak (indicated by gray bar) were chosen for further analysis. (C) SDS-PAGE (left), western blot (middle) and native gel electrophoresis (right) obtained from the SEC peak elution fraction (arrow in B). (D) Electron micrograph of the negatively stained AKAP18γ–PKAholo complexes (circles). Triangles indicate the three major densities of the AKAP18γ–PKAholo complex. Inset, shows labeling with a gold nanoparticle (arrow) conjugated to an AKAP18γ-streptavidin moiety (arrow). (E) Left, enlarged images of individual AKAP18γ–PKAholo complexes (denoted by asterisks in D). (E) Right, highlighted outline (yellow) of particle shapes. (F) Projection averages of the AKAP18γ–PKAholo complex classified into distinct conformations using ISAC (Yang et al., 2012). Unlabeled scale bars represent 25 nm.
Figure 2 with 2 supplements
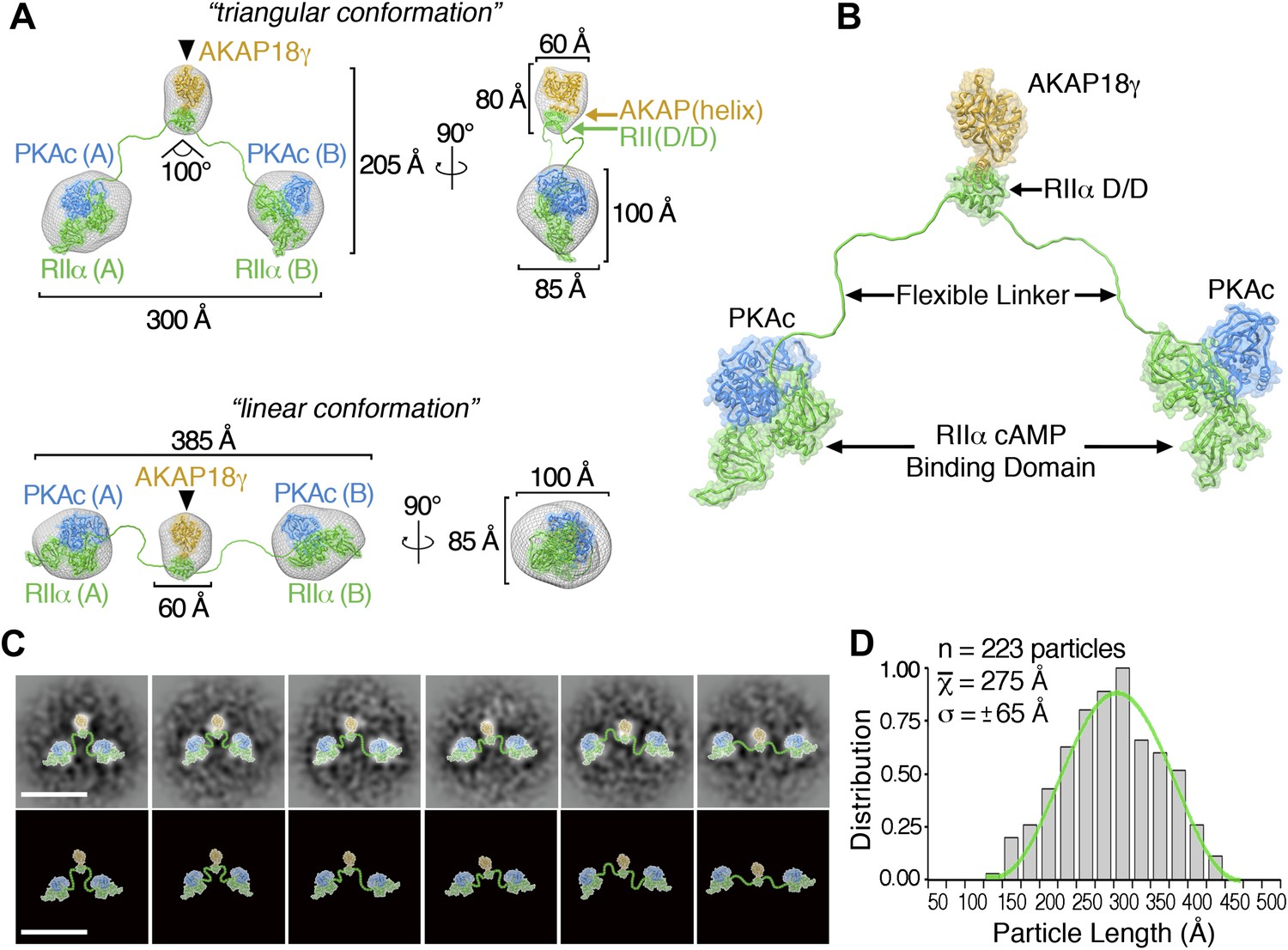
3D reconstructions and pseudo-atomic structure of the AKAP18γ–PKAholo complex.
