Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding
- Harvard Medical School, United States
- Korea Brain Research Institute, Republic of Korea
- Boston Children's Hospital, Harvard Medical School, United States
- Duksung Women's University, Republic of Korea
- Dana-Farber Cancer Institute, Harvard Medical School, United States
Figures
Figure 1 with 1 supplement
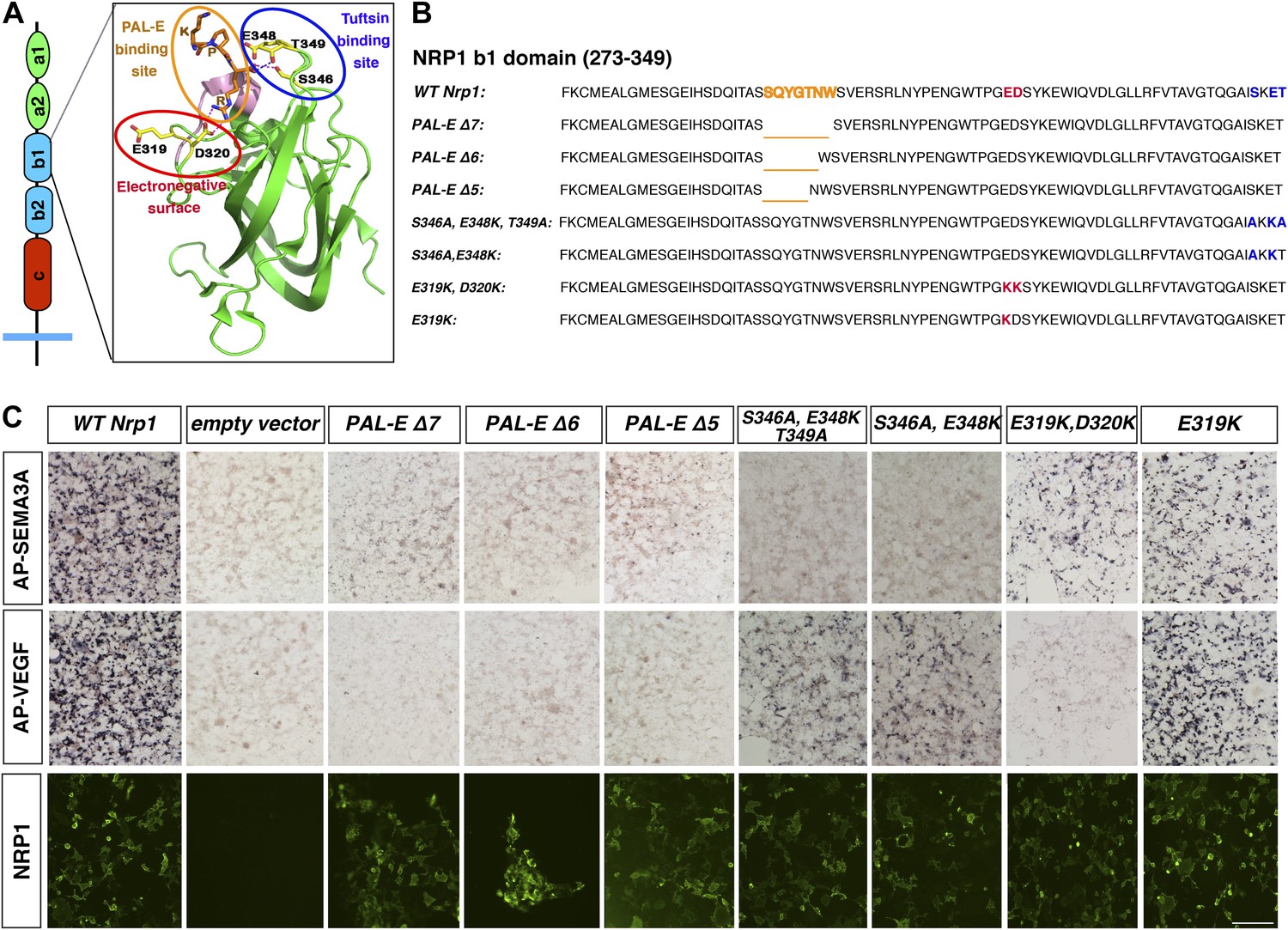
Design and assessment of Nrp1 variants harboring mutations in the VEGF-binding site.
(A) Schematic representation of the NRP1 b1 extracellular domain and crystal structure highlighting three potential mutagenesis sites: the PAL-E binding site (orange circle), tuftsin binding site (blue circle), and electronegative surface (red circle). (B) Sequence of the Nrp1 b1 domain indicating the deletion or mutation sites for the candidate constructs. (C) AP-SEMA3A (top row) or AP-VEGF (middle row) binding to COS-1 cells overexpressing the indicated constructs. Deletion of the entire PAL-E binding site (Nrp1PAL-EΔ7) or partial deletion of the PAL-E binding site (Nrp1PAL-EΔ6 and Nrp1PAL-E Δ5) eliminated both AP-SEMA3A and AP-VEGF binding. Likewise, mutations in the tuftsin binding site (S346A, E348A, T349A or S346A, E348A) abolished AP-SEMA3A binding and reduced AP-VEGF binding. Although mutations in the NRP1 electronegative surface (E319K, D320K) eliminated AP-VEGF binding and reduced AP-SEMA3A binding, the E319K mutation only slightly reduced AP-SEMA3A binding and maintained AP-VEGF binding. Antibody staining of unpermeabilized cells (lower row) demonstrated normal NRP1 surface expression. Scale bar: 50 μm.
