Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms
- University of Colorado, Boulder, United States
- BioFrontiers Institute, United States
- University of Colorado, Denver–Anschutz Medical Campus, United States
- Signalling and Gene Regulation Laboratory, Cecil H and Ida Green Center for Reproductive Biology Sciences, University of Texas Southwestern Medical Center, United States
Figures
Figure 1 with 1 supplement
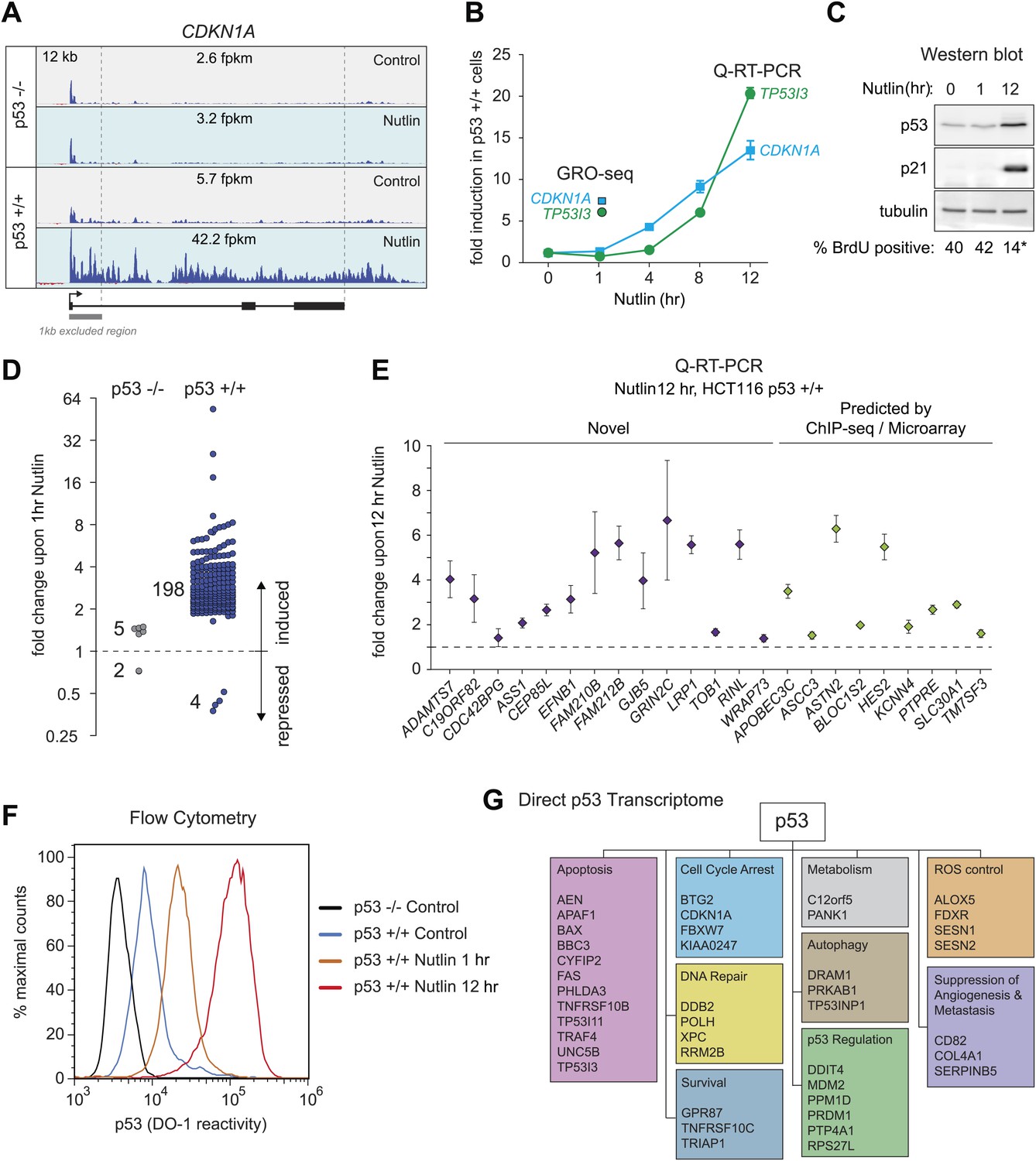
GRO-seq analysis of the p53 transcriptional program.
(A) GRO-seq results for the p53 target locus CDKN1A (p21). Isogenic p53 −/− and p53 +/+ HCT116 cells were treated for 1 hr with either 10 μM Nutlin-3a (Nutlin) or vehicle (DMSO, Control). Fragments per kilobase per million reads (fpkm) are shown for the intragenic region. The first kilobase downstream of the transcription start site (TSS) was excluded from the fpkm calculation to minimize effects of RNAPII pausing. The total genomic region displayed is indicated in the top left corner. Blue signals are reads mapping to the sense strand, red signals are reads mapping to the antisense strand. See Figure 1—figure supplement 1A for results of the TP53I3 locus. (B) GRO-seq detects transactivation of the canonical p53 target genes CDKN1A and TP53I3 at 1 hr of Nutlin treatment, prior to any detectable increase in steady state mRNA levels as measured by Q-RT-PCR. (C) A 1 hr time point of Nutlin treatment does not produce significant p53 accumulation, p21 protein induction or a decrease in number of S phase cells as measured by BrdU incorporation assays. * indicates p<0.05. See also Figure 1—figure supplement 1B for quantification data of BrdU assays. (D) Genome-wide analysis using the DESeq algorithm identifies 198 annotated gene loci transactivated upon Nutlin treatment only in HCT116 p53 +/+ cells. See Supplementary file 1 for a detailed annotation of these genes. (E) Q-RT-PCR validates induction of novel and predicted direct p53 target genes upon 12 hr of Nutlin treatment. mRNA expression was normalized to 18s rRNA values and expressed as fold change Nutlin/DMSO. Data shown are the average of three biological replicates with standard errors from the mean. (F) Flow cytometry analysis using the DO-1 antibody recognizing the MDM2-binding surface in the p53 transcactivation domain 1 (TAD1) reveals increased reactivity as early as 1 hr of Nutlin treatment, indicative of unmasking of the TAD1 at this early time point. (G) p53 directly activates a multifunctional transcriptional program at 1 hour of Nutlin treatment, including many canonical apoptotic genes. See Supplementary file 1 for a complete list and annotation.