(A) Three-dimensional (3D) EM reconstructions and 90° rotated views of the fitted molecular models for the AKAP18γ–PKAholo complex. High-resolution structures for regions of AKAP18γ (gold), RIIα (green) and C subunit of PKA (blue) that fit within the EM densities are indicated. Models are presented in the (top) compact triangular and (bottom) extended linear conformation. (B) Pseudo-atomic model of the AKAP18γ–PKAholo complex. (C) (top) Projection averages of the AKAP18γ–PKAholo complex with structural domains fitted into the EM densities and connected by lines representing the RIIα flexible linker regions. (bottom) Projection averages were removed for clarity. Scale bar represents 25 nm. (D) Statistical analysis of individual particle lengths in angstroms (Å) displays a Gaussian distribution (green line) with a mean value of 275 Å and a standard deviation (σ) ±65 Å.
Figure 2—figure supplement 1
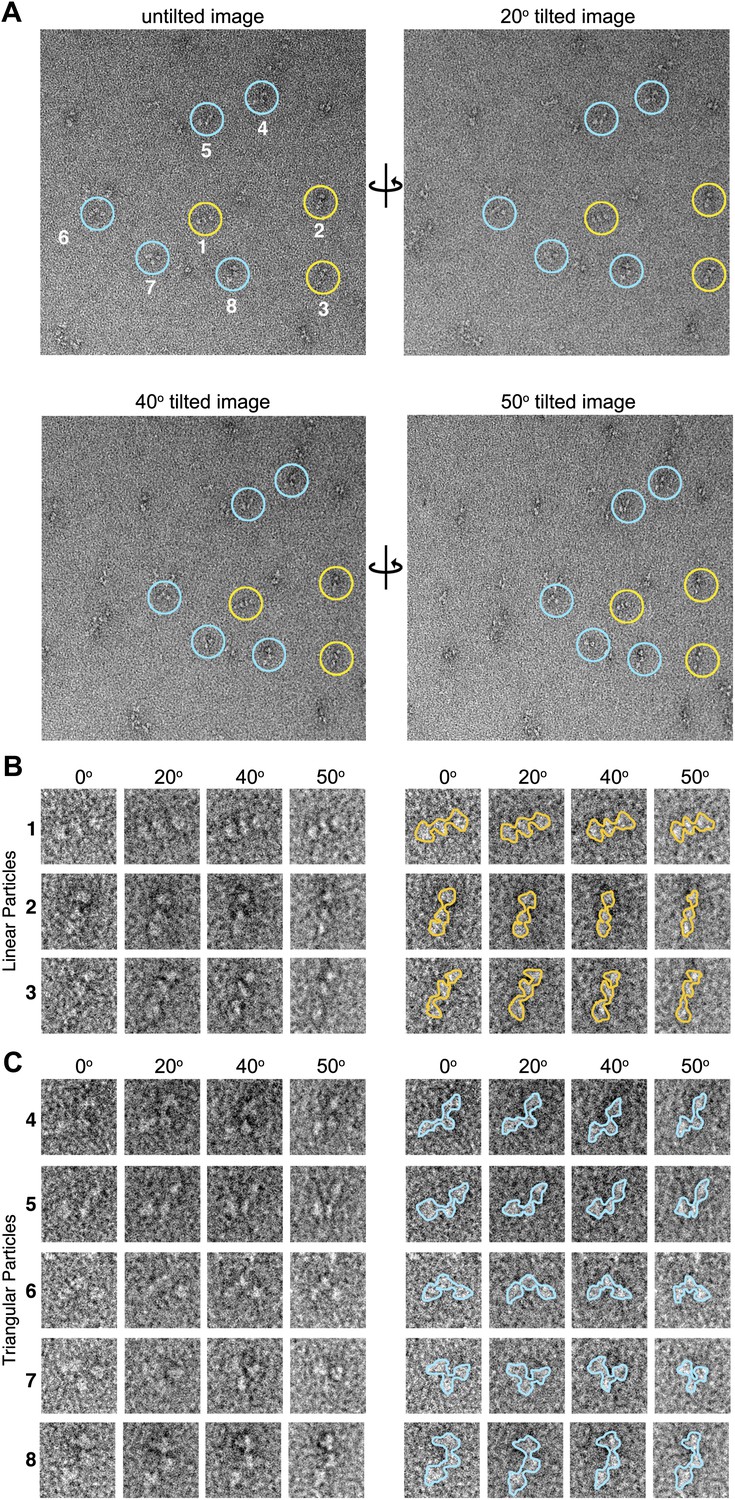
Tilted-series electron microscopy data.
(A) Example electron micrographs collected using a series of image tilts (α = 0°, 20°, 40°, and 50°). Colored circles indicate positions of the numbered particles in the tilt series. (B and C) Left, zoom-views of circled particles in (A). At all tilt angles particles are readily classified as either linear (B, yellow) or as non-linear ‘triangular’ (C, blue) conformations. Right, particle boundaries are outlined for clarity. The tilted-series data was used to provide unique particle ‘views’ used for angular reconstitution methods applied in IMAGIC (van Heel et al., 1996) for initial 3D reconstruction of the AKAP18–PKAholo models.
