In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse
- University of California, Los Angeles, United States
- Laboratory of Functional Genomics and Protein Engineering, Italy
- Howard Hughes Medical Institute, University of California, Los Angeles, United States
Figures
Figure 1 with 2 supplements
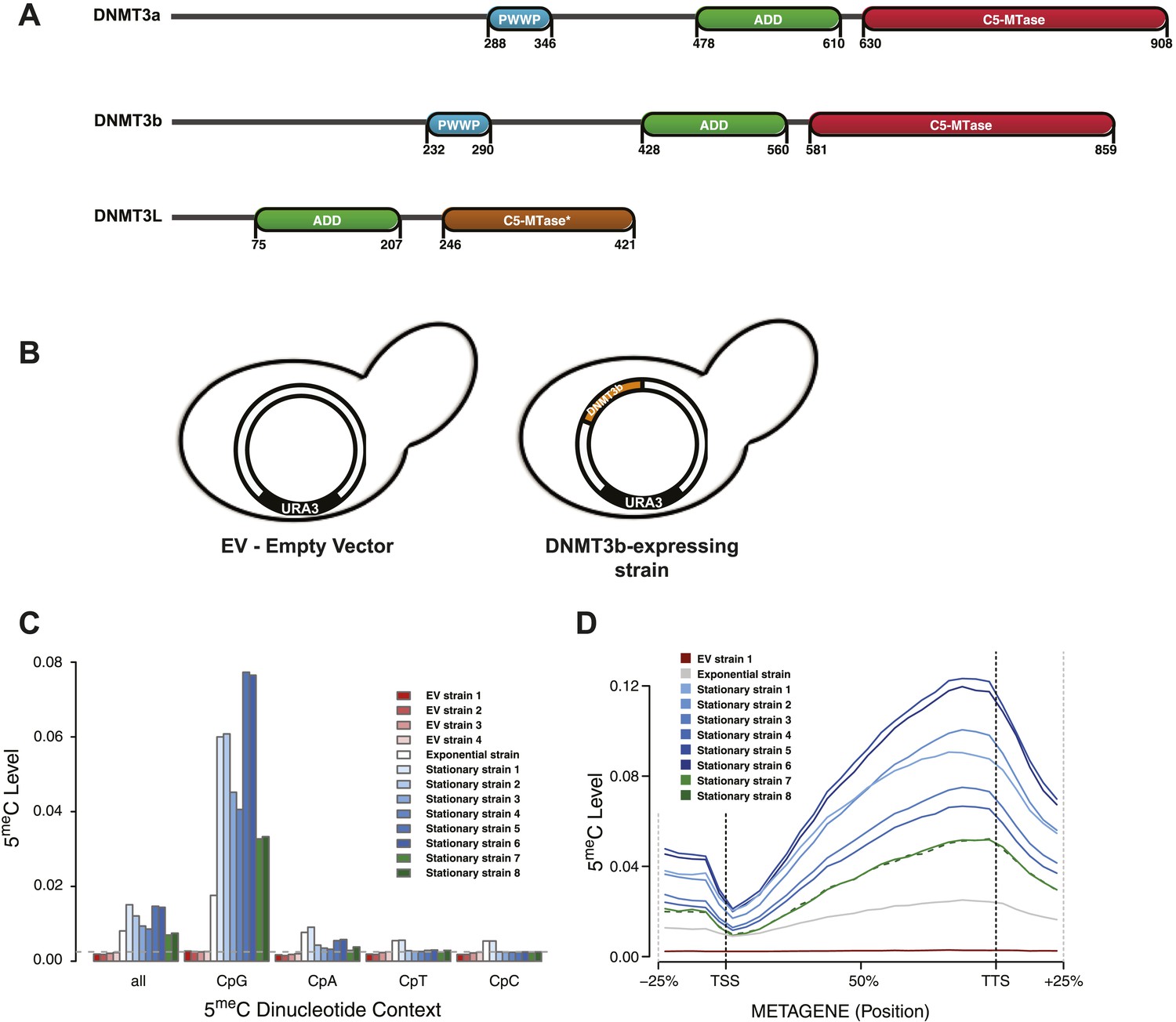
Distribution of induced DNA methylation in Saccharomyces cerevisiae.
(A) Murine DNMT3 proteins with known domains: PWWP, ADD (ATRX–DNMT3–DNMT3L), and C-5 methyltransferase domain (not functional in DNMT3L). Accession numbers: DNMT3a = O88508; DNMT3b = O88509; DNMT3L = Q9CWR8. (B) Constructs used in this study. The empty vector (EV) is pYES2 (Life Technologies). DNMT3b expression is controlled by the GAL1 promoter. (C) Levels of 5meC in different dinucleotide contexts. The gray dotted line represents the unconversion rate. (D) Metagene plot of CpG methylation in cells expressing DNMT3b during logarithmic and stationary phase. EV (strain not expressing DNMT3b). Exponential and stationary strains 1–6 are derived from the W303 strain, while stationary strains 7 and 8 are in a BY4741 background.
Figure 1—figure supplement 1
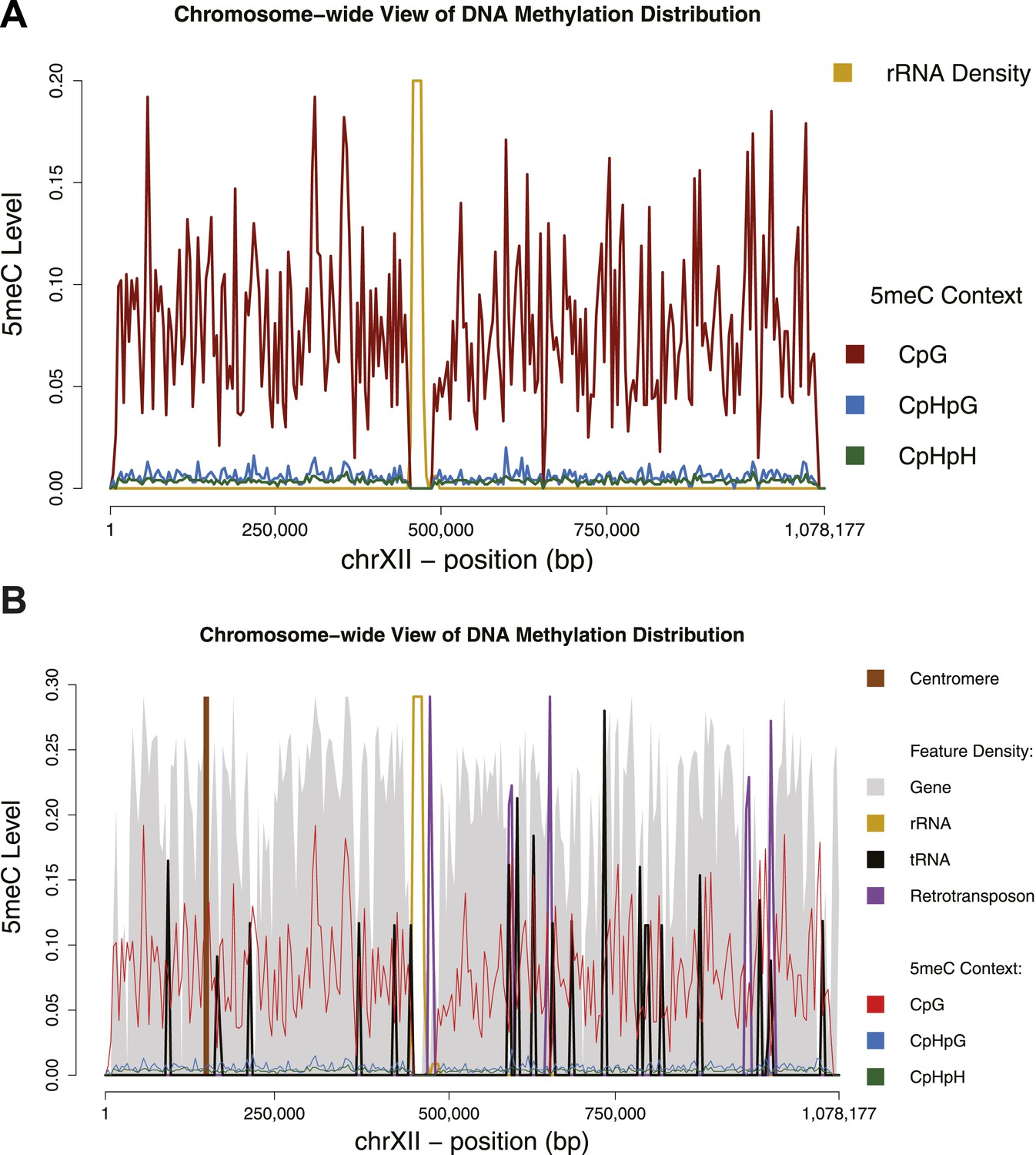
Chromosome-wide view of DNA methylation and genomic features.