Figure 1—figure supplement 1
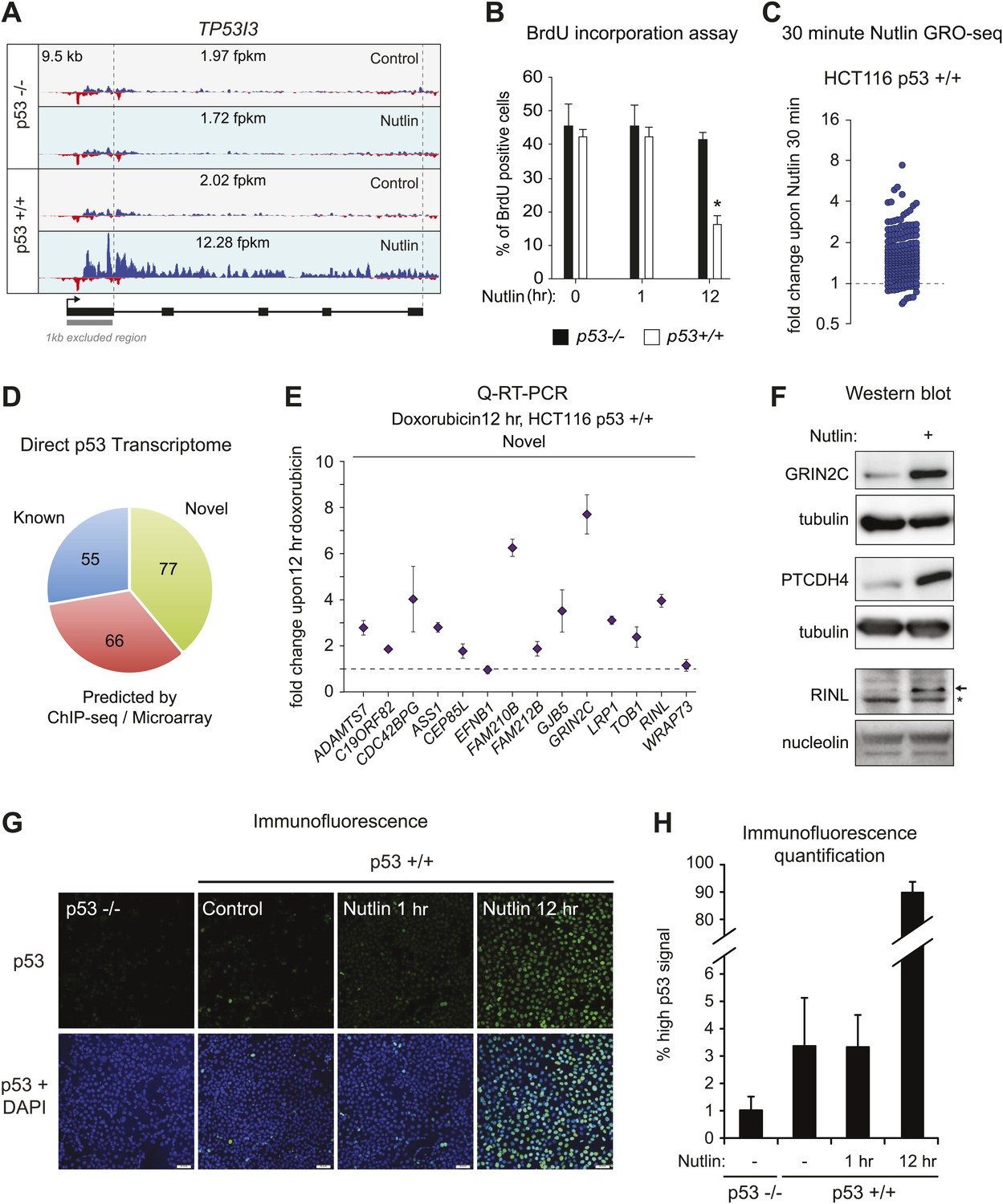
GRO-seq reveals the immediate direct p53 transcriptional response.
(A) GRO-seq results for the well characterized pro-apoptotic p53 target gene TP53I3 (PIG3). (B). BrdU incorporation assays for HCT116 p53 −/− and p53 +/+ cells upon Nutlin treatment for the 1 hr time point employed for GRO-seq and the 12 hr time point used for microarray experiments. One hour Nutlin does not induce significant cell cycle arrest. (C) GRO-seq shows that 176 of the 198 genes transactivated upon 1 hr of Nutlin are already increased above 1x as early as 30 min after addition of the drug to cell cultures. (D) Of the 198 genes transactivated by p53 at 1 hr of Nutlin treatment, 55 were known direct targets, 66 were predicted by published ChIP-seq/microarray studies and 77 are novel. See also Supplementary file 1. (E) Q-RT-PCR shows that most novel p53 targets identified by GRO-seq are also induced by doxorubicin, a genotoxic p53-activating agent. mRNA expression was normalized to 18s rRNA values and is expressed as fold change doxorubicin/DMSO. Data shown are the average of three biological replicates with standard errors from the mean. (F) Western blot assays show induction of the novel p53 target genes GRIN2C, PTCDH4 and RINL at the protein level upon Nutlin treatment of HCT116 p53 +/+ cells. Nucleolin and α-tubulin serve as loading controls. Asterisk denotes a non-specific band in the RINL blot. (G–H) Immunofluroescence staining of p53 shows a similar pattern for control cells (0 hr) and cells treated with Nutlin for 1 hr. Upon long exposure to Nutlin (12 hr), almost the entire cellular population exhibits strong nuclear signal for p53 staining. In H, 'high p53 staining' was defined as the average signal observed in the 12 hr Nutlin cell culture.
Figure 2 with 2 supplements
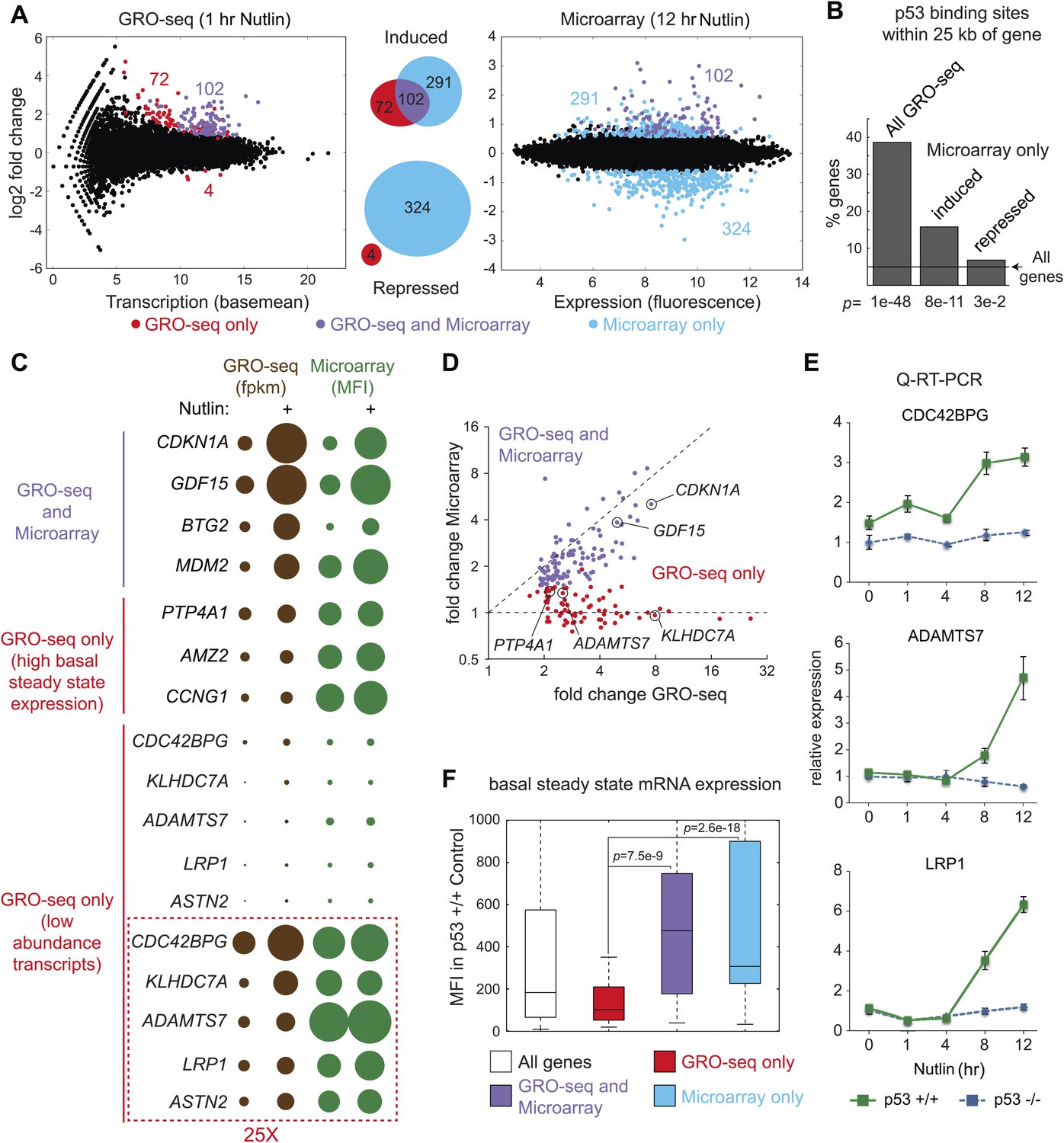
Global analysis of p53 effects on RNA synthesis vs steady state levels.