Figure 2—figure supplement 2

Three-dimensional EM maps of the AKAP18γ–PKAholo complex.
Rotated views of the 3D reconstructions for the AKAP18γ–PKAholo complex determined to 35 Å resolution. (A) In the triangular conformation, the three spherical densities are arranged in a pseudo-symmetric fashion, with the two peripheral densities extending 300 Å apart and at an angle of 100° with respect to the central density. The smaller central density is 60 × 60 × 80 Å and the two larger peripheral densities are 100 × 100 × 85 Å. (B) In the linear conformation, the three spherical densities are arranged in a linear fashion, with the two peripheral domains extending 385 Å apart. (C and D) Back-projections (right) of the calculated 3D maps are compared to class averages (left) used for generating the initial reconstructions in IMAGIC (van Heel et al., 1996).
Figure 3
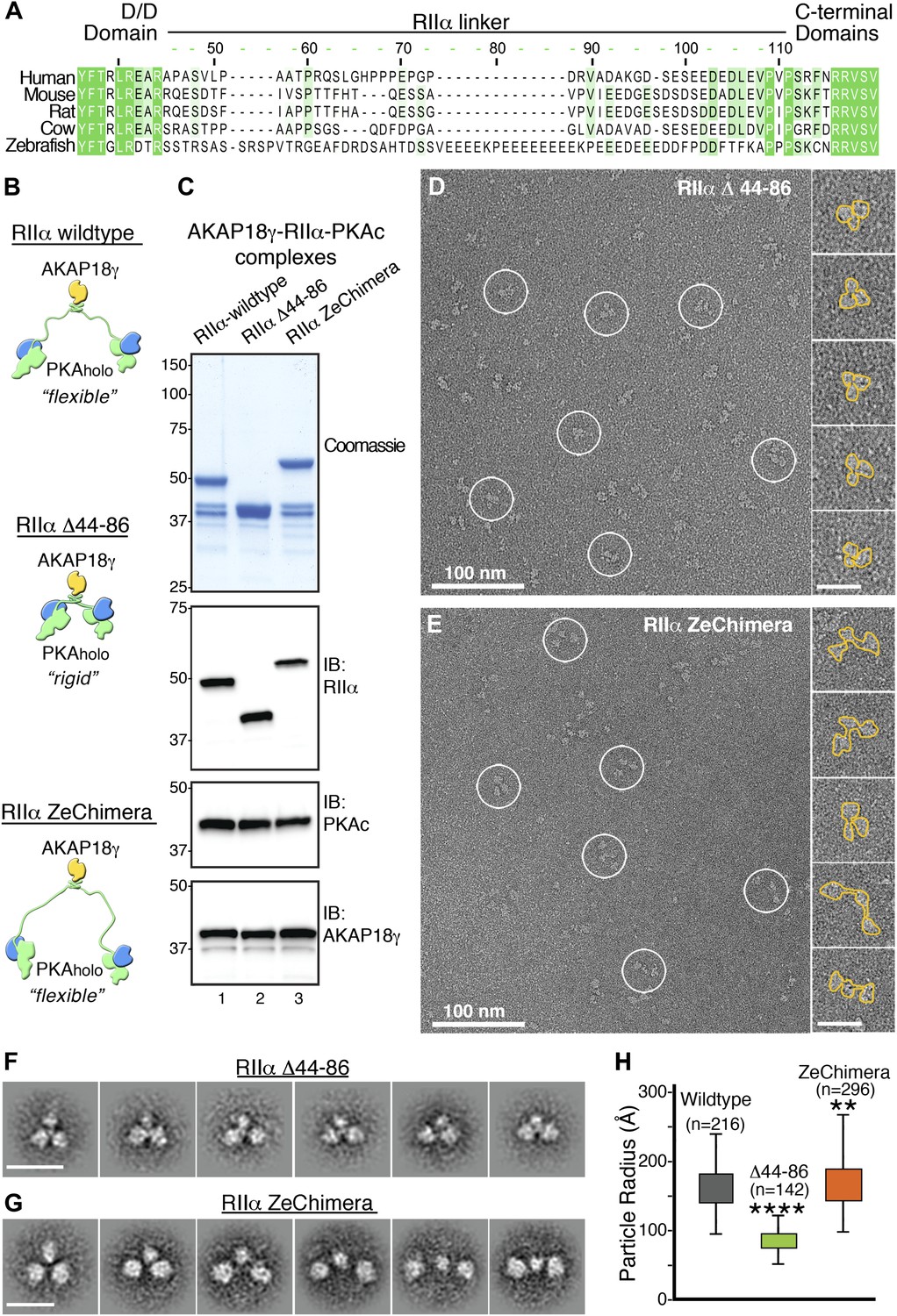
Flexibility within RIIα constrains the configuration of the anchored kinase assembly.