Distribution of DNA methylation on chromosome XII of S. cerevisiae (A and B). In (B) the density of other genomic features is shown (arbitrary units). Averages for DNA methylation and genomic features are calculated on 4 Kb bins. Areas of repetitive sequences (such as rRNA and transposable elements) show very little to no coverage. Gene-rich bins also correspond to peaks in DNA methylation levels.
Figure 1—figure supplement 2
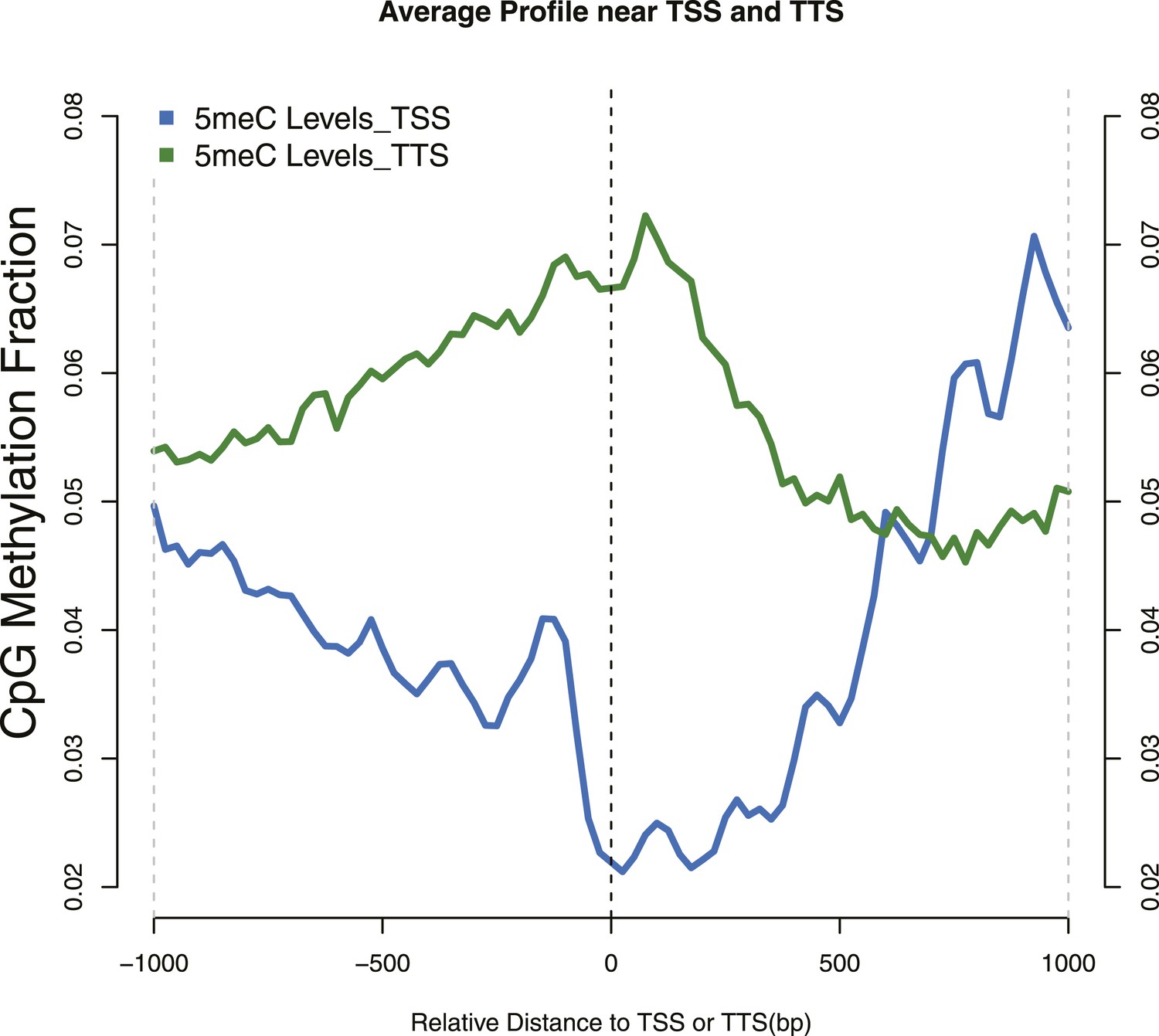
Distribution of 5meC around TSS and TTS.
CpG methylation levels around (TSSs—blue) and (TTSs—green) of yeast genes. Periodic peaks of DNA methylation are evident at the TSS, where nucleosomes form a well positioned array.
Figure 2 with 1 supplement

Influence of nucleosome positioning on DNA methylation.
Average distribution of nucleosomes and DNA methylation (CpG context) around (A) Transcriptional Start Site (TSS), (B) Transcriptional Termination Site (TTS), and (C) nucleosome centers. (D) Meta-nucleosome plot of CpG methylation. a.u. = Arbitrary units.
Figure 2—figure supplement 1
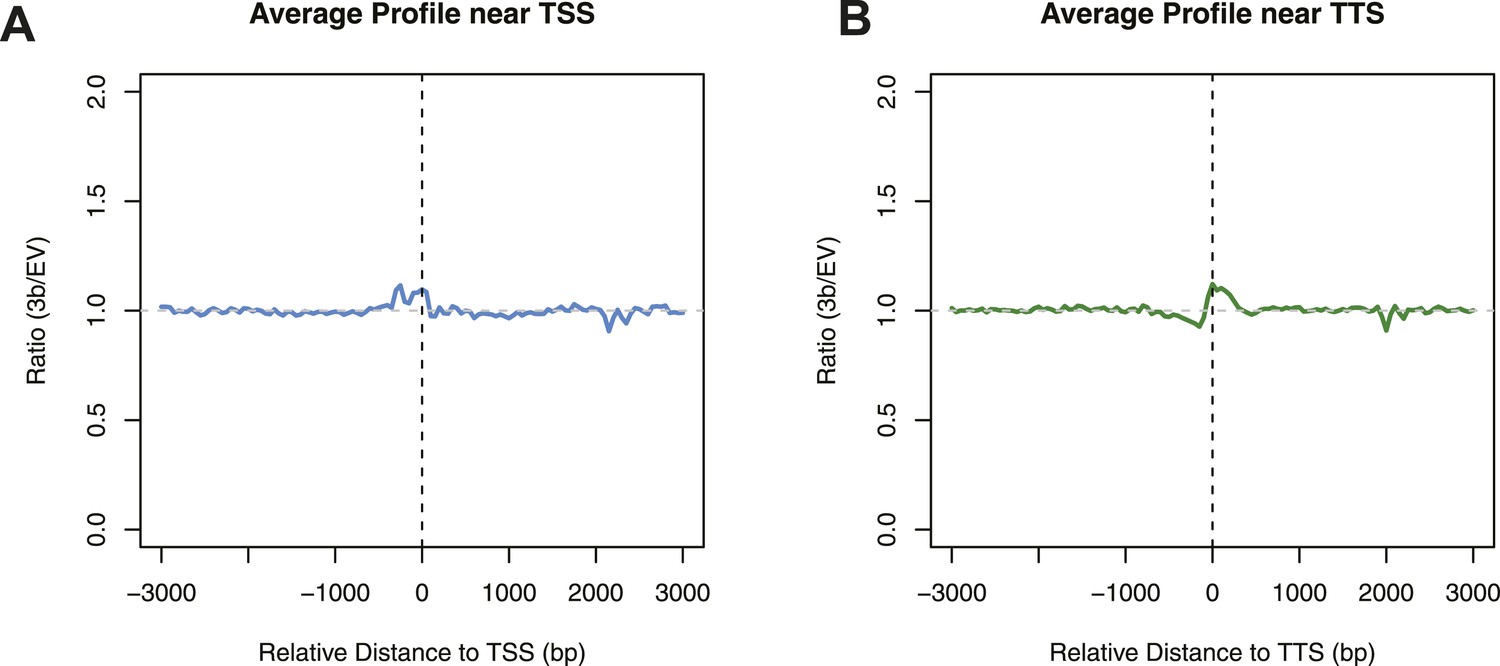
Differences in nucleosome occupancy between DNMT3b-expressing and non-expressing yeast strains.