(A) MAplots for GRO-seq and microarray gene profiling experiments in HCT116 p53 +/+ cells after 1 hr and 12 hr of Nutlin treatment, respectively. Colors indicate whether genes scored as statistically different in both platforms (purple), in the GRO-seq only (red) or the microarray experiment only (blue). (B) Few genes downregulated in the microarray experiment show p53 binding within 25 kb of the gene, suggestive of indirect regulation. (C) Bubble plots displaying relative signals derived from the GRO-seq and microarray experiments illustrate how genes with very high basal expression or very low transcription are not significantly affected at the steady state level as measured by microarray. For the CDC42BPG, KLHDC7A, ADAMTS7, LRP1 and ASTN2 loci, the signals were replotted at 25-fold magnification. (D) Scatter plot showing comparative fold induction for p53 target genes transactivated at 1 hr Nutlin treatment between the GRO-seq and microarray experiments. (E) Q-RT-PCR indicates that many low abundance transcripts upregulated by GRO-seq are indeed induced at the steady state level. (F) Box and whisker plots showing the expression of various gene sets as detected by microarray.
Figure 2—figure supplement 1
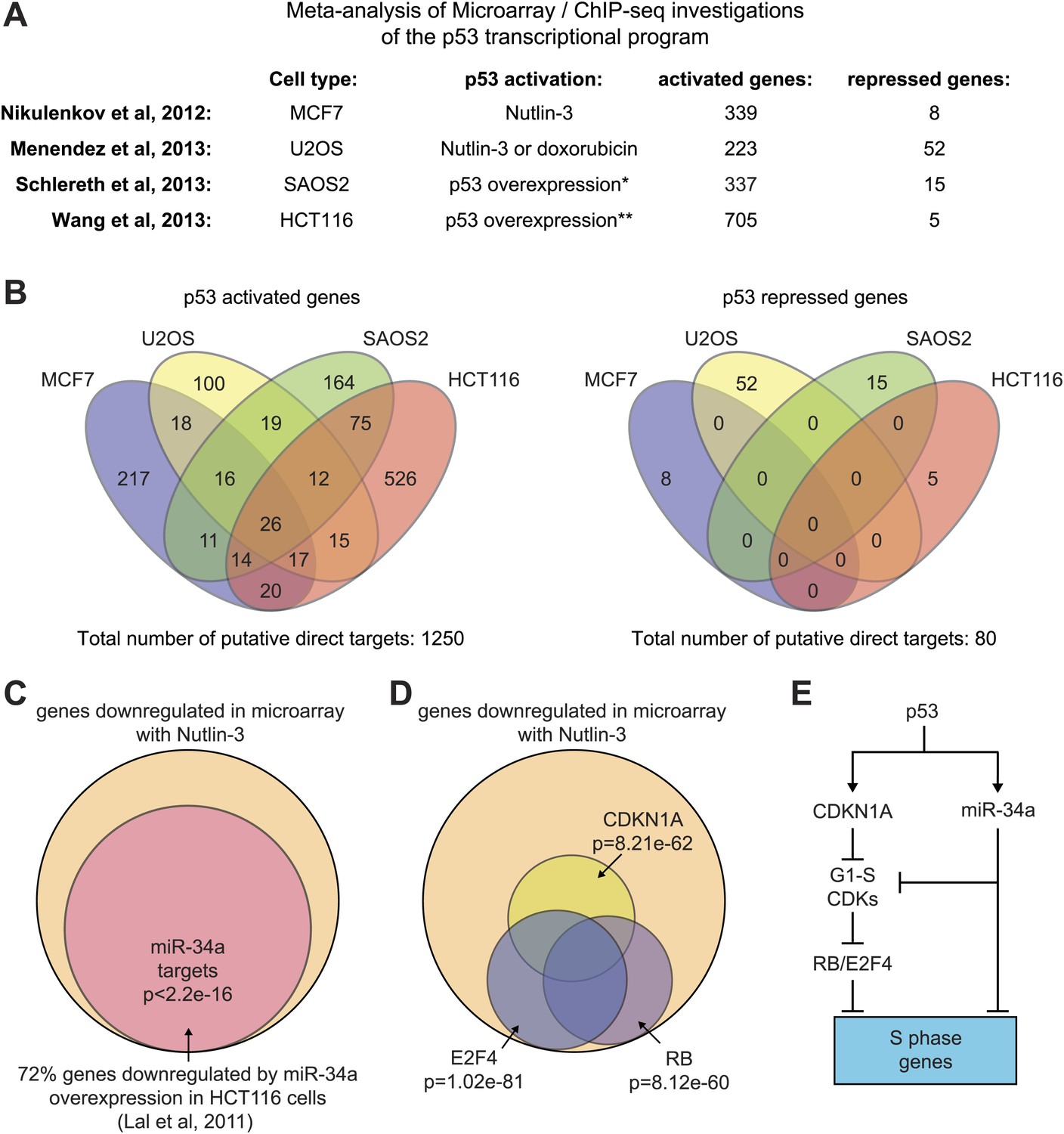
Mechanisms of indirect gene repression by p53.
(A and B) Meta-analysis of four recent investigations of the p53 transcriptional program using microarray analysis of RNA steady state levels and ChIP-seq measurements of p53 binding reveals little overlap between the experiments. See Supplementary file 2 for the various gene lists used to generate the Venn diagrams using Venny (http://bioinfogp.cnb.csic.es/tools/venny/). *This study employed SAOS2 cells, which are p53 null, with overexpression of various p53 isoforms. ** This study employed HCT116 p53 −/− cells with overexpression of natural p53 polymorphic variants. (C) 72% of genes downregulated upon 12 hr of Nutlin treatment in HCT116 cells were found to be repressed in this same cell type by overexpression of miR-34a, a p53 inducible miRNA. (D) Ingenuity Pathway Analysis of genes downregulated upon 12 hr Nutlin treatment indicates that the top three regulators of this gene set are E2F4, CDKN1A and RB. (E) Model of indirect gene repression by p53 via upregulation of CDKN1A and miR-34a, both of which inhibited G1-S CDK complexes.
Figure 2—figure supplement 2
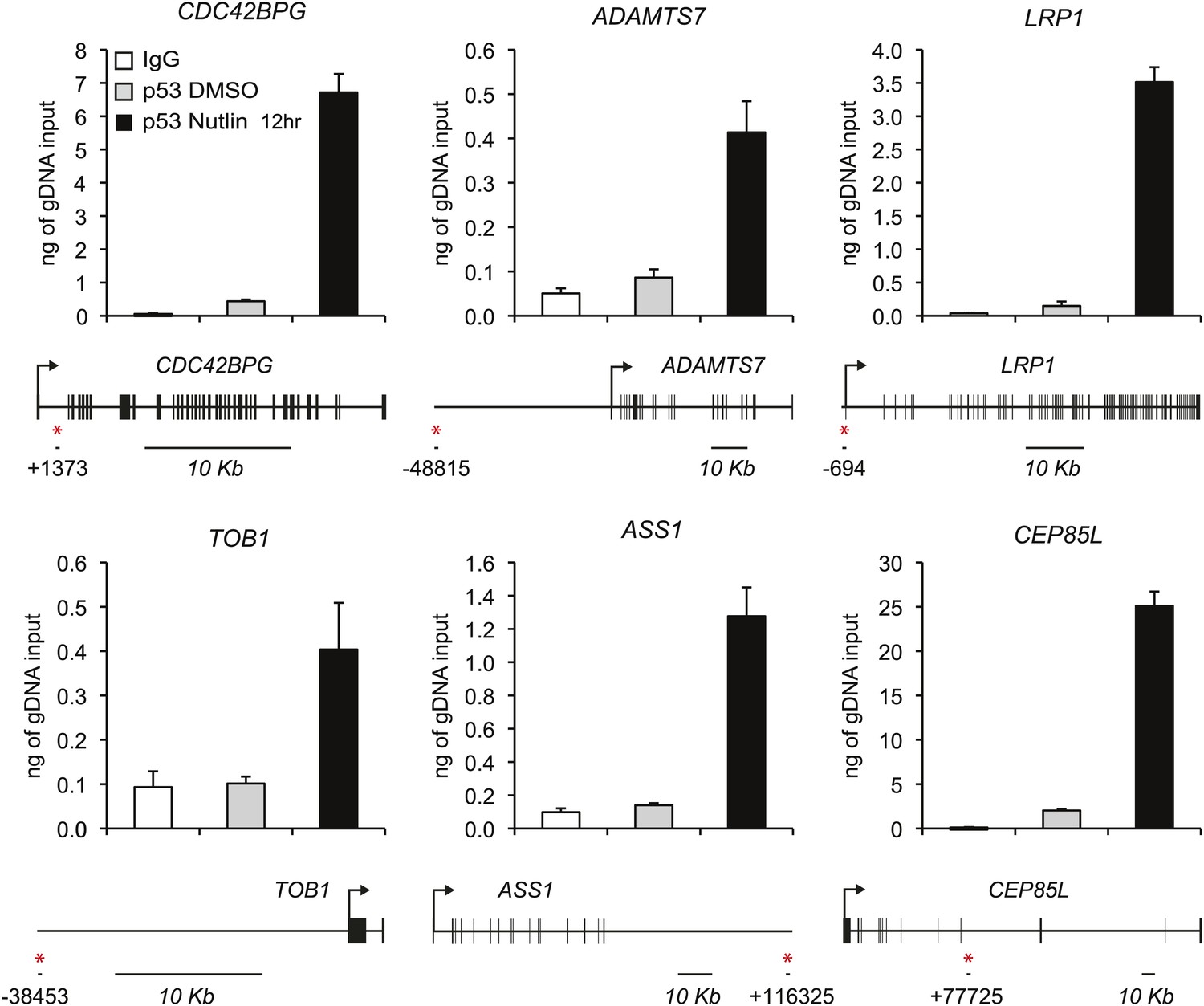
ChIP analysis of novel p53 target genes.