Figure 1—figure supplement 1
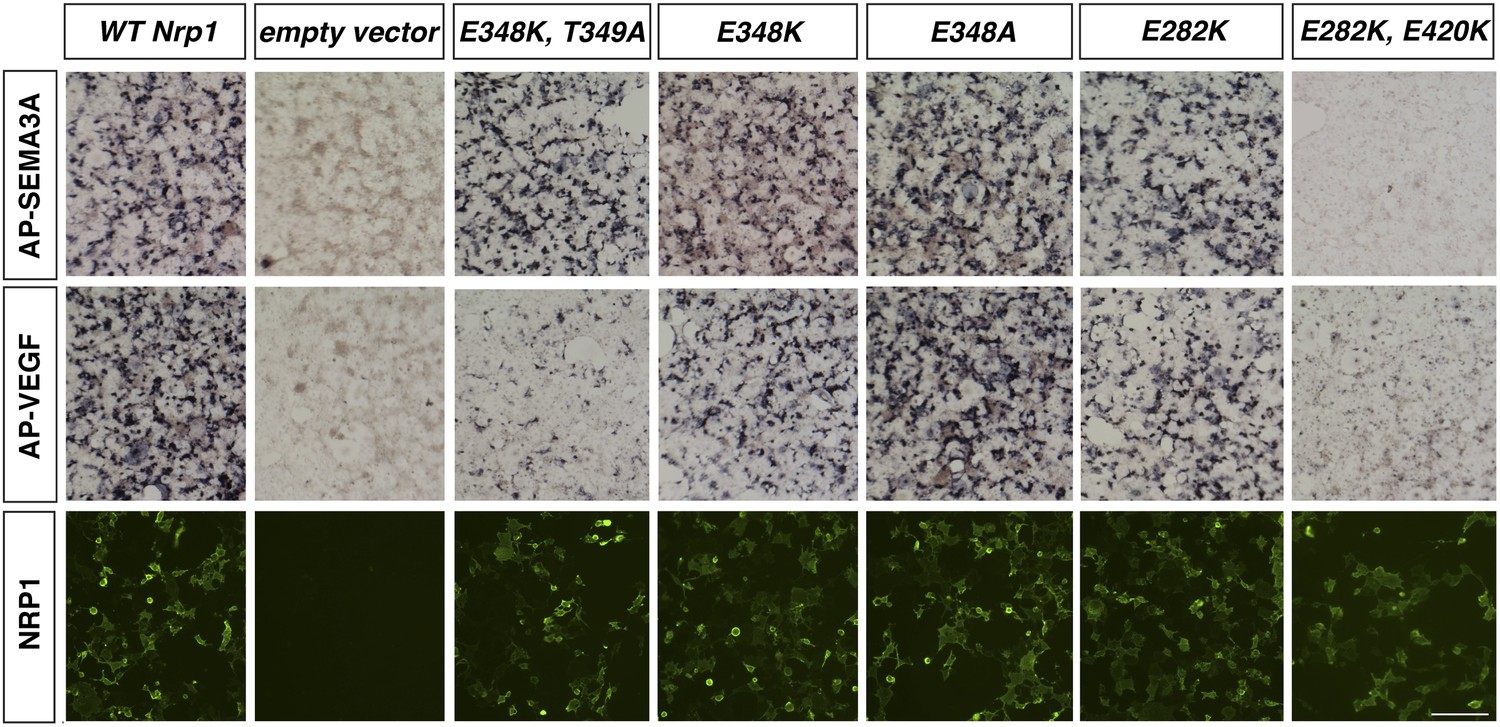
Assessment of additional Nrp1 variants containing mutations in the VEGF-binding site.
AP-SEMA3A or AP-VEGF was applied to COS-1 cells overexpressing the indicated construct (top and middle row). Non-permeabilized antibody staining was performed with a polyclonal anti-NRP1 antibody to detect NRP1 surface expression (bottom row). Scale bar: 50 μm.
Figure 2 with 1 supplement
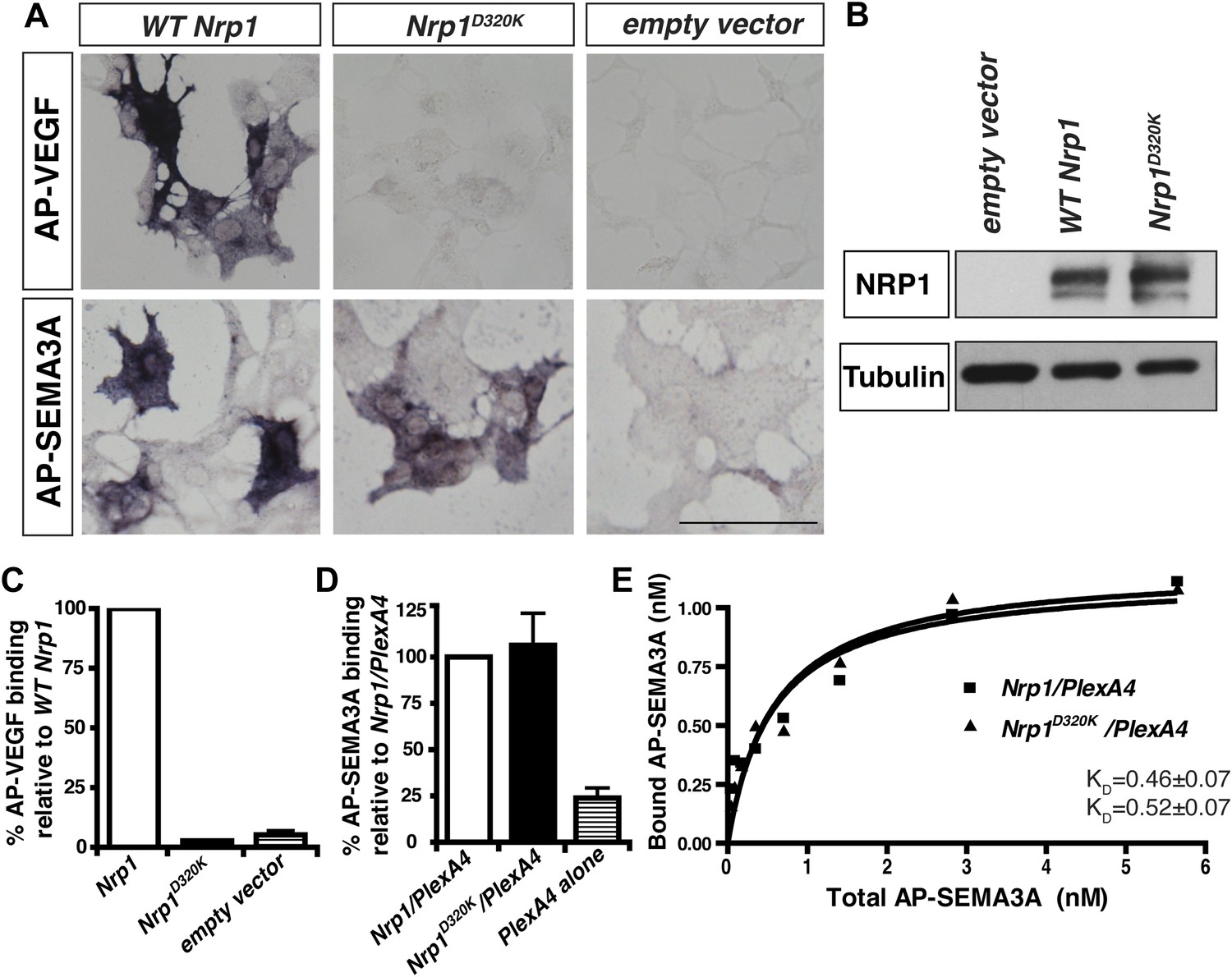
The Nrp1D320K mutation selectively eliminates VEGF-NRP1 binding in vitro.