Ratio of nucleosome occupancy between DNMT3b-expressing (3b) and non-expressing (EV) yeast strains at TSS (A) and TTS (B).
Figure 3 with 2 supplements
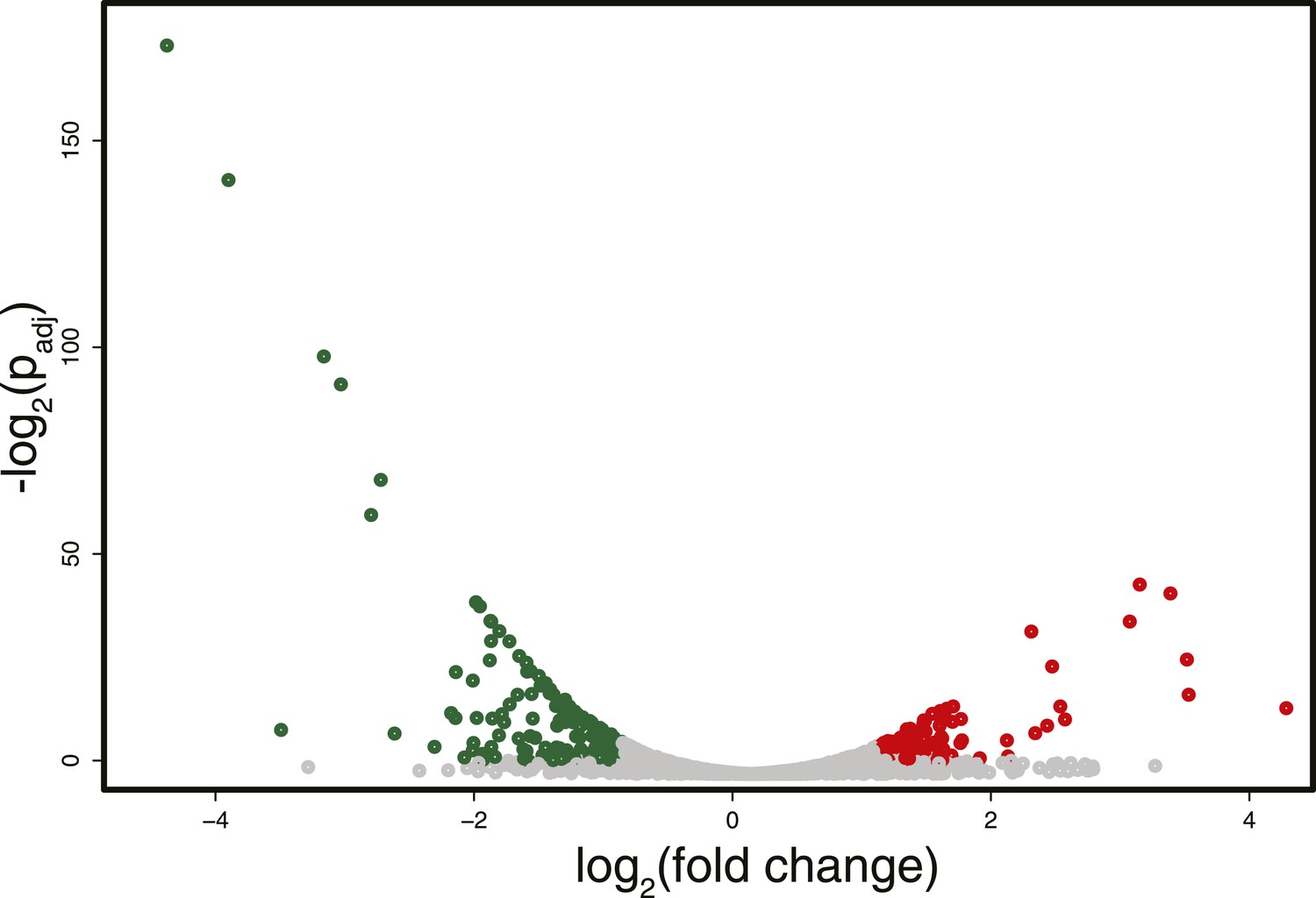
Differences in RNA expression between DNMT3b-expressing and non-expressing yeast strains.
The expression difference in RNA expression between DNMT3b and EV strains is plotted on the x axis, and false discovery rate (FDR)-adjusted significance is plotted on the y-axis (–log2 scale). Upregulated and downregulated RNAs shown in red and green, respectively. Significantly expressed RNAs have a fold change bigger than two with a FDR smaller than 0.1.
Figure 3—figure supplement 1
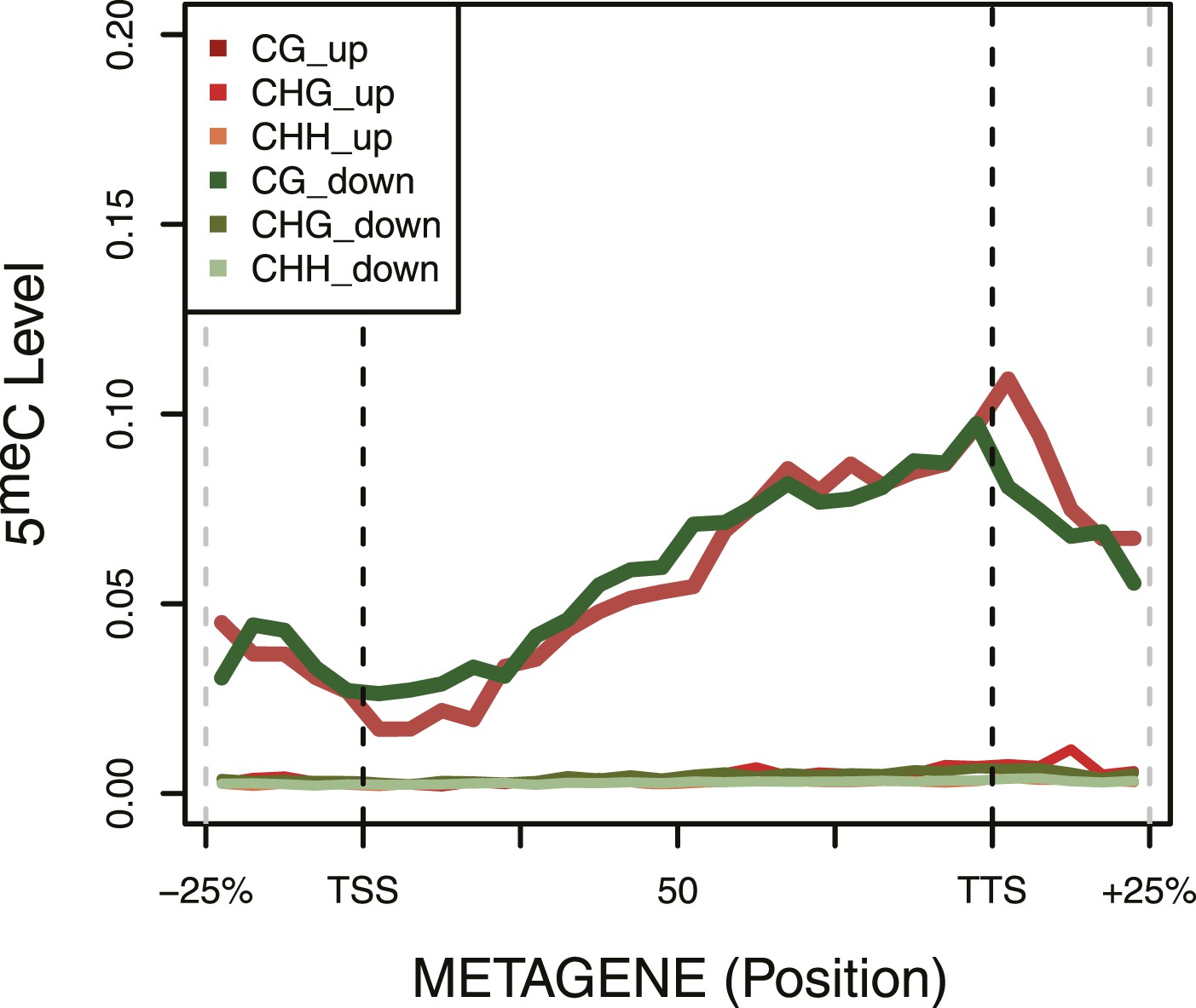
DNA Methylation in up- and down-regulated genes.