Analysis of p53 ChIP-seq datasets indicated high confidence p53 binding events in the vicinity of novel p53 target genes identified by GRO-seq at the genomic positions labeled with a red asterisk. ChIP-Q-PCR assays in HCT116 p53 +/+ cells confirm p53 binding at these locations above background levels (IgG control).
Figure 3 with 2 supplements
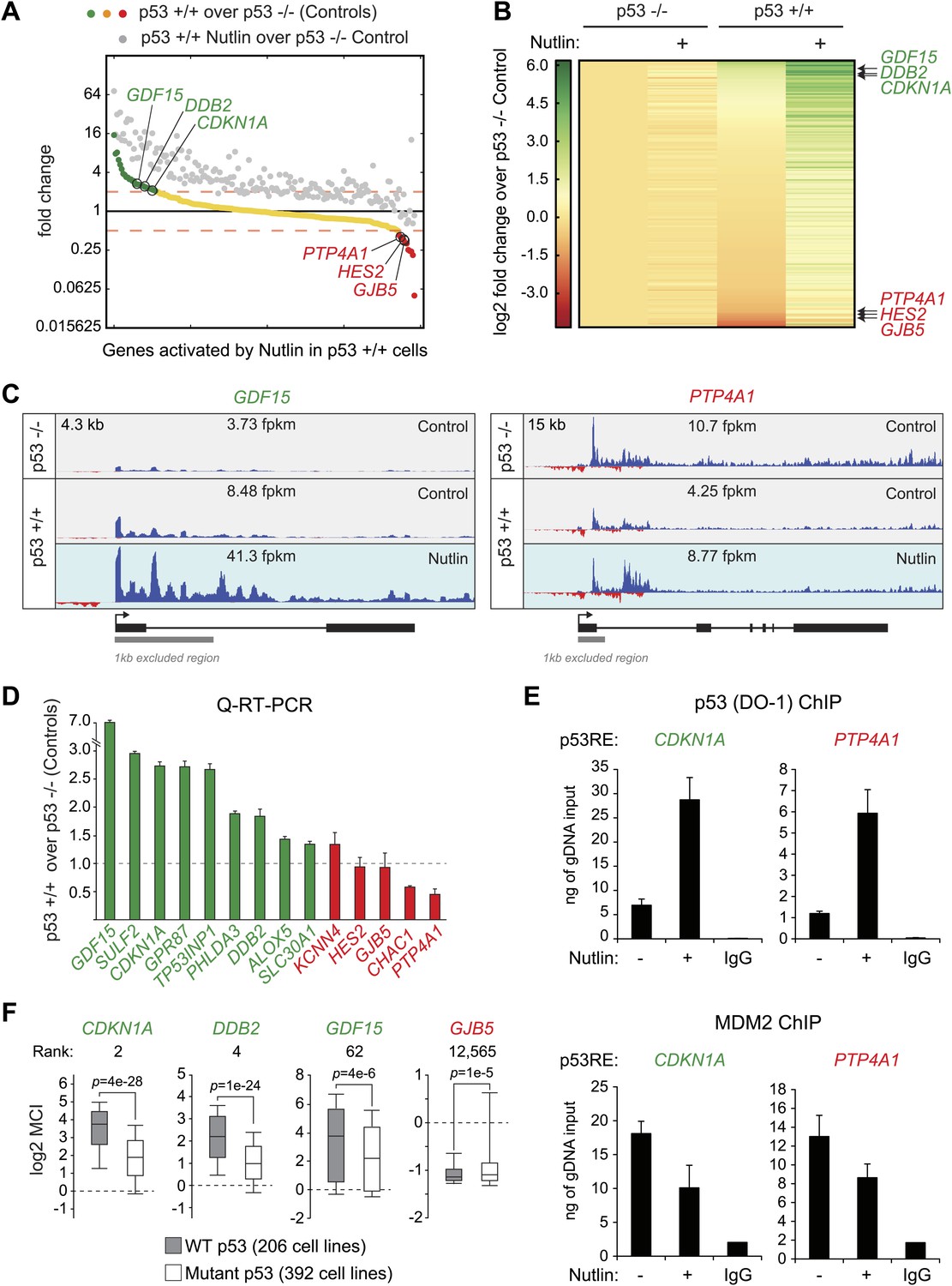
p53 exerts varying activating and repressing effects on its target genes prior to MDM2 inhibition.
(A) 198 genes activated upon 1 hr Nutlin treatment in HCT116 p53 +/+ cells are ranked from left to right based on their basal transcription in p53 +/+ cells over p53 −/− cells. Green indicates genes whose basal transcription is greater than twofold in p53 +/+ cells, red indicates lesser than twofold. Grey dots display the transcription of the same genes in Nutlin-treated p53 +/+ cells. (B) Heatmap displaying relative transcriptional activity of direct p53 target genes identified by GRO-seq relative to control p53 −/− cells. Genes are sorted based on their transcription in control p53 +/+ cells. (C) Genome browser views of representative genes whose basal transcription is higher (GDF15) or lower (PTP4A1) in the presence of MDM2-bound p53. See Figure 3—figure supplement 1A for matching RNAPII ChIP data. (D) Q-RT-PCR measurements of genes whose basal transcription was found to be 2x higher (green) or lower (red) in the presence of MDM2-bound p53. (E) ChIP assays show binding of p53 and MDM2 to the p53REs in the CDKN1A and PTP4A1 gene loci (−2283 bp and +1789 relative to TSS, respectively), prior to inhibition of the p53-MDM2 interaction by Nutlin. Nutlin treatment leads to increased p53 signals with the DO-1 antibody recognizing the p53 TAD1, concurrently with a decrease in MDM2 signals. MDM2 ChIP was performed in SJSA cells carrying a MDM2 gene amplification F. Oncomine gene expression analysis of 598 cancer cell lines of varied p53 status shows that CDKN1A, DDB2 and GDF15 are more highly expressed in wild type p53 cell lines, whereas GJB5 is more highly expressed in mutant p53 cell lines. The ranking position of these genes is also indicated.