(A) AP-VEGF binding in COS-1 cells overexpressing the indicated Nrp1 construct. WT NRP1 bound AP-VEGF strongly, while AP-VEGF binding to NRP1D320K was abolished. Scale bar: 100 μm (B) Western blot shows that equivalent levels of NRP1 protein in COS-1 cells transfected with the WT Nrp1 and Nrp1D320K. (C) Quantification of the binding assay shows that AP-VEGF-NRP1D320K binding was abolished even after normalization for protein content and NRP1 expression. (D) Quantification of AP-SEMA3A binding shows comparable AP-SEMA3A binding to WT NRP1 and NRP1D320K. (E) Measurement of the dissociation constant (KD) of AP-SEMA3A demonstrates that AP-SEMA3A bound to the NRP1D320K/PlexA4 complex with the same affinity as the NRP1/PlexA4 complex.
Figure 2—figure supplement 1
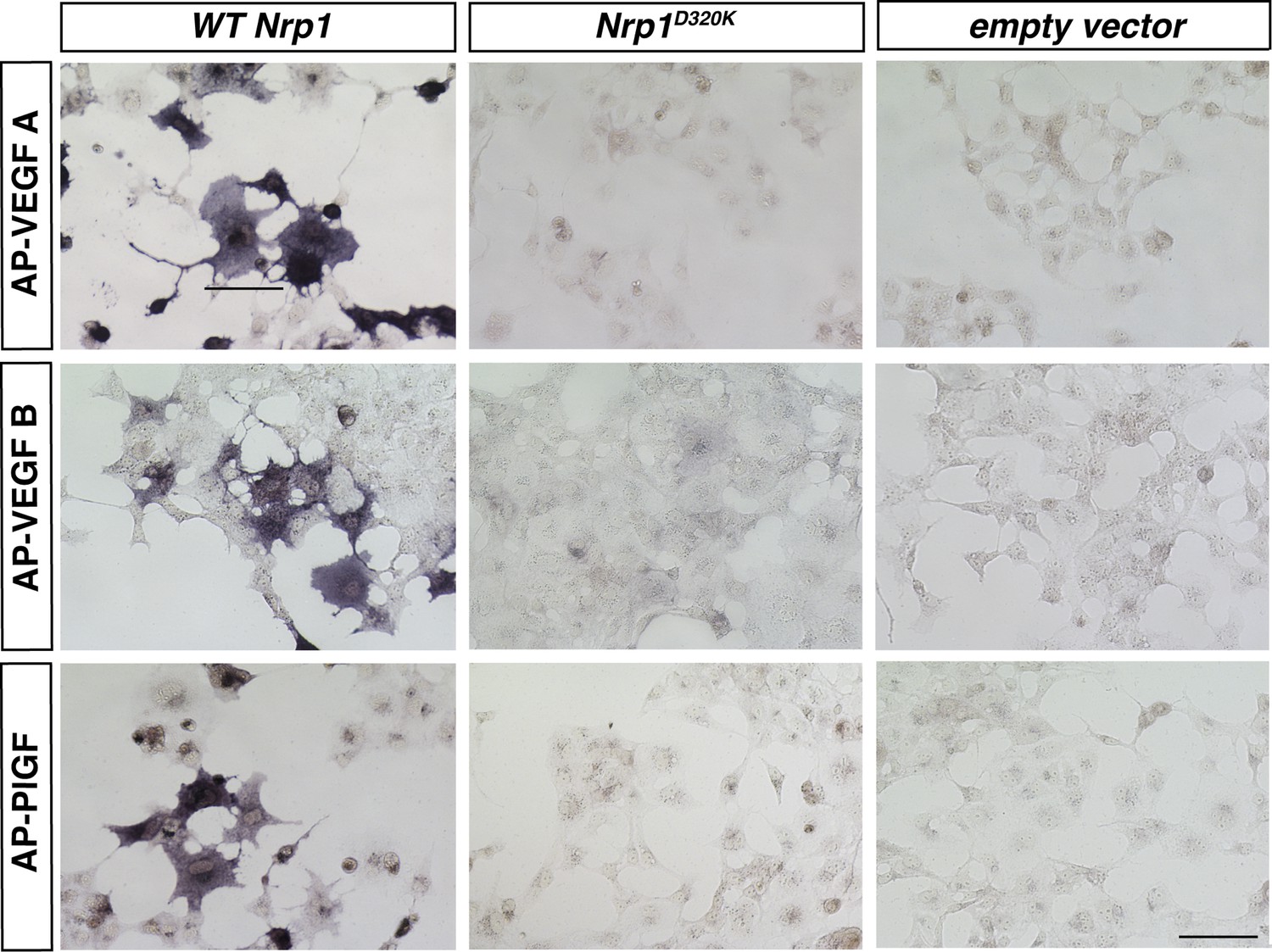
VEGFA, VEGFB, and PLFG binding to NRP1 was abolished in the Nrp1D320K mutant.
Nrp1 constructs were overexpressed in COS-1 cells, and AP-VEGFB or AP-PlGF was applied to cells to observe ligand binding. WT NRP1 bound AP-VEGFB and AP-PlGF strongly, while these ligands did not bind to NRP1D320K. Scale bar: 100 μm.
Figure 3 with 2 supplements
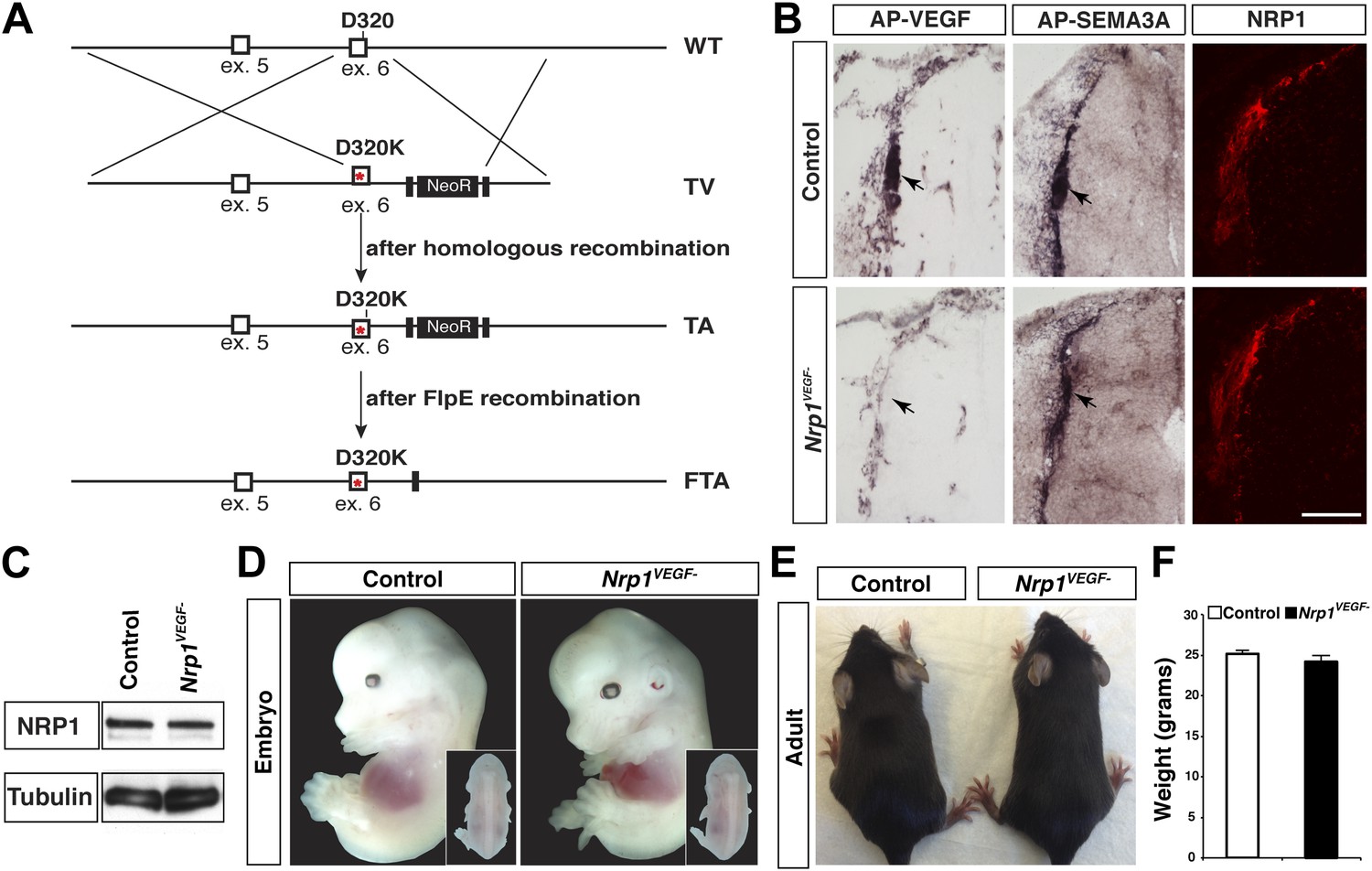
Nrp1VEGF- mice selectively abolish VEGF-NRP1 binding in vivo.