Metagene plot of 5meC in different contexts (CpG, CpHpG, CpHpH) of upregulated (red) and downregulated (green) genes.
Figure 3—figure supplement 2
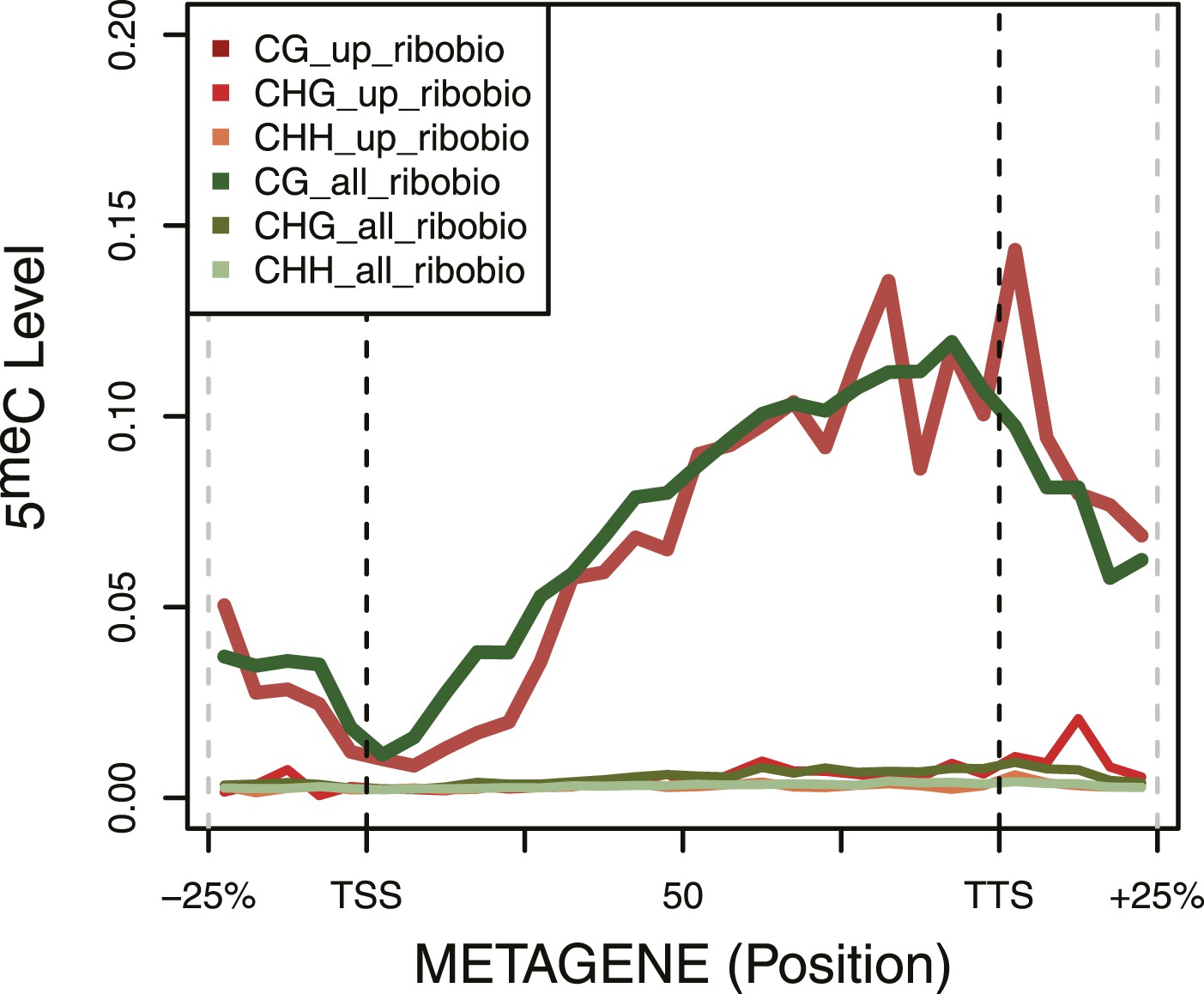
DNA Methylation in ribosomal biogenesis genes.
Metagene plot of 5meC in different contexts (CpG, CpHpG, CpHpH) of upregulated ribosomal biogenesis genes (red) compared to all the genes of the same class (green).
Figure 4 with 5 supplements
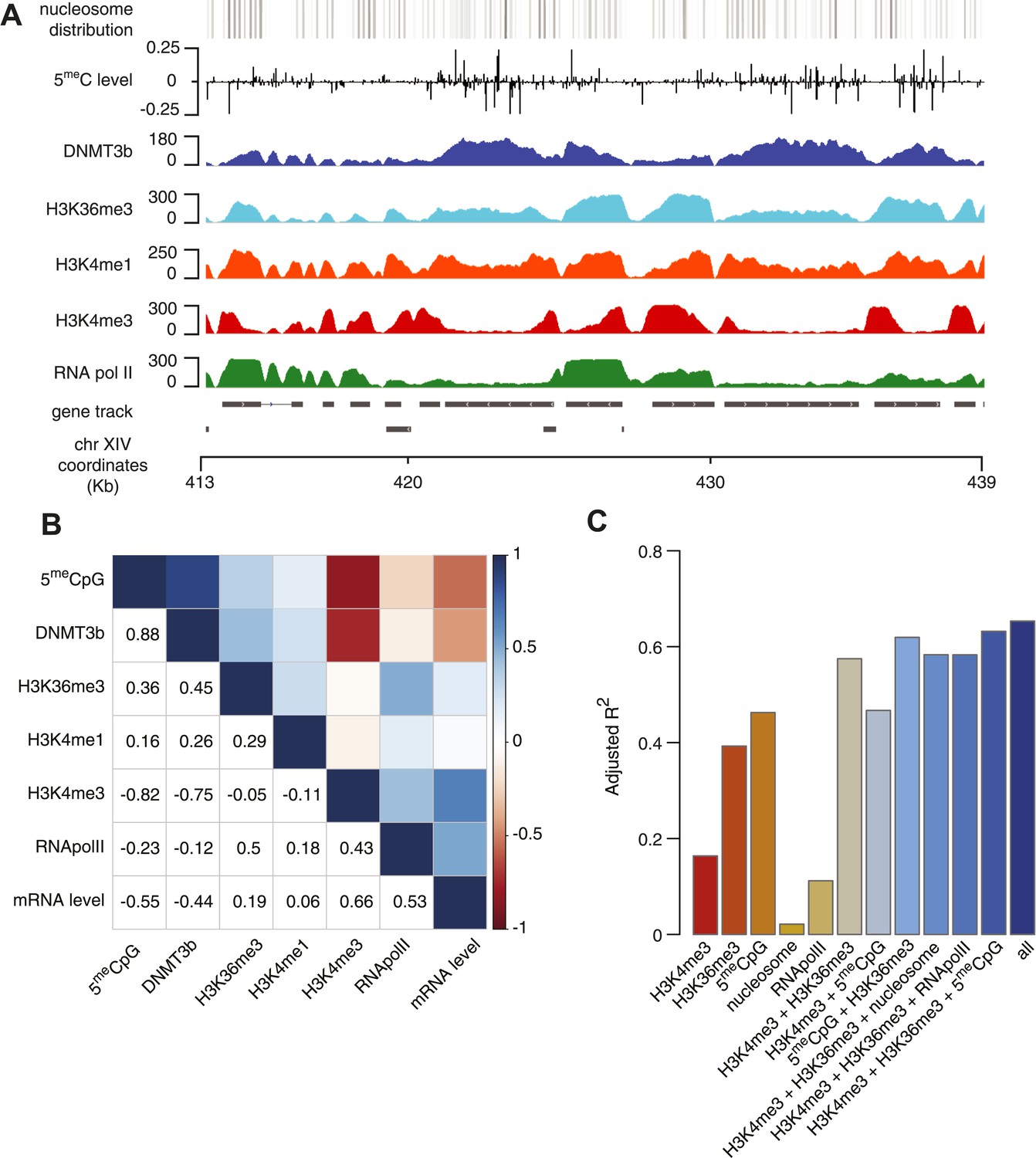
Correlation between histone marks and DNA methylation.