Figure 3—figure supplement 1
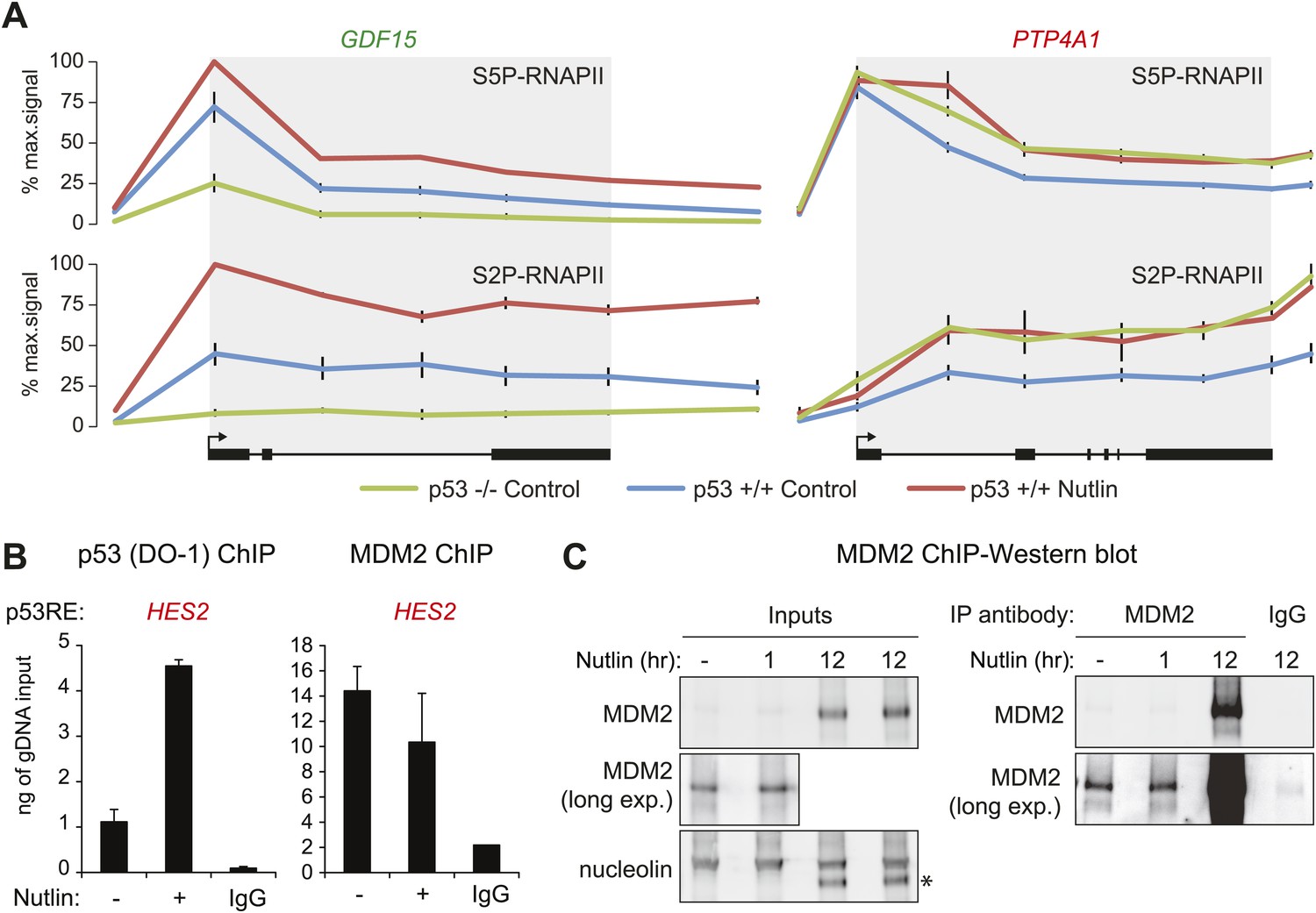
Differential effects of p53 on the basal transcription of its target genes.
(A) ChIP analysis of the ‘basally activated’ GDF15 and ‘basally repressed’ PTP4A1 gene loci using antibodies against the Serine 5- and Serine 2-phosphorylated forms of the C-terminal domain repeats of RBP1, the largest subunit of RNAPII. (B) ChIP assays for p53 and MDM2 at the p53RE in the HES2 locus (+5160 relative to TSS) performed as in Figure 3E. (C) Western blots for MDM2 using the protein extracts from SJSA cells employed for MDM2 ChIP assays. Long exposures show detectable amounts of MDM2 in both the input and MDM2-ChIP samples. A 12 hr or Nutlin treatment was included as positive control.
Figure 3—figure supplement 2
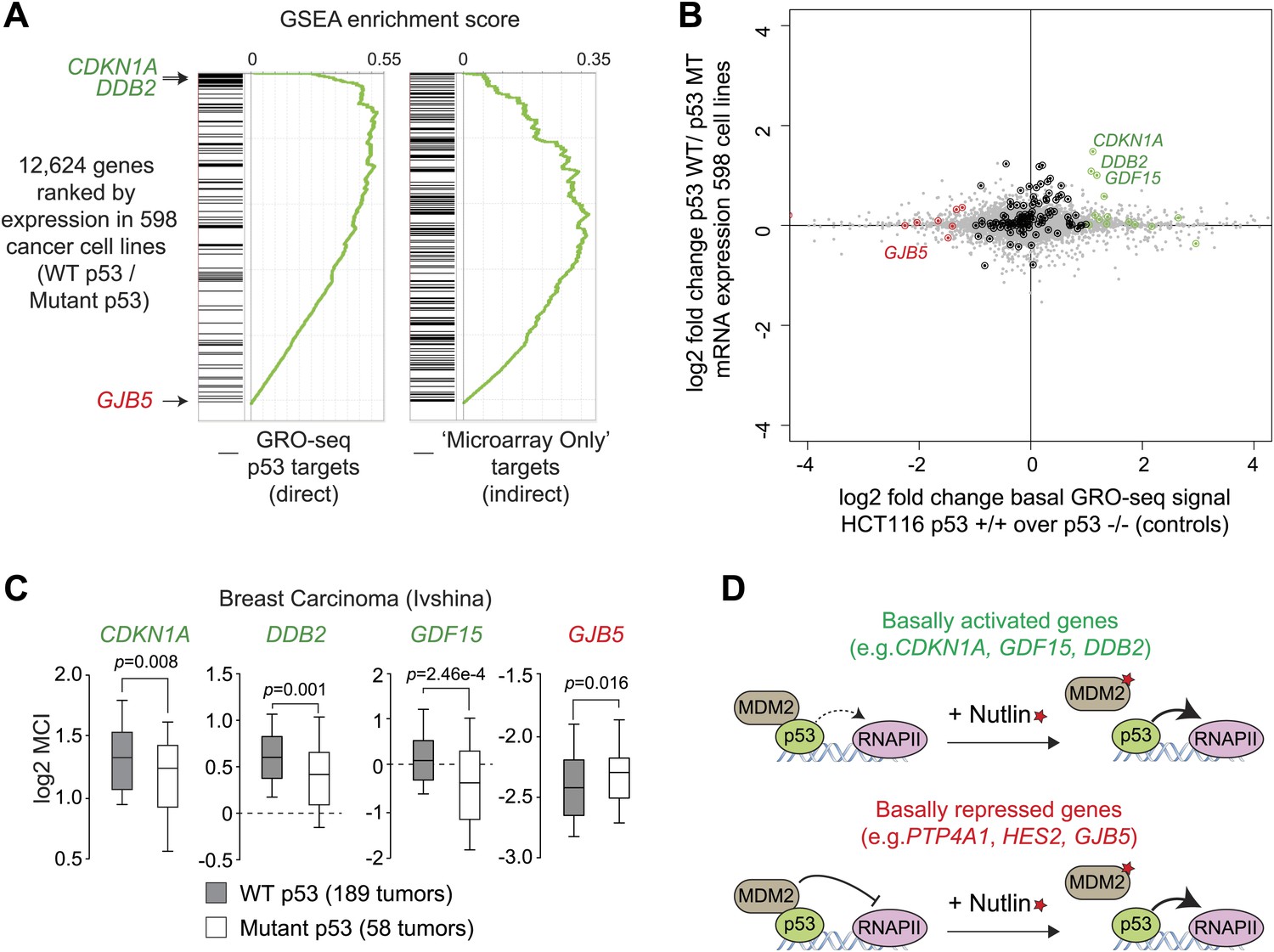
p53 mutational status affects the basal expression of its target genes.