(A) Amino acid sequence alignment of the linker region in RIIα that connects the conserved N-terminal D/D domain to the C-terminal autoinhibitor and cAMP binding domains. This region shows low sequence homology and is likely to be structurally disordered. (B) Schematic representations of modified AKAP18γ–PKAholo complexes with AKAP18γ depicted in yellow, RIIα in green and PKAc in blue. (C) Biochemical analysis of the purified AKAP18γ–RIIα-PKAc complexes assembled with the wild-type RIIα subunit, the RIIα Δ44–86 mutant where the linker region was deleted, and the RIIα ZeChimera mutant where the mouse RIIα linker region was replaced with the corresponding and extended sequence from zebrafish. Top panel shows SDS-PAGE and Coomassie blue staining of the protein components. The next three panels show western blotting for RIIα, PKAc subunit and AKAP18γ, respectively. (D) Electron micrograph of negatively stained AKAP18γ–PKAholo complexes (circles) assembled with the RIIα Δ44–86 construct. (E) Electron micrograph of negatively stained AKAP18γ–PKAholo complexes (circles) assembled with the RIIα ZeChimera construct. Insets in (D and E) show enlarged views of individual particles outlined in gold for clarity. (F) Projection averages of the AKAP18γ–PKAholo complexes assembled with a truncated RIIα Δ44–86 construct using ISAC (Yang et al., 2012). (G) Projection averages of AKAP18γ–PKAholo complex assembled an RIIα ZeChimera construct. Scale bars in (F) and (G) represent 25 nm. (H) Statistical analysis of particle radius in angstroms (Å) for each AKAP18γ–PKAholo complexes. Box plot displays second and third quartile values, tails corresponding to minimum and maximum distances, (**) indicates p<0.01; (****) indicates p<0.0001.
Figure 4 with 1 supplement
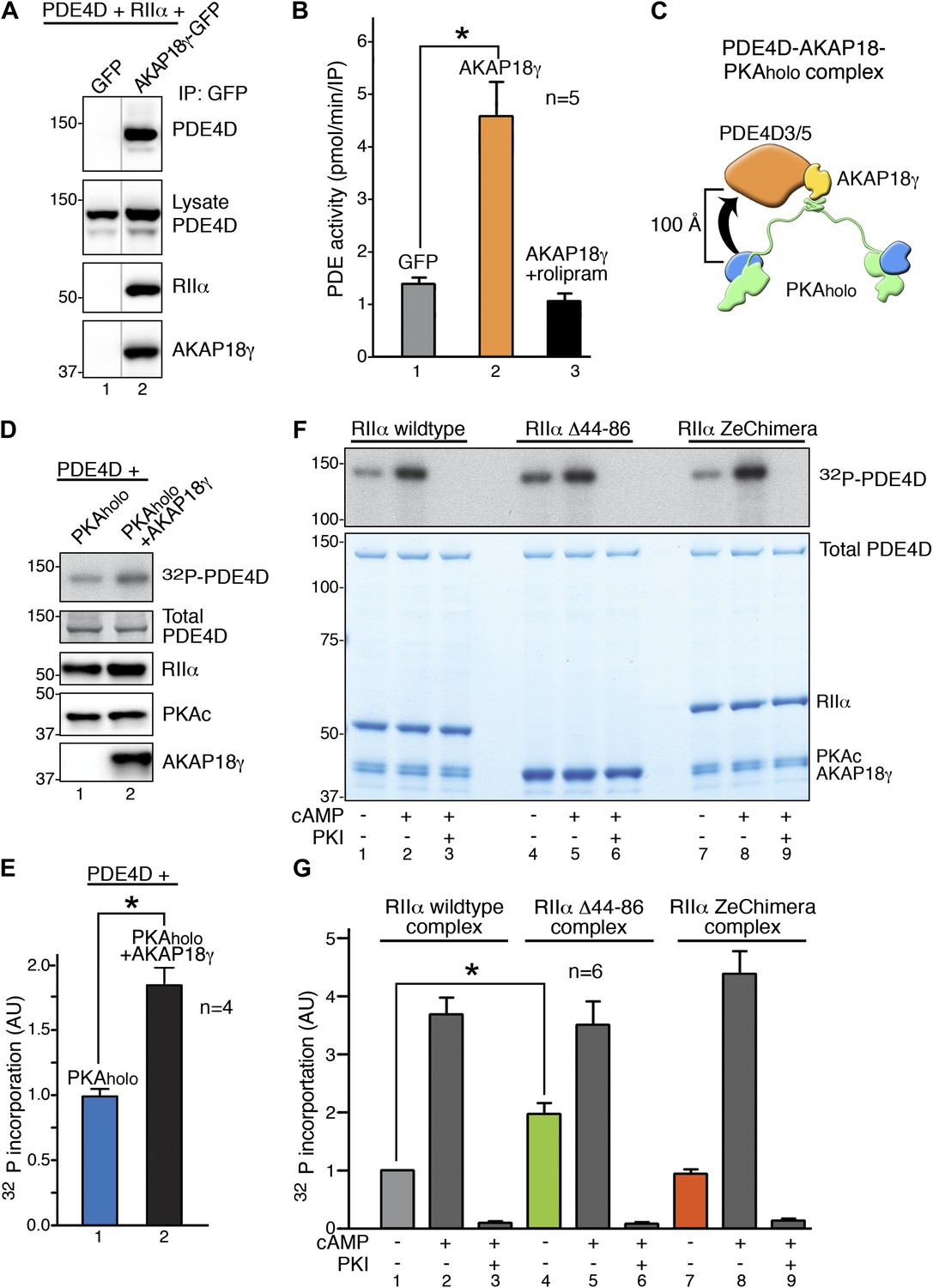
RIIα linker length influences basal PKA phosphorylation of associated substrates.