(A) Targeting vector design for the generation of Nrp1VEGF− mice. The WT genomic region contained residue D320 in exon 6 of Nrp1. The targeting vector (TV) introduced the D320K mutation along with an Frt-flanked NeoR cassette to form the targeted allele (TA). After FlpE-mediated excision of the NeoR cassette, the final targeted allele (FTA) had the D320K mutation as well as one remaining Frt site. (B) Section binding assays demonstrated that AP-VEGF binding to the dorsal root entry zone (DREZ) was abolished in the Nrp1VEGF− mutants (arrows, left panels) while AP-SEMA3A binding to the DREZ appeared similar between Nrp1VEGF− and control animals (arrows, middle panels). Scale bar: 100 μm. (C) Western blot from E14.5 lung tissue shows that NRP1 protein level was not affected in Nrp1VEGF− animals. (D and E) Nrp1VEGF− mutants appear indistinguishable from controls littermates at embryonic (E14.5) and adult stages. (F) Nrp1VEGF− mutants exhibit normal body weight in adulthood (n = 7, males).
Figure 3—figure supplement 1
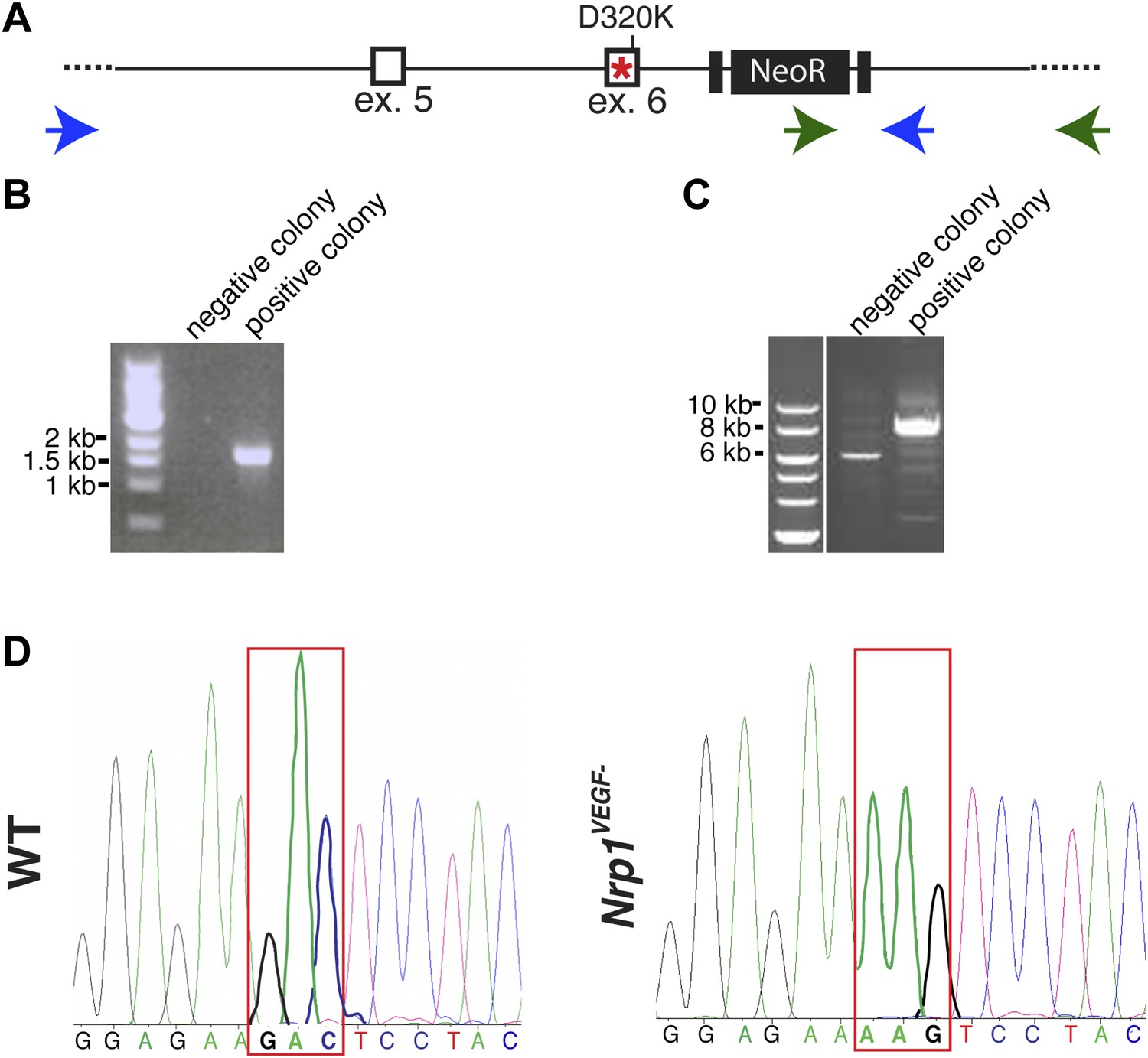
Screening and verification of ES cells for the generation of the Nrp1VEGF− mutant.