(A) Genome-wide distribution of nucleosome, 5meC, DNMT3b, H3K36me3, H3K4me1, H3K4me3, and RNA polymerase II. (B) Spearman correlation coefficients between 5meC, histone marks, RNA polymerase II, DNMT3b and mRNA average levels for protein coding genes. (C) Prediction of DNMT3b levels using DNA methylation, H3K4 and H3K36 trimethylation, RNA polymerase II and nucleosome distribution as predictors. The y-axis shows the adjusted R2 value between the predicted linear model and observed values.
Figure 4—figure supplement 1
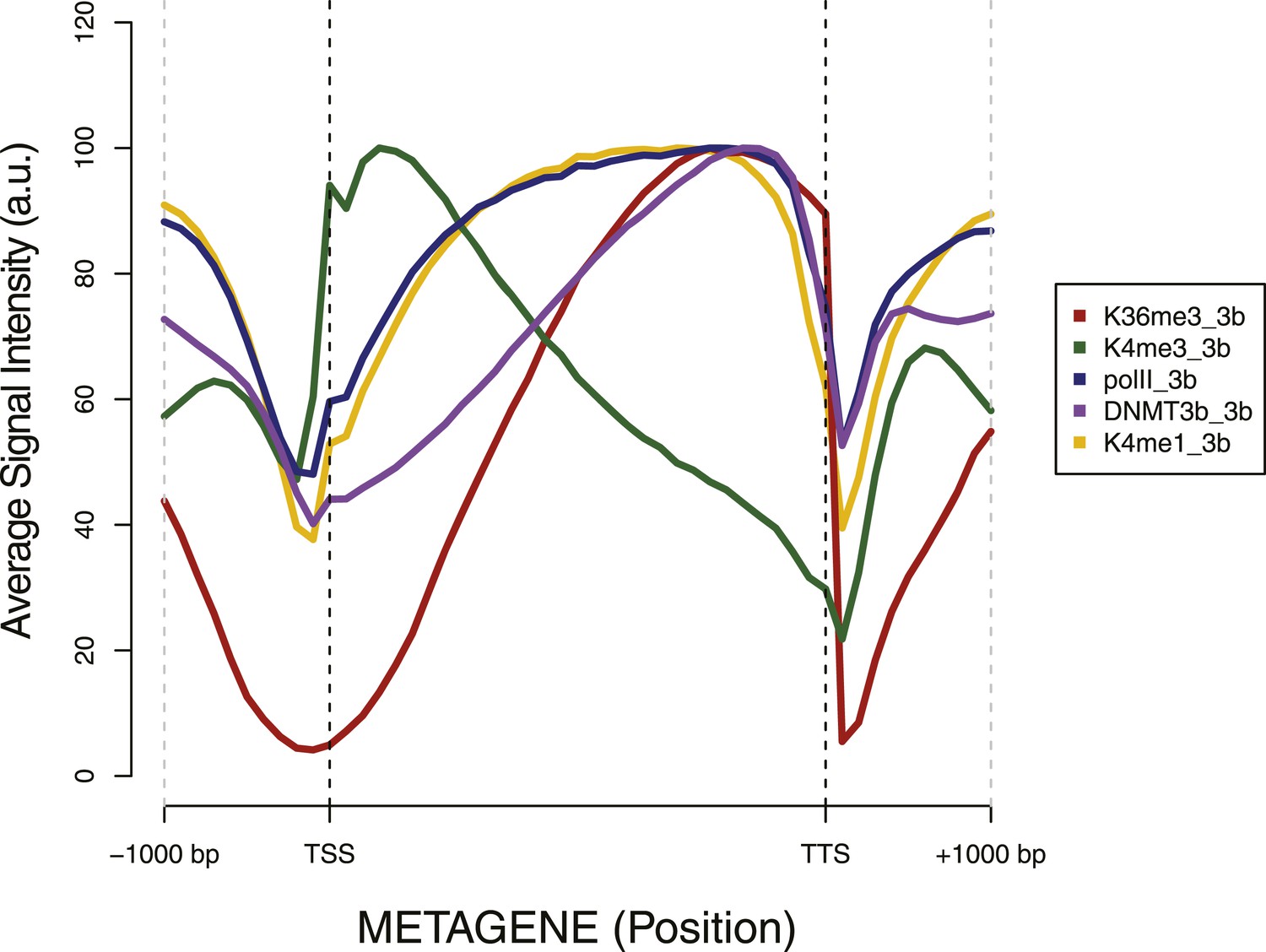
Metagene plot of ChIP sequencing in a DNMT3b-expressing strain.
ChIP-seq reads average intensity across yeast genes and 1 Kb upstream and downstream.
Figure 4—figure supplement 2
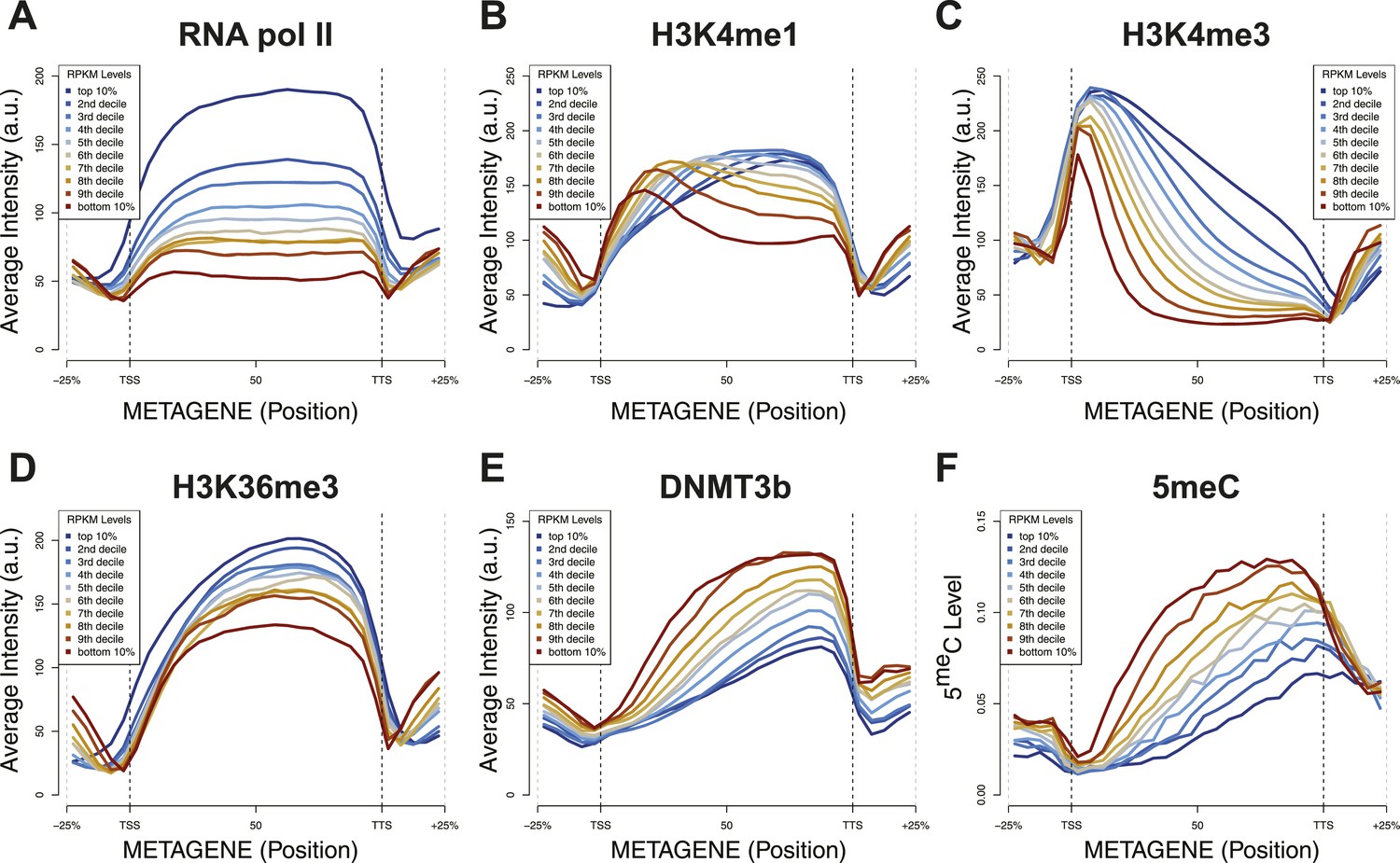
Relationship between transcription and 5meC or histone marks levels.