(A) PDE4D isoforms associate with AKAP18 in cells. HEK293 cells expressing AKAP18–GFP or GFP alone along with PDE4D and RIIα were subjected to immunoprecipitation with anti-GFP antibodies. Immunocomplexes were separated by SDS-PAGE and immunoblotted for PDE4D and RIIα. AKAP18 was detected by RII overlay analysis. (B) Similar immunocomplexes as in (A) were used for phosphodiesterase activity assays. Inclusion of the small molecule rolipram (10 μM, bar 3) inhibited associated PDE4D activity. (C) Schematic of AKAP18–PKA holoenzyme-PDE4D3/5 complexes used in subsequent in vitro substrate phosphorylation assays. Based on our structural data, the PKA catalytic subunit is expected to be positioned within ∼100 Å of its substrate PDE4D. (D) Anchoring of PKA and PDE4D stimulates cAMP-independent phosphorylation of the phosphodiesterase. (Top panel) Basal 32P incorporation into PDE4D (detected by autoradiograph) is shown in the absence or presence of AKAP18γ. (Bottom panels) Levels of PDE4D, RIIα, PKAc and AKAP18γ were assessed by immunoblot. (E) Densitometeric quantification of phospho-PDE4D in panel (D), n = 4 (p<0.05). (F–G) Deletion of the flexible linker augments cAMP-independent phosphorylation of PDE4D by 1.97 ± 0.18-fold (p<0.05). (F) (Top panel) Autoradiograph showing incorporation of 32P into PDE4D in each complex. (Bottom panel) Coomassie blue staining of the SDS-PAGE gel showing components of the assay. The PDE4D is at the top, while the different complexes are shown at the bottom. (G) Densitometric quantification of phospho-PDE4D levels in (F), n = 6.
Figure 4—figure supplement 1
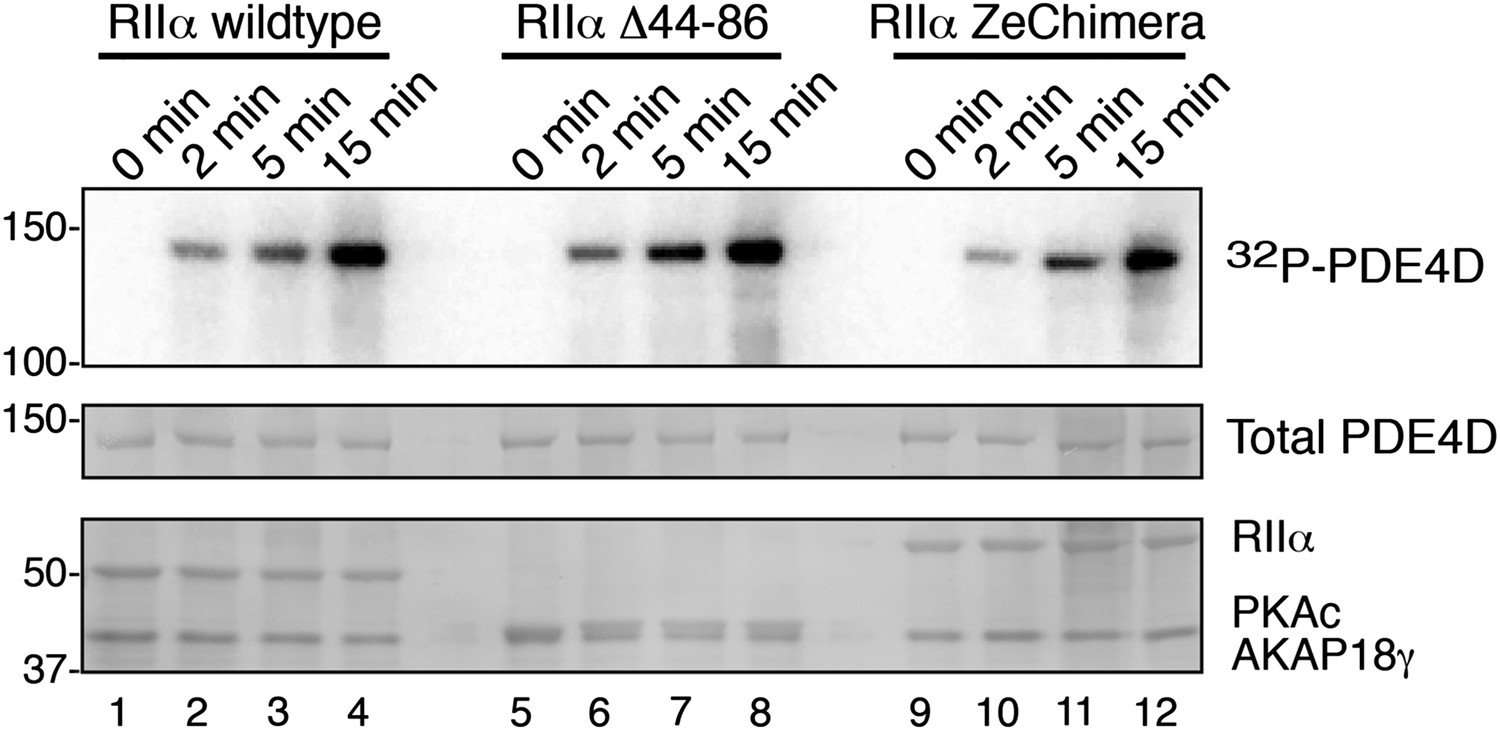
Phosphorylation of PDE4D is time-dependent.