(A) Diagram of the Nrp1 genomic region following successful homologous recombination to insert the targeting vector. The green arrows indicate the primers used in (B), while the blue arrows represent the primers used in (C). (B) PCR screening for the proper insertion of the 3′ homology arm. The 5′ primer was located in the NeoR sequence while the 3′ primer bound to an area outside of the targeting vector. Therefore, WT colonies did not produce a band, while correctly targeted clones produced a band of 1.7 kb. (C) PCR screening for the proper insertion of the 5′ homology arm. The 5′ primer was located outside of the targeting vector area and the 3′ primer was located within the genomic sequence present in the 3′ homology arm. Thus, PCR from a properly targeted clone produced a fragment that was 1.5 kb larger than a negative colony. (D) Sequencing of the D320K region in WT and Nrp1VEGF− homozygous mutants. The boxed region indicates the altered codon.
Figure 3—figure supplement 2
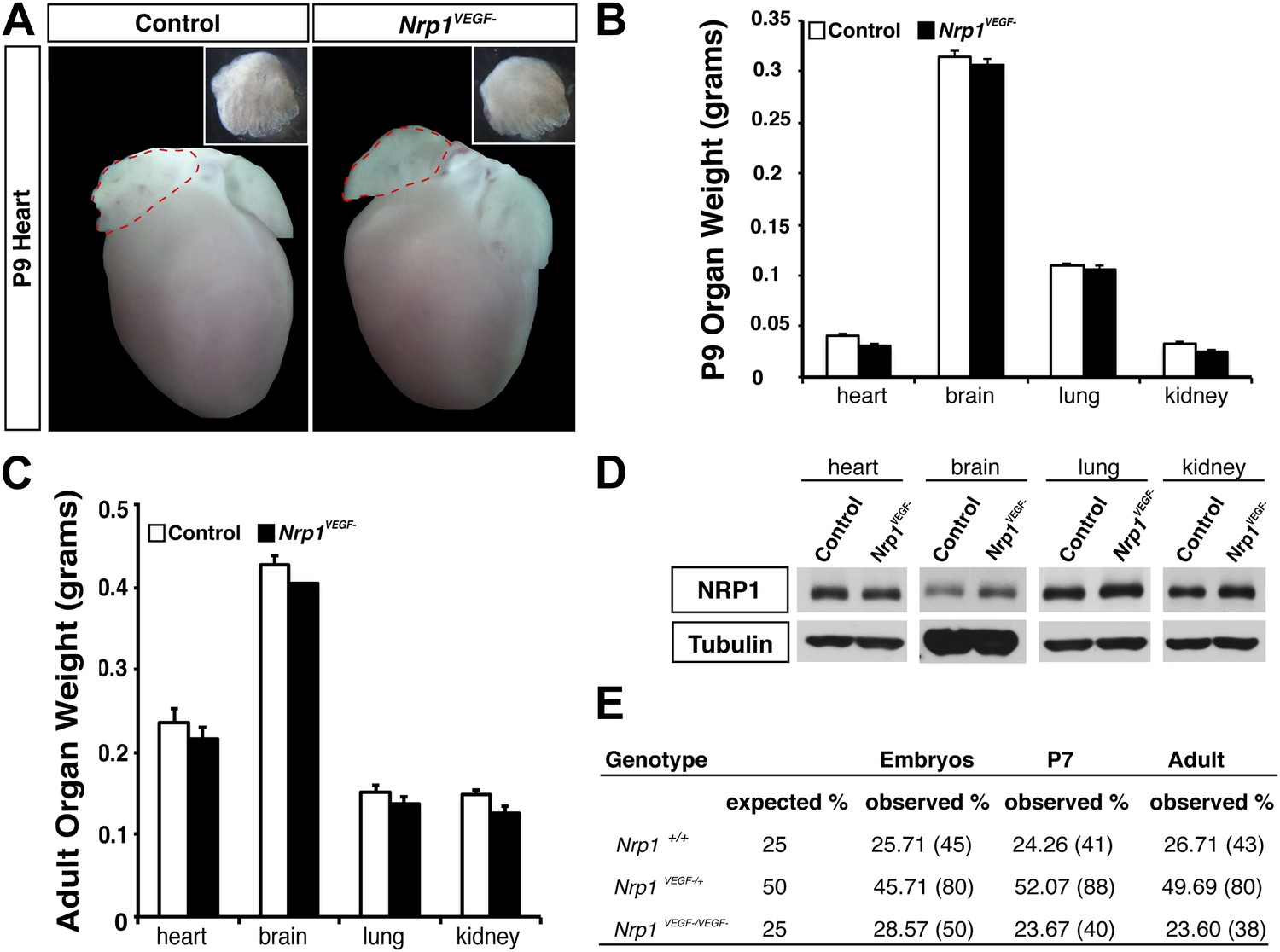
The Nrp1VEGF− mutant mice exhibit normal gross morphology.
(A) Whole-mount images of the heart at P9 show the normal cardiac morphology of the Nrp1VEGF− mutants. (B and C) Organ weights measured at P9 (B) and adulthood (C) demonstrate that the heart, brain, lung, and kidney undergo appropriate growth in Nrp1VEGF− animals, n ≥ 5. (D) Western blots from adult heart, brain, lung, and kidney tissue demonstrate that NRP1 protein levels were not affected in Nrp1VEGF− animals. (E) Viability table depicts the predicted and observed frequencies for each genotype at the indicated developmental stages. The table values represent the percentage of the total number of animals genotyped per age while the total number of animals is shown in parentheses.