Average ChIP-seq intensity (A–E) or 5meC levels (F) across yeast genes divided in deciles based on RNA values (RPKM). a.u. = Arbitrary units.
Figure 4—figure supplement 3

Relationship between DNA methylation and histone marks levels.
Average ChIP-seq (A–E) or DNA methylation (F) distribution across yeast genes divided in deciles based on average 5meCpG intragenic levels. a.u. = Arbitrary units.
Figure 4—figure supplement 4
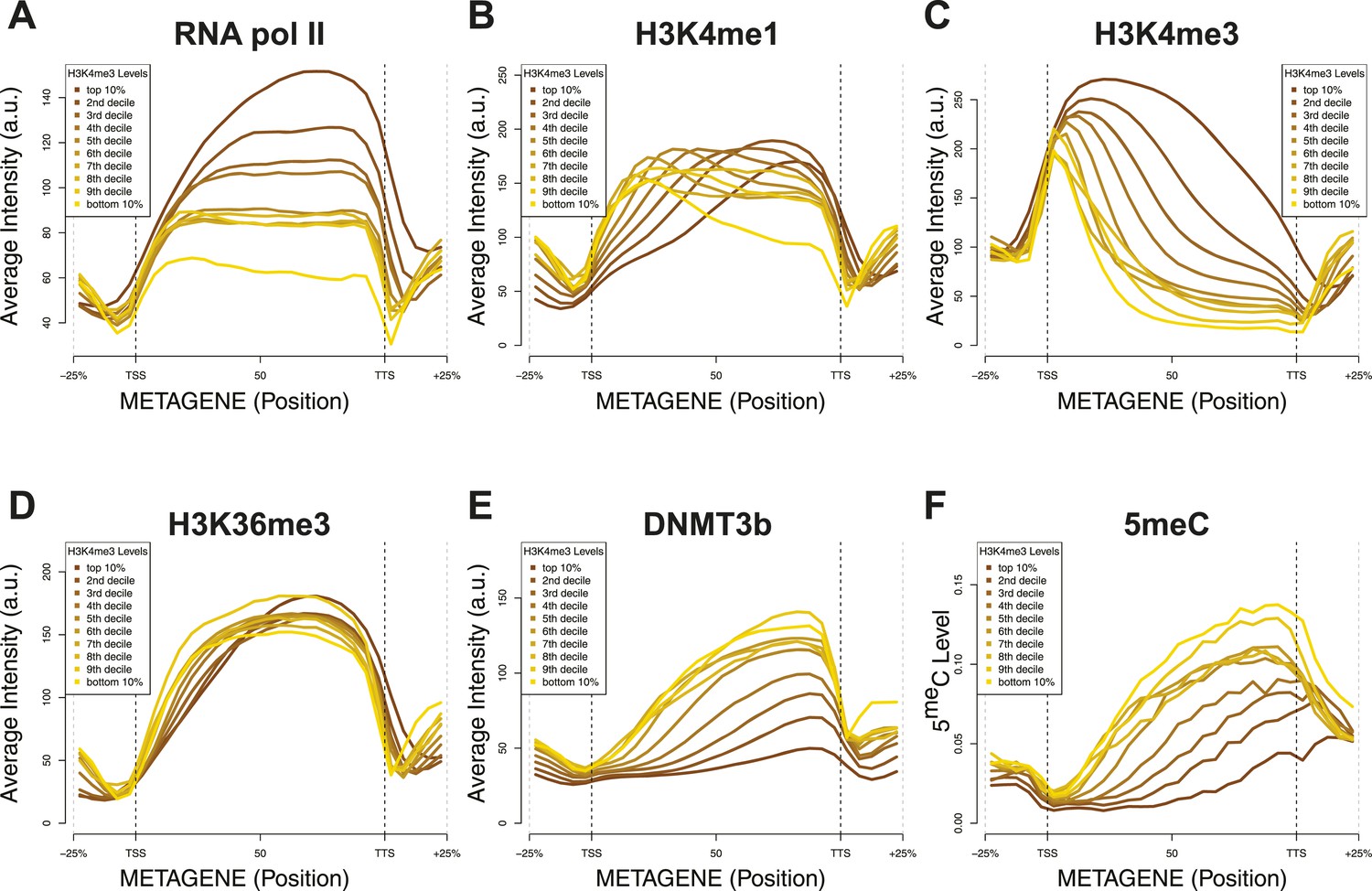
Relationship between H3K4me3 and 5meC or histone marks levels.
Average ChIP-seq intensity (A–E) or 5meC levels (F) across yeast genes divided in deciles based on H3K4me3 average in the last third of each gene. a.u. = Arbitrary units.
Figure 4—figure supplement 5
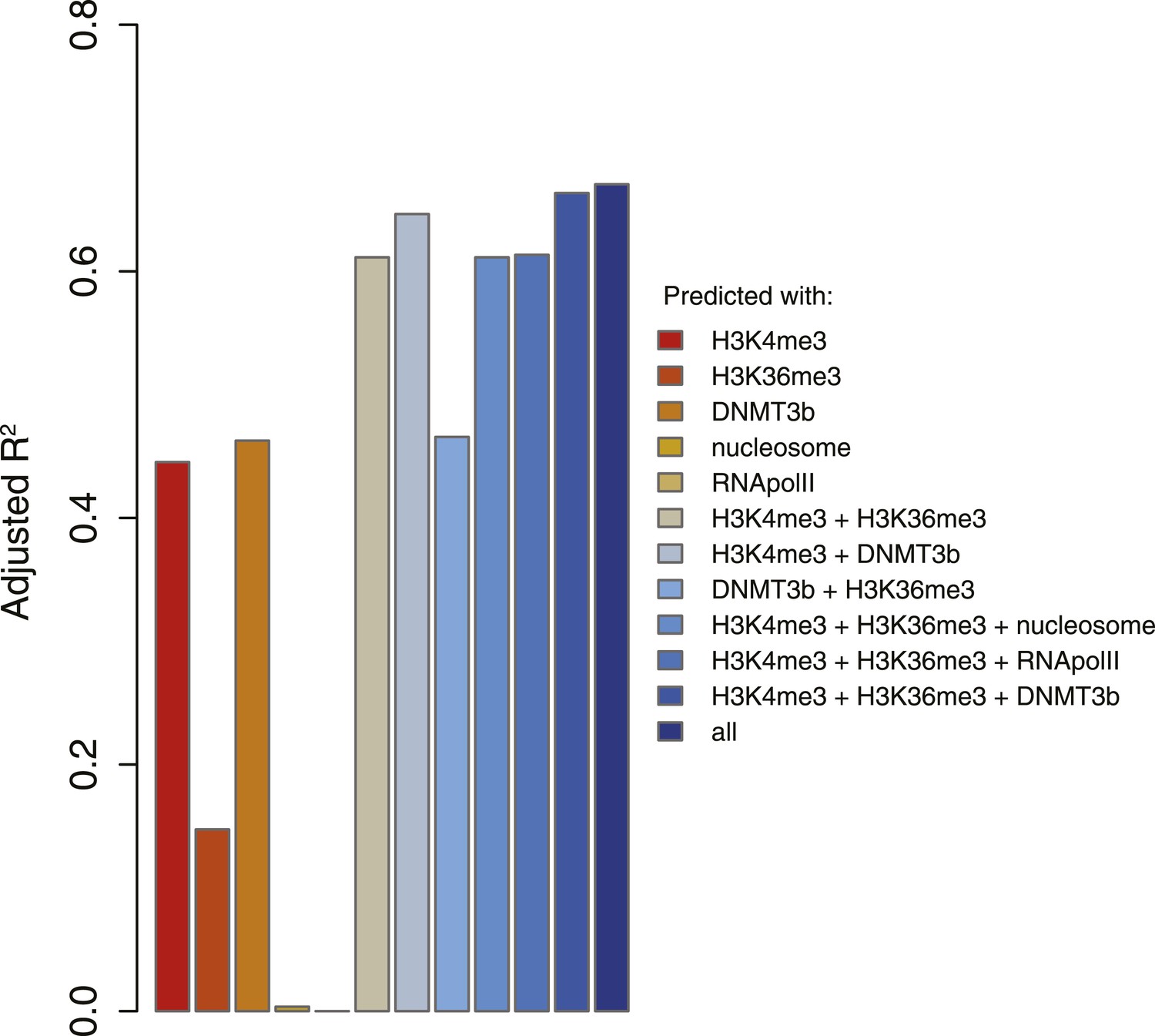
5meC levels prediction using chromatin marks.