AKAP18 complexes formed with either RIIα or each variant were incubated with PDE4D3 and γ-32P-ATP for the times indicated. (Top panel) Autoradiograph showing incorporation of 32P into PDE4D in each complex. (Middle and bottom panels) India ink staining of the membrane, showing components of the assay. Total PDE4D3 is in the middle, and RII, PKAc and AKAP18γ are shown in the bottom panel.
Figure 5 with 1 supplement
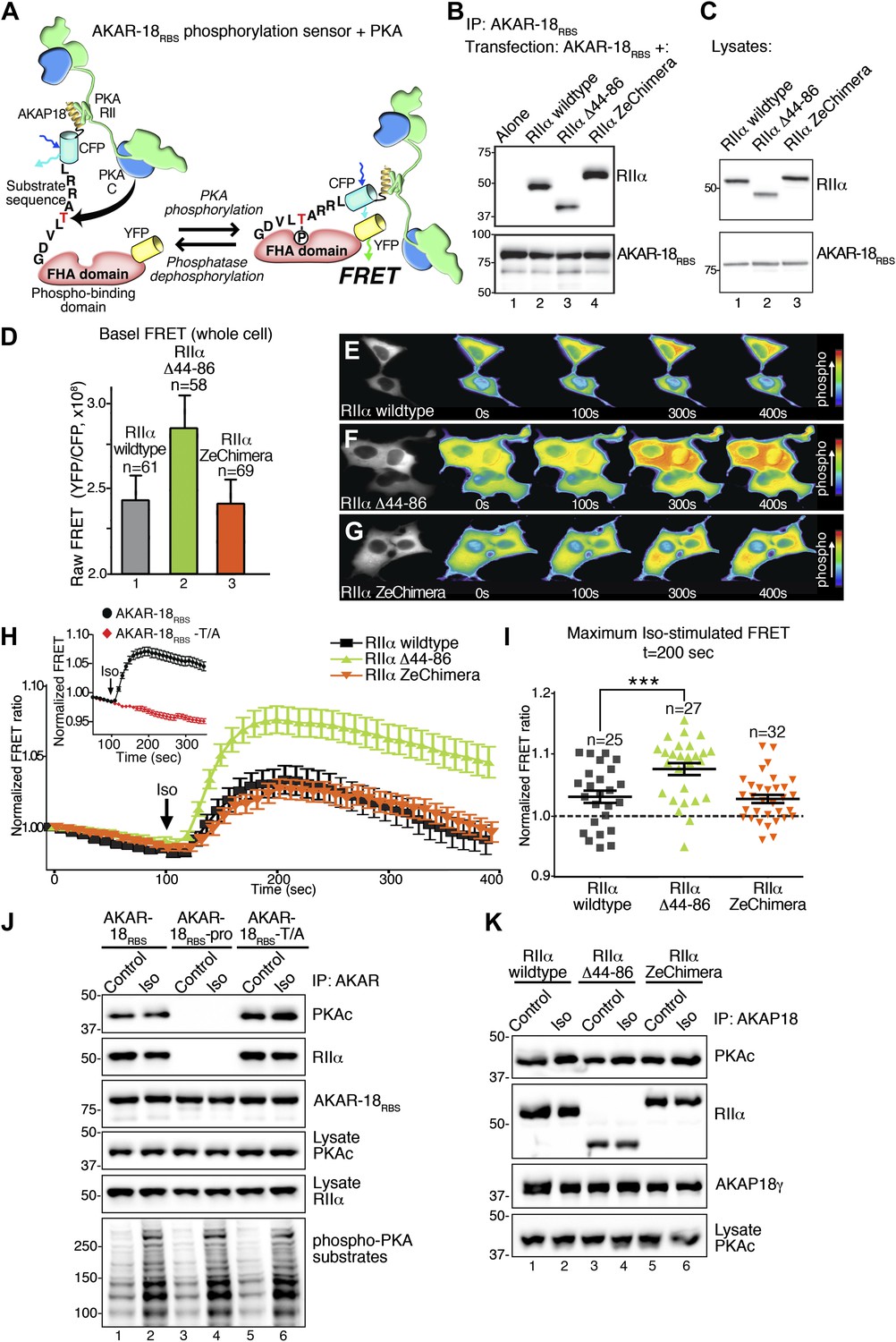
Flexibility within the anchored PKA holoenzyme impacts cAMP responsive signaling inside cells.