Figure 4 with 1 supplement
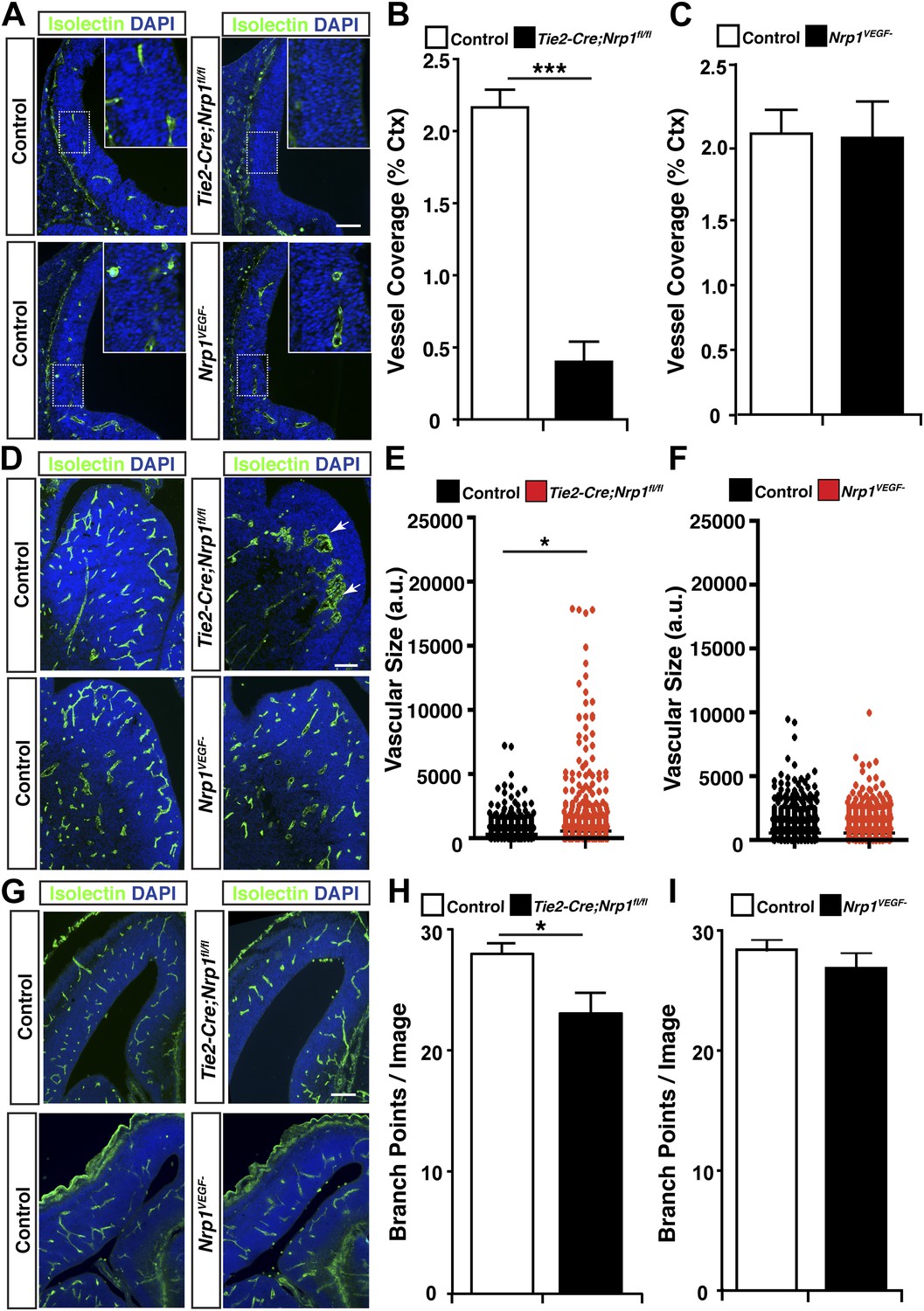
VEGF-NRP1 binding is not required for developmental angiogenesis.
(A) Vessel staining with isolectin (green) revealed that Tie2-Cre;Nrp1fl/fl mutants had delayed vessel ingression into the cerebral cortex at E11.5 while the Nrp1VEGF− mutants exhibited normal ingression. DAPI was used to stain the nuclei (blue). (B and C) Quantification of cortical vessel ingression shown in A, n = 3. (D) Tie2-Cre;Nrp1fl/fl mutants exhibited large vessel clumps in the brain (particularly in the striatum) at E14.5, a phenotype not observed in the Nrp1VEGF− mutants. (E and F) Quantification of vessel size in E14.5 striatum shown in D, n = 3. (G) Tie2-Cre;Nrp1fl/fl mutants had reduced vessel branching in the cerebral cortex while the Nrp1VEGF− mutants displayed normal vessel branching at E14.5. (H and I) Quantification of vessel branching in E14.5 cortex shown in G, n = 4. Scale bar: 200 μm.
Figure 4—figure supplement 1
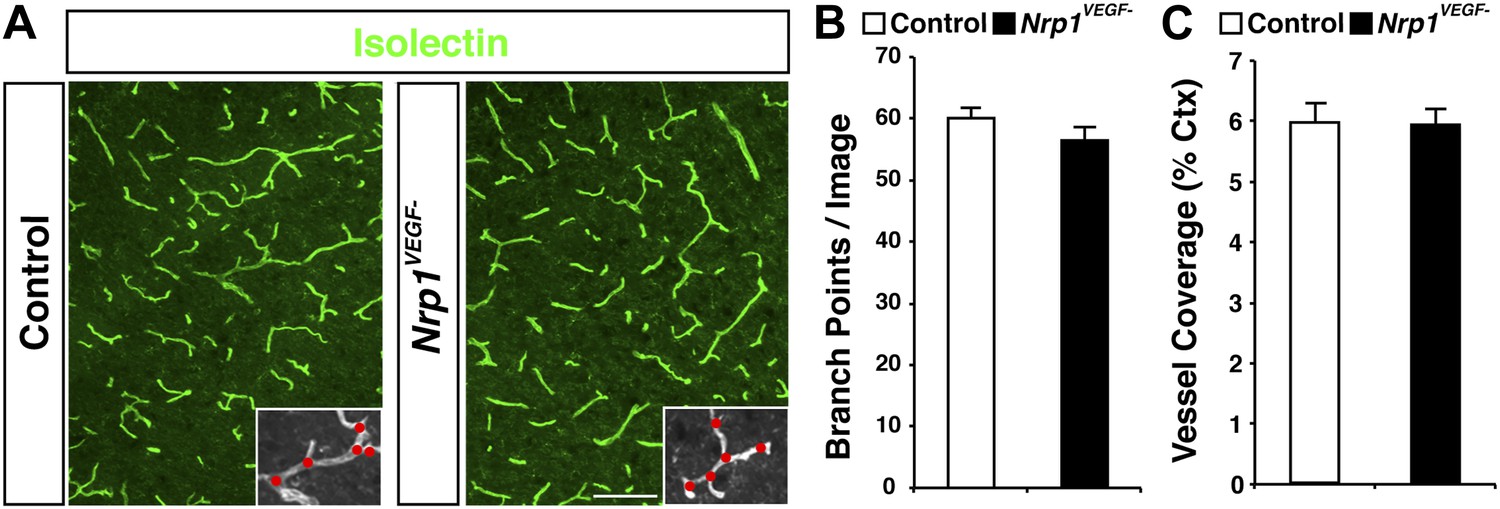
The Nrp1VEGF− mutant mice display normal vessel branching and coverage at postnatal stages.
(A) Vessel staining with isolectin (green) demonstrates that the Nrp1VEGF− mutants have normal vessel coverage and branching in the cerebral cortex at P7. (B and C) Quantification of vessel coverage and branching in P7 cortex shown in A, n = 3. Scale bar: 200 μm.