(A) Gene set enrichment analysis (GSEA) of direct p53 target genes identified by GRO-seq (left) or genes upregulated upon Nutlin treatment in the microarray experiment only (right) relative to a ranked list of 12,624 genes analyzed via Oncomine for their relative expression in WT p53 vs mutant p53 cell lines. (B) Scatter plot comparing relative transcription as measured by GRO-seq in HCT116 p53 +/+ over p53 −/− cells vs relative mRNA expression in p53 WT vs p53 mutant cell lines. Red dots are genes whose transcription was lesser than twofold in p53 +/+ cells, green dots are genes whose transcription was greater than twofold in p53 +/+ cells, all other direct p53 targets in HCT116 cells are highlighted in black. (C) Oncomine gene expression analysis of 247 breast carcinomas of varied p53 status shows that CDKN1A, DDB2 and GDF15 are more highly expressed in wild type p53 tumors, whereas GJB5 is more highly expressed in mutant p53 tumors. (D) Model depicting the differential effects of MDM2-p53 complexes on the basal expression of p53 target genes.
Figure 4 with 2 supplements
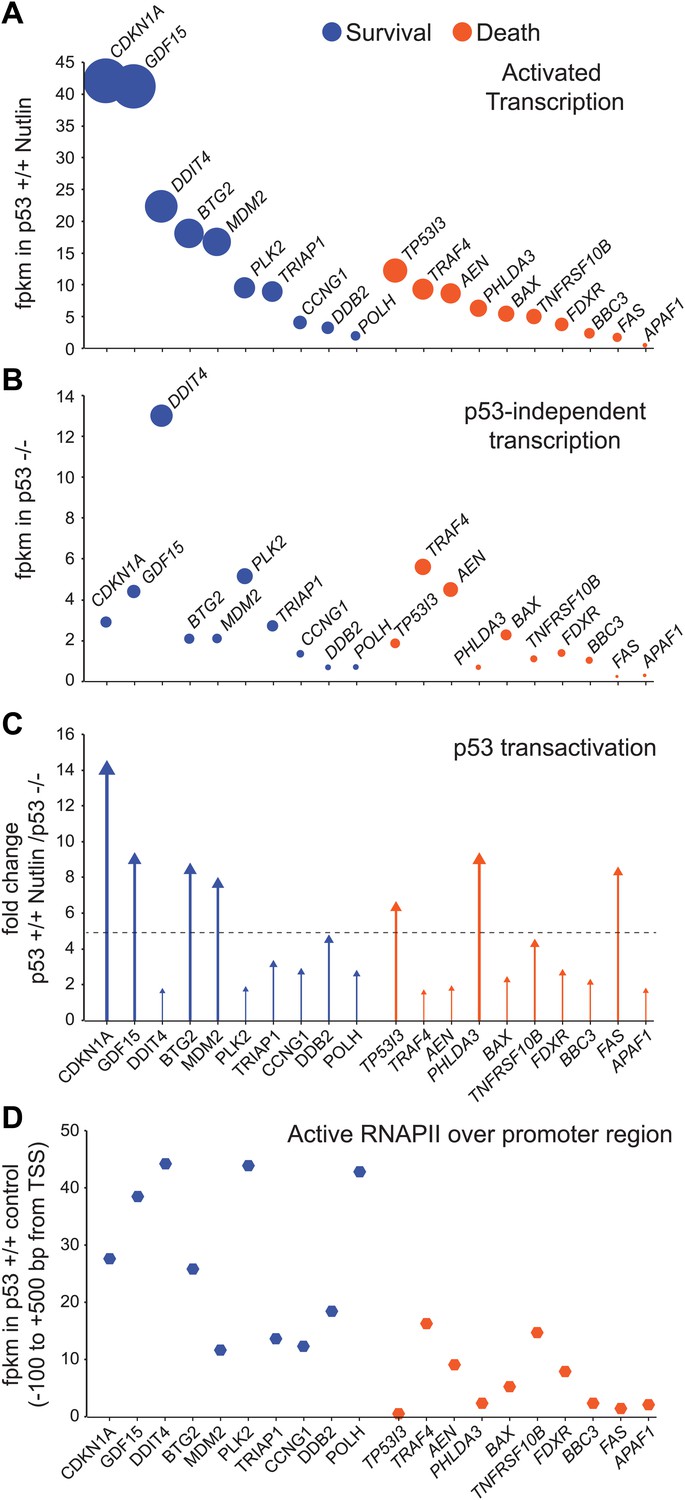
GRO-seq analysis of key survival and death genes within the p53 network.
(A) The 10 most transcribed pro-survival and pro-apoptotic genes identified by GRO-seq ranked by decreasing transcriptional output in Nutlin-treated p53 +/+ cells. The surface of the bubbles represents the GRO-seq signal output relative to the CDKN1A locus. (B) Transcriptional output of same genes shown in A in p53 −/− cells. (C) Fold change analysis showing the overall effect of p53 on the transcription of its survival and apoptotic targets. (D) Survival genes within the p53 network tend to carry more proximally bound, transcriptionally engaged RNAPII over their promoter regions than apoptotic genes.
Figure 4—figure supplement 1
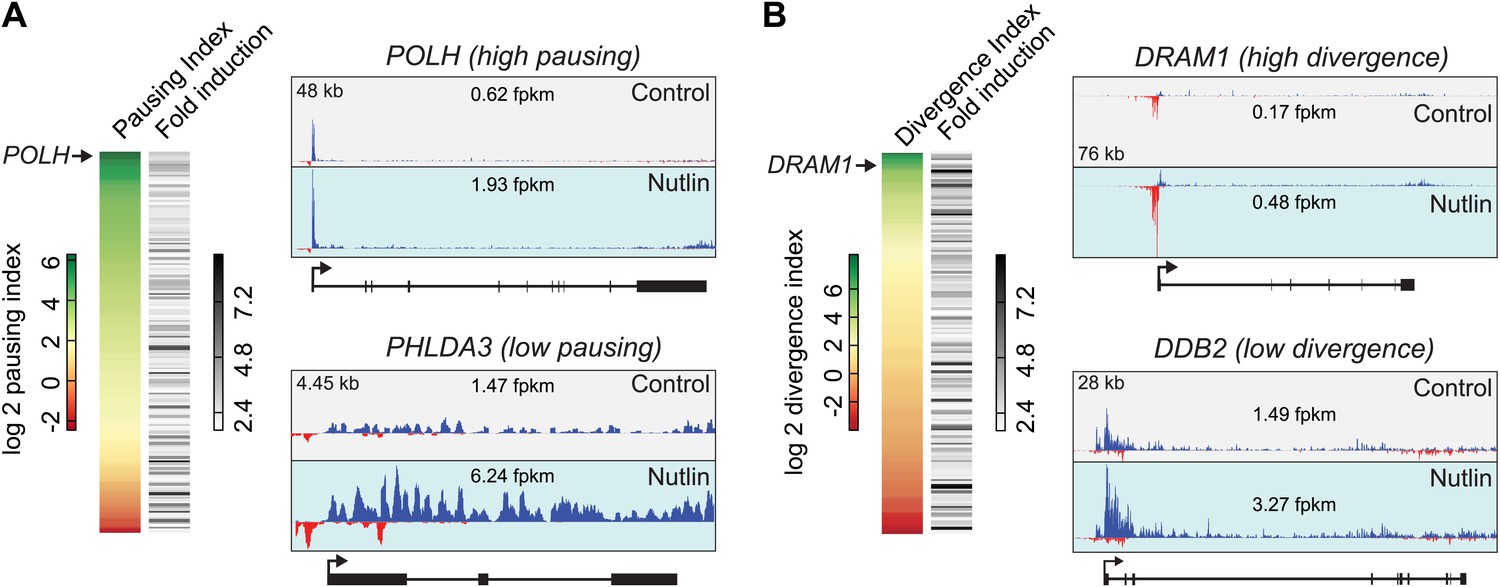
p53 target genes display a wide range of RNAPII pausing and promoter divergence.