Prediction of 5meC levels across the genome divided in 200-bp bins with a linear multivariate regression method using several combinations of chromatin marks. On the y-axis the adjusted R2 value is reported.
Figure 5 with 1 supplement
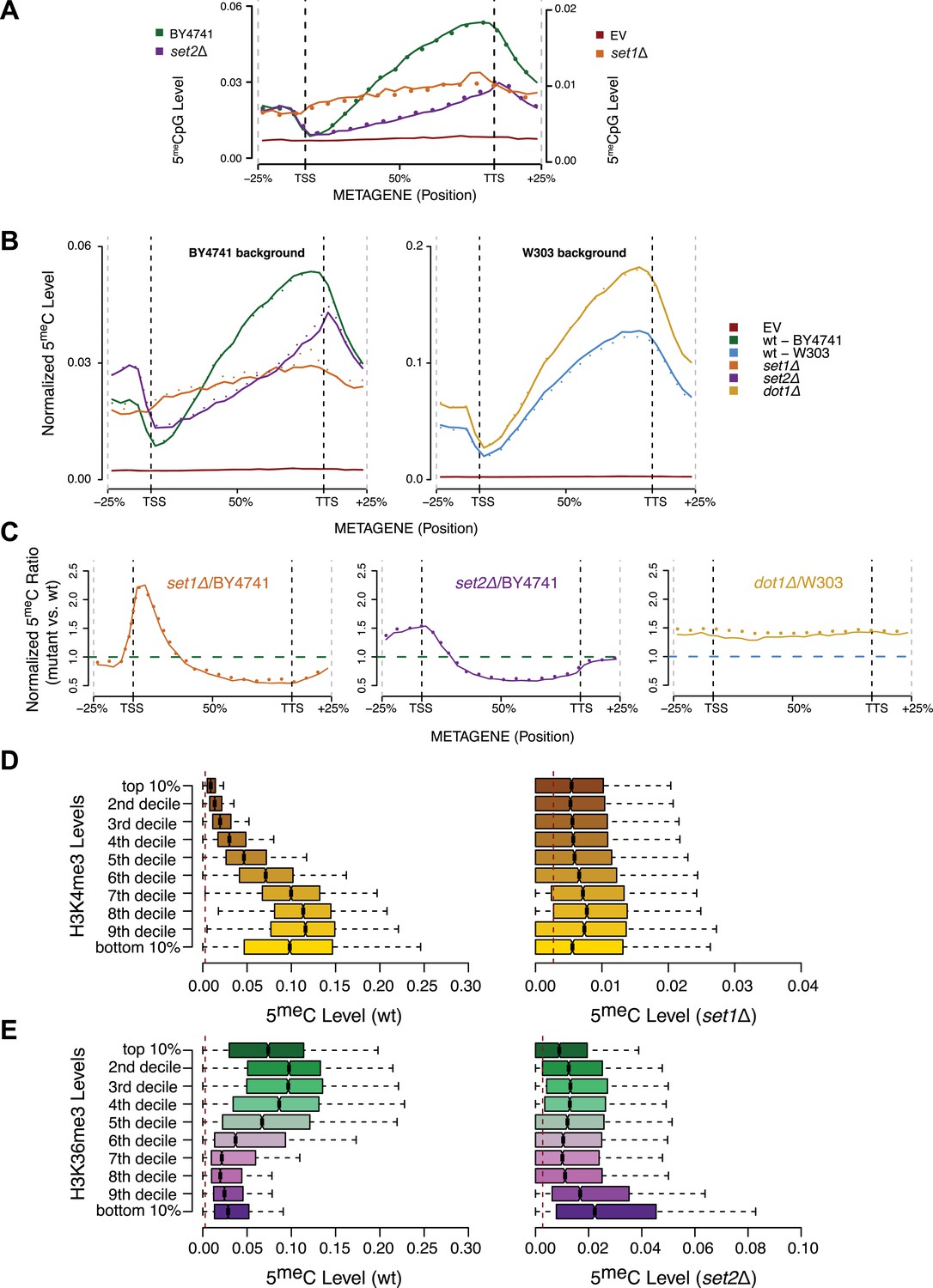
Effect of histone lysine methyltransferase deletions on the distribution of DNA methylation.
(A) Metagene plot of CpG methylation in set1Δ and set2Δ cells expressing DNMT3b. Differently from Figure 5B, 5meC levels are not normalized. Replicates of the same strain are represented as dotted lines. Data from BY4741-derived strains. BY4741 = Wild type (wt); EV = Empty vector. (B) Metagene plot of CpG methylation in set1Δ, set2Δ, and dot1Δ cells expressing DNMT3b. set1Δ, set2Δ are in a BY4741 background, while dot1Δ is in a W303 background. 5meC levels are normalized by DNMT3b expression measured by RT-qPCR. Two replicates for each strain are shown (solid and dotted line). (C) Metagene plots of CpG methylation ratio between the mutant and its wt counterpart. Two replicates for each mutant strain are shown (solid and dotted line). Wt ratios (=1) are represented by the horizontal dashed line (green or blue). (D) Boxplots showing levels of DNA methylation in the wt (left) and set1Δ strain (right) of 200-bp genome bins sorted into deciles by H3K4me3 level. (E) Boxplots showing levels of DNA methylation in the wt (left) and set2Δ strain (right) of 200-bp genome bins sorted into deciles by H3K36me3 level. The dashed red line represents background levels of DNA methylation due to incomplete bisulfite conversion (>99.7%).