Figure 5 with 2 supplements
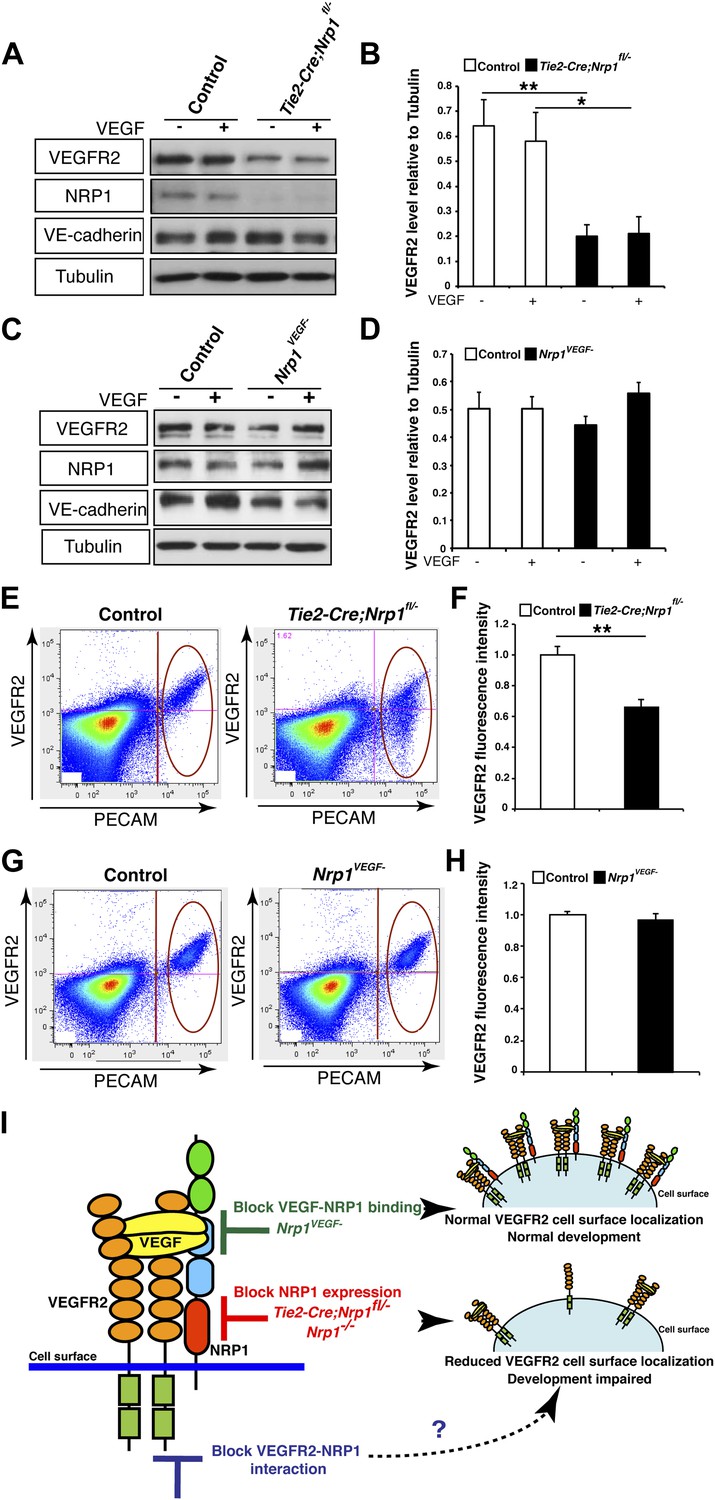
NRP1 regulates VEGFR2 expression at the cell surface independent of VEGF-NRP1 binding.
(A) Western blot from E14.5 lung tissue treated with 50 ng/ml VEGF for 15 min revealed that VEGFR2 was reduced in Tie2-CreNrp1fl/− mutants while VE-cadherin expression remained at control levels. Western blot for NRP1 demonstrates that the Tie2-Cre allele successfully knocked down NRP1 expression. (B) Quantification of VEGFR2 expression shown in A, n = 4. (C) Western blot from E14.5 lung tissue treated with 50 ng/ml VEGF for 15 min demonstrates that VEGFR2, NRP1, and VE-cadherin expression were unperturbed in the Nrp1VEGF− mutants. (D) Quantification of VEGFR2 expression shown in C, n = 5. (E) FACS analysis plots illustrate a reduction in VEGFR2 surface expression in endothelial cells isolated from Tie2-Cre;Nrp1fl/− mice. (F) Quantification of the VEGFR2 fluorescence intensity from the FACS analysis shown in E, n = 5. (G) FACS analysis plots demonstrate that VEGFR2 surface expression in endothelial cells isolated from Nrp1VEGF− mice remained at control levels. (H) Quantification of the VEGFR2 fluorescence intensity from the FACS analysis shown in G, n ≥ 7. (I) Schematic of VEGFR2 and NRP1 at the cell surface illustrates VEGF ligand binding to both VEGFR2 and NRP1. In the Nrp1VEGF− mutants, VEGF-NRP1 binding is abolished, VEGFR2 has normal cell surface localization, and vascular development proceeds appropriately. However, in Nrp1−/− mutants, VEGFR2 cell surface localization is reduced and vascular development is impaired.
Figure 5—figure supplement 1

VEGF-NRP1 binding is not required for NRP1-VEGFR2 complex formation in vitro and in vivo.
(A) HEK293T cells transfected with Vegfr2 and either WT Nrp1 or Nrp1D230K exhibited normal NRP1-VEGFR2 complex formation. (B) Lung lysates generated from the Nrp1VEGF− mutants also displayed normal NRP1-VEGFR2 complex formation comparable to littermate controls.
Figure 5—figure supplement 2

VEGF-induced VEGFR2 phosphorylation is reduced in both the Nrp1VEGF− and Tie2-Cre;Nrp1fl/− mutants.
(A) Western blot from E14.5 lung tissue shows that VEGFR2 phosphorylation upon VEGF treatment was diminished in the Nrp1VEGF− mutant. (B) Quantification of VEGFR2 phosphorylation shown in A, n = 7. (C) Western blot from E14.5 lung tissue demonstrates that VEGFR2 phosphorylation is significantly reduced in the Tie2-Cre;Nrp1fl/− mutants. (D) Quantification of VEGFR2 phosphorylation shown in B, n = 5.