(A) Schematic of the modified AKAR-18RBS FRET reporter used in these studies. The PKA RII binding site common to all AKAP18 isoforms was fused to the N-terminus of AKAR2 to create a FRET-based kinase activity sensor that anchors type II PKA holoenzyme. Phosphorylation of a consensus site Thr by PKA induces recruitment of the adjacent Forkhead homology-associated (FHA) phospho-Thr binding domain. Subsequent rearrangement brings together the ECFP and YFP (citrine) moieties to produce an increase in FRET signal as readout of kinase activity. (B) Co-immunoprecipitation of AKAR-18RBS-PKA complexes with holoenzymes composed of three different RII forms. AKAR-18RBS was immunoprecipitated with anti-GFP antibodies and bound PKA subunits were detected by western blotting. (C) Lysates from cells transfected with the AKAR-18RBS reporter and each of the three RIIαforms were immunoblotted to confirm similar expression levels in each cohort. (D) Basal (unstimulated) raw FRET signals from cells expressing RIIα (gray), RIIα Δ44–86 (green) and RIIα ZeChimera (orange). (E and G) Time course of AKAR-18RBS activation in response to the β-adrenergic agonist isoproterenol in cells co-expressing the FRET reporter with (E) RIIα wild type, (F) RIIα Δ44–86, or (G) RIIα ZeChimera. Isoproterenol (Iso, 10 μM) was added at t = 100 s and FRET was recorded for 5 min post-stimulation. Warmer colors indicate increasing phosphorylation as shown in the pseudo-color scale. (H) Amalgamated traces of the Iso stimulated changes in FRET in each cohort (0–400 s). Data are normalized to unstimulated basal FRET level for each respective RIIα form. Changes in the AKAR-18RBS normalized FRET ratio are shown from cells expressing RIIα wild type (black), RIIα Δ44–86 (green) and RIIα ZeChimera (orange). Mutation of the phosphoacceptor threonine in the FRET reporter to create AKAR-18RBS–T/A blocks phosphorylation and abolishes FRET in response to Iso treatment (Inset). (I) Scatter plot representation of peak FRET signals from all cells at 200 s. This plot indicates that AKAR-18RBS-PKA complexes formed with RIIα wild type (black) or the RIIα ZeChimera (orange) had similar peak responses, while complexes formed with RIIα Δ44–86 displayed significantly greater peak FRET responses (p<0.001). This plot also shows that in all cases, some cells fail to respond entirely; these non-responders are included in the amalgamated data analysis presented in (H). (J) C subunit of PKA association with AKAR-18RBS, AKAR-18RBS–pro or AKAR-18RBS–T/A complexes. AKAR-18RBS and AKAR-18RBS–T/A co-precipitate RIIα and endogenous PKA catalytic subunit; AKAR-18RBS–pro, a mutant that cannot bind RII subunits, fails to co-precipitate PKA complexes (top panels). Iso treatment (1 μM, 5 min) does not cause dissociation of PKAc from the AKAR-18RBS or AKAR-18RBS–T/A complexes (top panel, lanes 1–2 and 5–6). Control immunoblots show equivalent levels of AKAR-18RBS, AKAR-18RBS–pro and AKAR-18RBS–T/A in immunoprecipitates as well as RIIα and PKAc expression in cell lysates (middle panels). Immunoblotting for phospho-PKA substrates (R-X-X-pS/T motif) confirms that Iso treatment activates endogenous β-ARs and initiates downstream phosphorylation events (bottom panel). (K) Immunoprecipitation of full-length AKAP18 complexes following Iso treatment. Cells expressing AKAP18γ and RIIα variants were treated with vehicle or Iso (1 μM, 5 min) and AKAP18γ complexes were immunoprecipitated. Immunoblotting shows that Iso has no effect on the amount of PKA catalytic subunit in complexes formed with RIIα wild type, RIIα Δ44–86, or RIIα ZeChimera.