(A) RNAPII pausing is not a pre-requisite for rapid activation among p53 target genes. A ranking of pausing indices show no correlation with fold induction among p53 targets. POLH and PHLDA3 are representative examples of genes with high and low pausing indices. (B) p53 target genes show a great variation in the amount of promoter divergence, but a ranking of divergence indices shows no correlation with fold induction. DRAM1 and DDB2 are examples of p53 targets with high and low divergence, respectively.
Figure 4—figure supplement 2
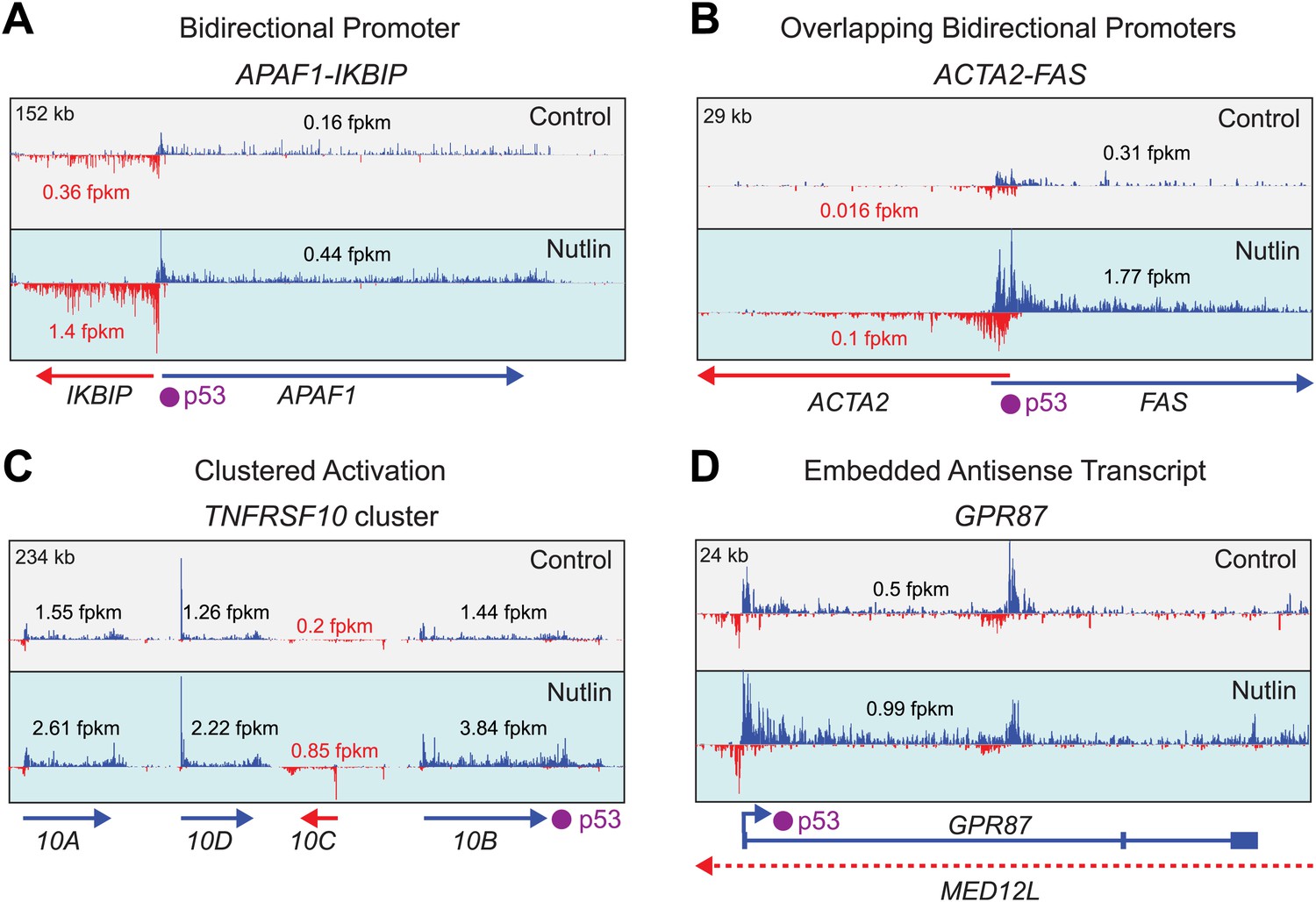
Examples of gene-specific features affecting key pro-apoptotic and survival p53 target genes.
See main text for details.
Figure 5 with 1 supplement
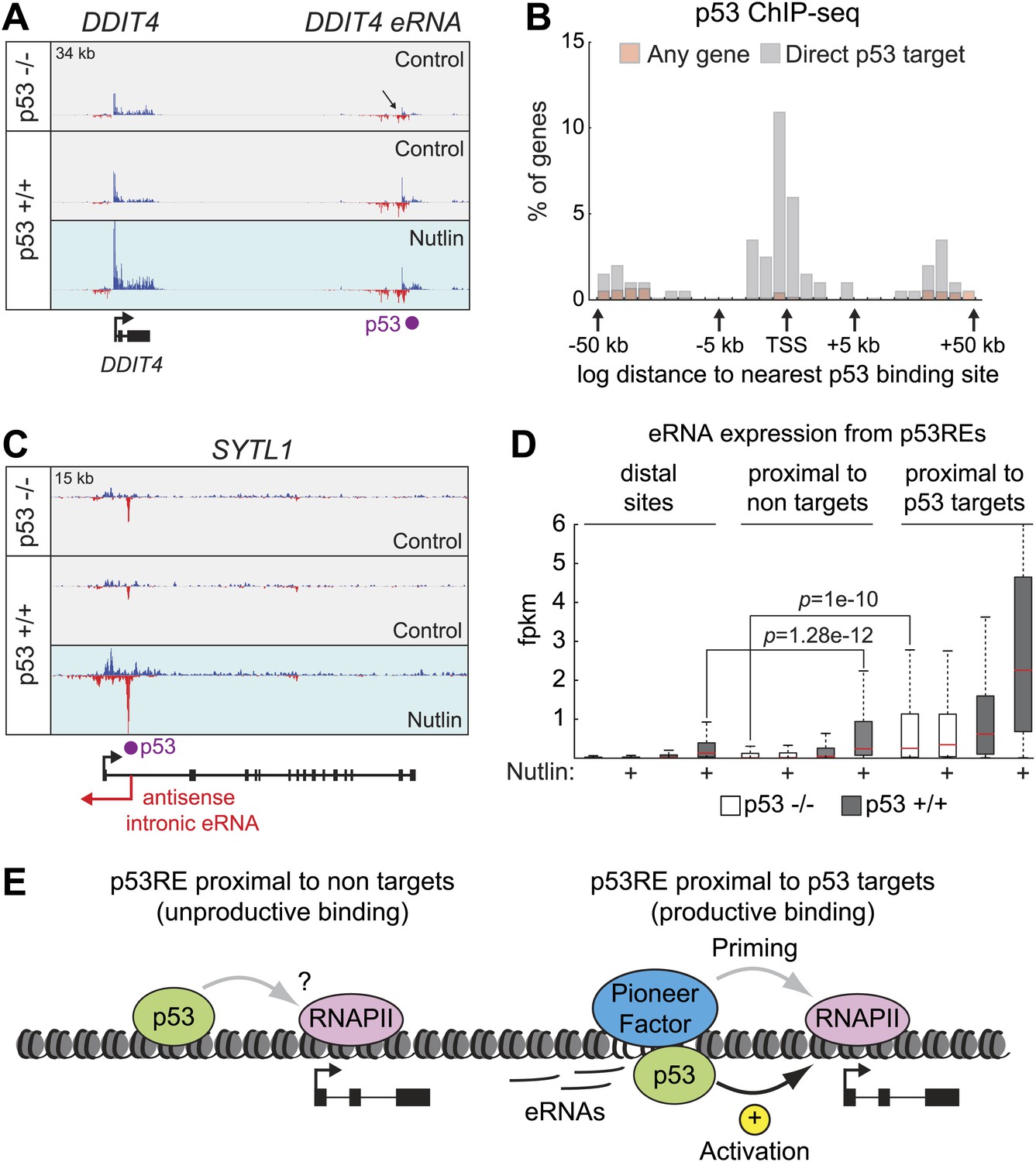
Direct p53 target genes harbor pre-activated enhancers.