Figure 6 with 1 supplement
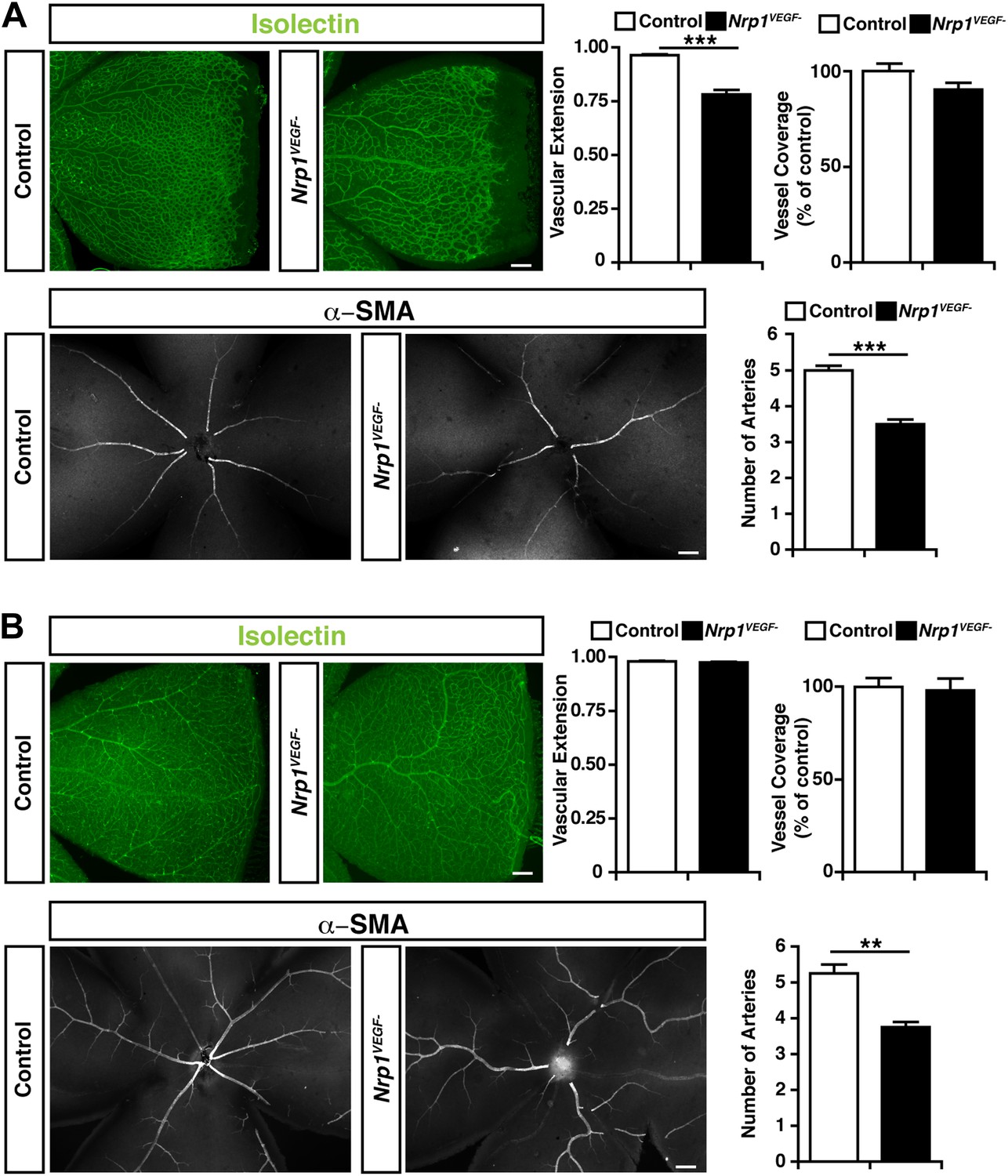
Retinal angiogenesis is perturbed in the Nrp1VEGF− mutant.
(A) Isolectin and α-SMA staining on P9 retinal flat-mounts revealed a significant reduction in vascular extension and artery number in Nrp1VEGF− mutants. However, vessel coverage in the retina was unperturbed in the Nrp1VEGF− mutants, n = 6. (B) In the adult, isolectin and α-SMA staining showed that the number of retinal arteries remained lower in the Nrp1VEGF− mutants than littermate controls while vascular extension and vessel coverage in the retina were normal, n = 4. Scale bar: 200 μm.
Figure 6—figure supplement 1
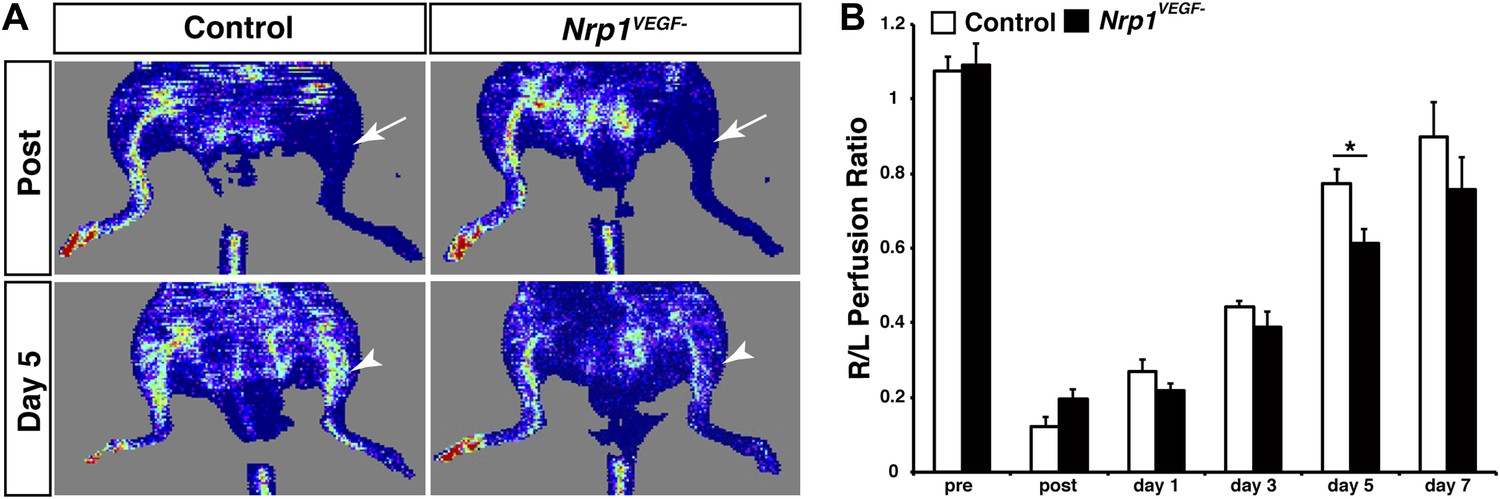
The Nrp1VEGF− mutants have delayed blood flow recovery following femoral artery ligation.
(A) Laser doppler imaging demonstrates severe hindlimb ischemia directly after femoral artery ligation in both control and Nrp1VEGF− animals (arrows). Five days after surgery, blood flow recovery in the injured hindlimb was significantly greater in control vs Nrp1VEGF− animals (arrowheads). (B) Quantification of blood flow recovery following femoral artery ligation, n = 7.
Additional files
-
Supplementary file 1
Primers used for generating, genotyping, and sequencing the Nrp1VEGF- knock-in mouse line.
- https://doi.org/10.7554/eLife.03720.017
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding
eLife 3:e03720.
https://doi.org/10.7554/eLife.03720




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}