Figure 5—figure supplement 1
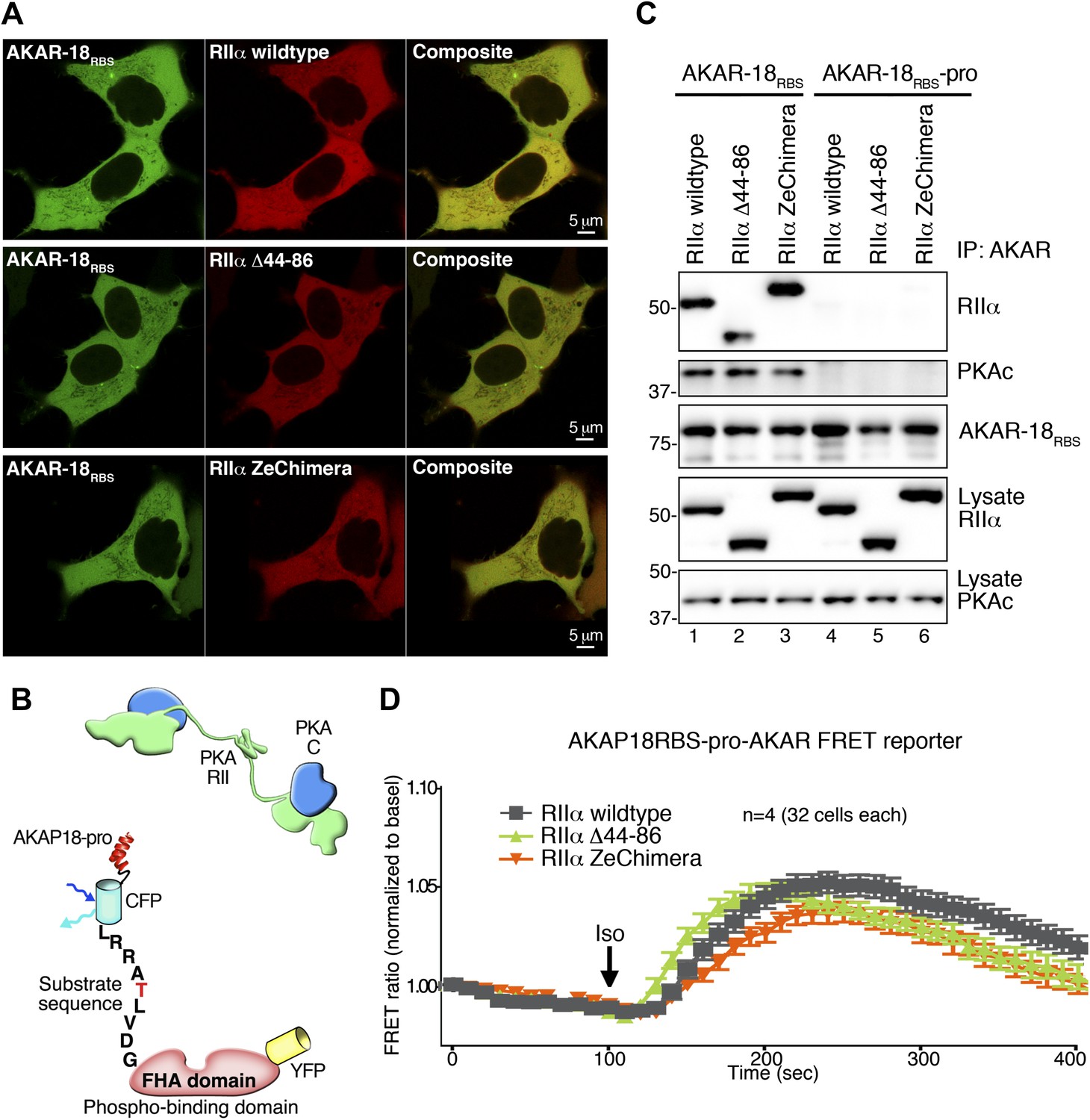
Comparison of AKAR-18RBS and AKAR-18RBS–pro anchoring and FRET controls.
(A) Representative confocal micrographs of live cells show comparable expression levels and the intracellular localization of AKAR-18RBS (green) and mCherry-RIIα (red). Composite images are also shown. HEK293 cells were transiently transfected with AKAR-18RBS and either PKA RIIα wild type (upper panels), RIIα Δ 44–86 (middle panels) or RIIα ZeChimera (lower panels). Scale bars represent 5 μm. (B) Schematic of AKAR-18RBS–pro that contains two helix-breaking proline substitutions in the PKA anchoring site that abolish RII binding. (C) AKAR-18RBS co-precipitates all three RIIα variants as well as endogenous PKA catalytic subunit from transiently transfected HEK293 cells (top panels). AKAR-18RBS–pro no longer anchors RII and does not co-precipitate RIIα or PKAc (top panels). Control immunoblots show equivalent levels of AKAR-18RBS and AKAR-18RBS–pro in immunoprecipitates as well as RIIα and PKAc expression in cell lysates. (D) FRET recordings using AKAR-18RBS–pro. Traces show Iso stimulated changes in FRET in each cohort (0–400 s). Data are normalized to unstimulated basal FRET level for each respective RIIα form. The increase in the AKAR-18RBS normalized FRET ratio in cells co-expressing RIIα Δ44–86 (Figure 5H) is no longer present when the reporter is unable to bind PKA.
Videos
Video 1
Conformational dynamics of the wild-type AKAP18γ–PKA holoenzyme complex.
A montage of projection averages obtained for the wild-type AKAP18γ–PKA holoenzyme complex displays the variety of topological configurations sampled by the dynamic signaling particle.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation
eLife 2:e01319.
https://doi.org/10.7554/eLife.01319




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}