(A) GRO-seq results for the DDIT4 locus, representative of p53 target genes that display bidirectional eRNA transcription (arrow) arising near sites of p53 binding (indicated by a purple dot). (B) Analysis of nearest p53 binding events relative to the transcription start site (TSS) of direct p53 target genes detected by GRO-seq (grey bars) vs all RefSeq genes (pink). (C) GRO-seq results for the SYTL1 locus, representative of p53 target genes that display intronic antisense eRNA transcription arising near sites of p53 binding. (D) Analysis of eRNA transcription at distal p53 binding sites (>25 kb of any gene), proximal sites associate with a gene not activated by p53 (<25 kb of non-target), and those proximal to a p53 target gene identified by GRO-seq. (E) p53 binding sites near target genes have higher transcription levels than sites near other genes even in p53 null cells, indicating the likely action of pioneer factors.
Figure 5—figure supplement 1
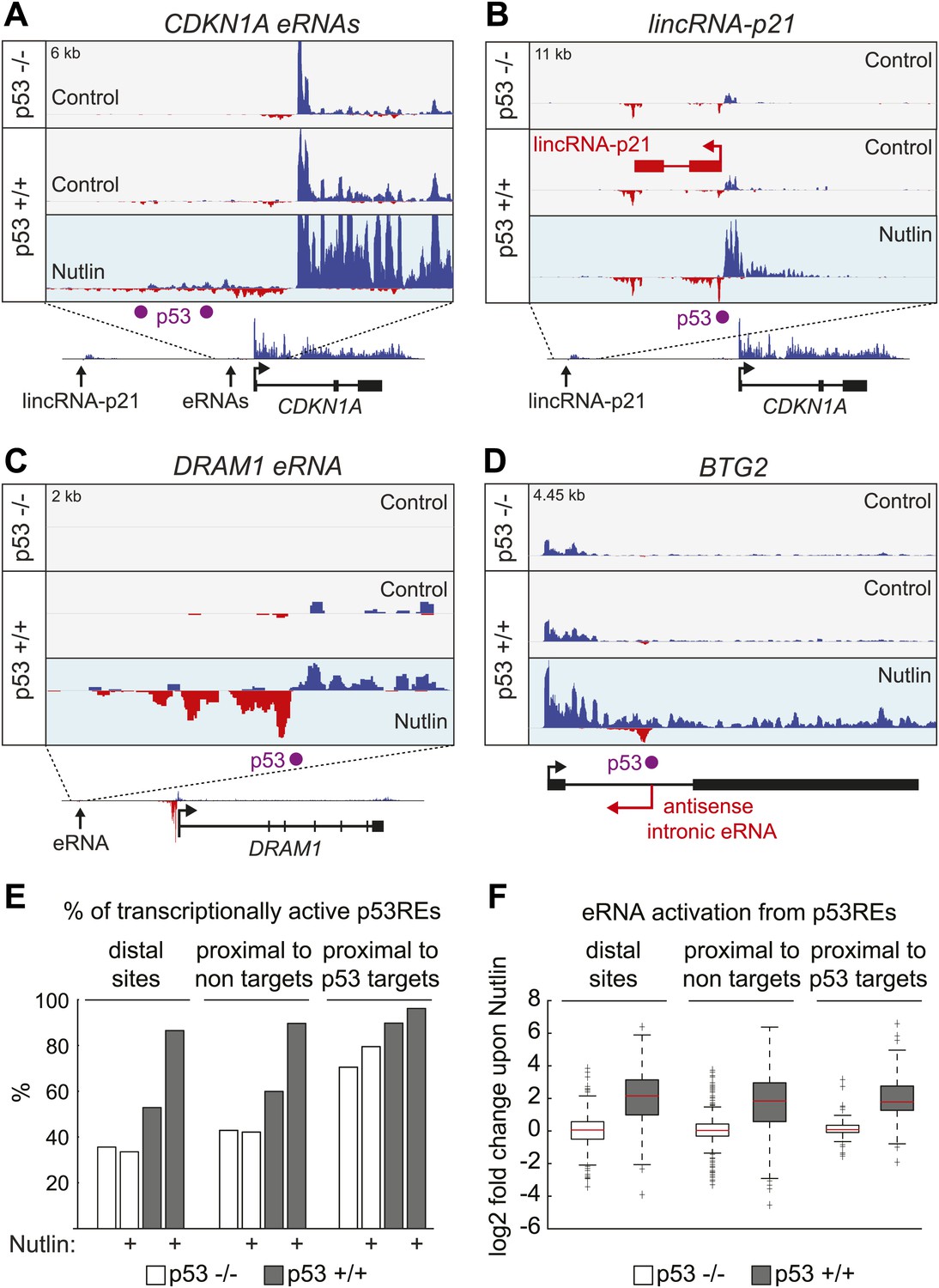
p53 stimulates eRNA production at extragenic and intragenic locations.
(A and B) GRO-seq results for the region upstream of CDKN1A showing eRNAs derived from the p53REs at −1.3 and −2.4 kb (A) and from the region encoding lincRNA-p21 (B). (C) GRO-seq results for the DRAM1 locus, a rare example of a p53 target gene whose enhancer is not obviously transcribed in p53 null cells. (D) GRO-seq results for the BTG2 locus, representative of p53 target genes that display intronic antisense eRNA transcription arising near sites of p53 binding. (E) Percentage of p53 binding sites that are transcribed as defined by Core et al. (2008). The distal sites are transcribed less often than the sites proximal to target genes. A higher percentage of sites near p53 target genes are transcribed than sites that are distal or near non-targets. (F) The fold change in transcription after addition of Nutlin is similar across p53 binding sites in various locations. Expectedly, in p53 −/− cells, there is no change in transcription after Nutlin treatment. In p53 +/+ cells eRNAs are increased in transcription is approximately fourfold and this is true for distal sites, sites proximal to non-targets and sites proximal to targets.
Additional files
-
Supplementary file 1
Genes upregulated at the transcriptional level in HCT116 p53 +/+ cells treated with 10 μM Nutlin-3a for 1 hr as detected by GRO-seq (198 genes). DeSeq algorithm was used to detect annotated gene loci whose GRO-seq signal was statistically different between DMSO- and Nutlin-treated cells (adjusted p<0.1). Columns in this table indicate: (a) Gene name, (b) Whether the gene was previously identified as a direct p53 target gene in the literature, (c–f) Whether the gene was predicted to be a direct p53 target gene by one or more recent studies employing ChIP-seq and microarrays (Figure 2—figure supplements 1 and 2), (g) fpkm in p53 +/+ control, (h) fpkm in p53 +/+ Nutlin-3, (i) Fold induction, (j) Protein Function, (k) Putative downstream pathway within the p53 network, (l) References describing the gene as a direct target, putative target or establishing gene function.
- https://doi.org/10.7554/eLife.02200.016
-
Supplementary file 2
Lists of genes bound by p53 as defined by ChIP-seq and concurrently upregulated or downregulated as defined by microarray measurements of RNA steady state levels. Related to Figure 2—figure supplement 1A,B. See ‘Materials and methods’, ‘Computational Analysis–Meta-analysis of published investigations of the p53 transcriptional program using a combination of microarray and ChIP-seq data’ for details.
- https://doi.org/10.7554/eLife.02200.017
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms
eLife 3:e02200.
https://doi.org/10.7554/eLife.02200




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}