Figure 5—figure supplement 1
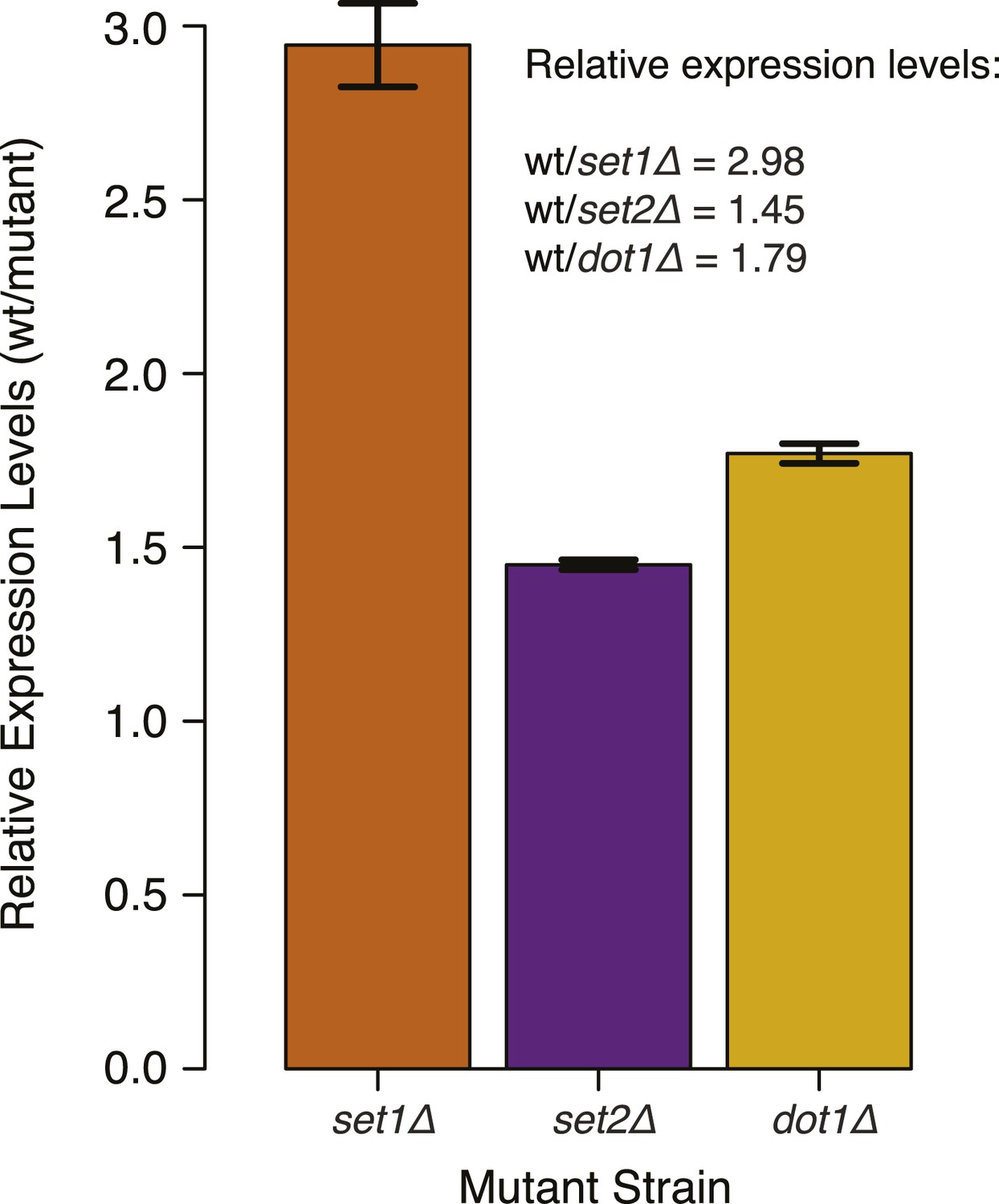
DNMT3b transcript levels in different yeast strains.
Expression levels of DNMT3b of the wild-type strain compared to yeast mutants (set1Δ, set2Δ, and dot1Δ). The relative levels were calculated using the ΔΔCt method (Schmittgen and Livak, 2008) using TDH1 gene as reference. DNA methylation levels of each mutant used to produce Figure 5B,C were linearly scaled according to the reported averages of RT-qPCR replicates.
Figure 6
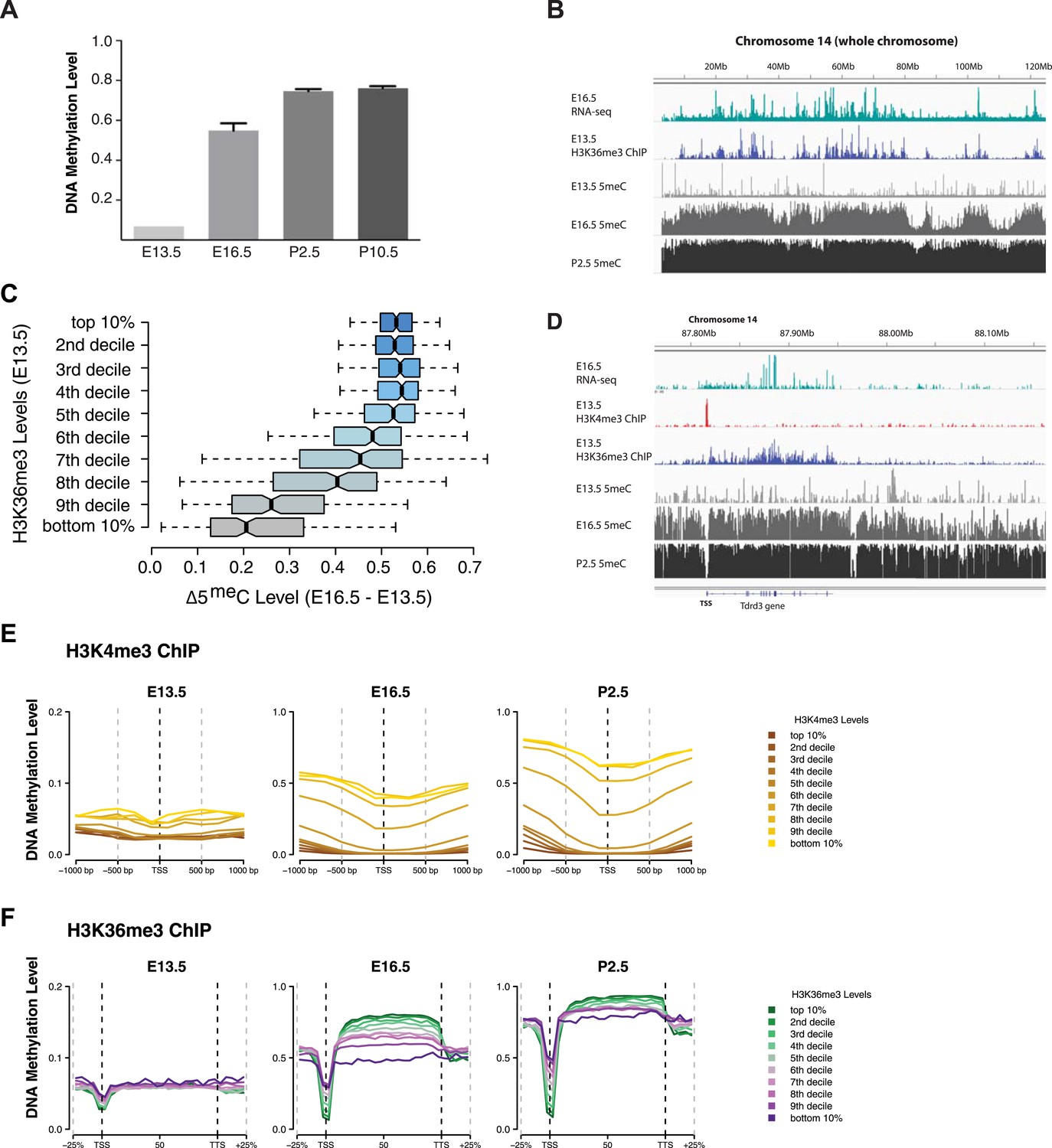
H3K4me3 and H3K36me3 distribution predicts de novo DNA methylation pattern in male germline.
(A) Genome-wide CG methylation levels during murine development as measured by bisulfite sequencing. (B) RNA-seq, and ChIP read abundance and relative DNA methylation levels are plotted across chromosome 14. Note the correspondence between RNA-seq and H3K36me3 ChIP levels and rapid DNA methylation between E13.5 and E16.5. (C) Boxplots showing the difference of DNA methylation levels between E13.5 and E16.5 of 1 Mb genome bins sorted into deciles by H3K36me3 level. (D) RNA-seq and ChIP read abundance and DNA methylation levels are plotted relative to transcriptionally active genes. The gene promoters contain high H3K4me3 and are not methylated, while the gene bodies contain high H3K36me3 and are methylated rapidly. (E) Metaplots showing DNA methylation level ±1000 bp relative to the TSS of genes sorted into deciles by H3K4me3 level. (F) Metagene plots showing DNA methylation across gene bodies sorted into deciles by H3K36me3 level.
Figure 7 with 1 supplement
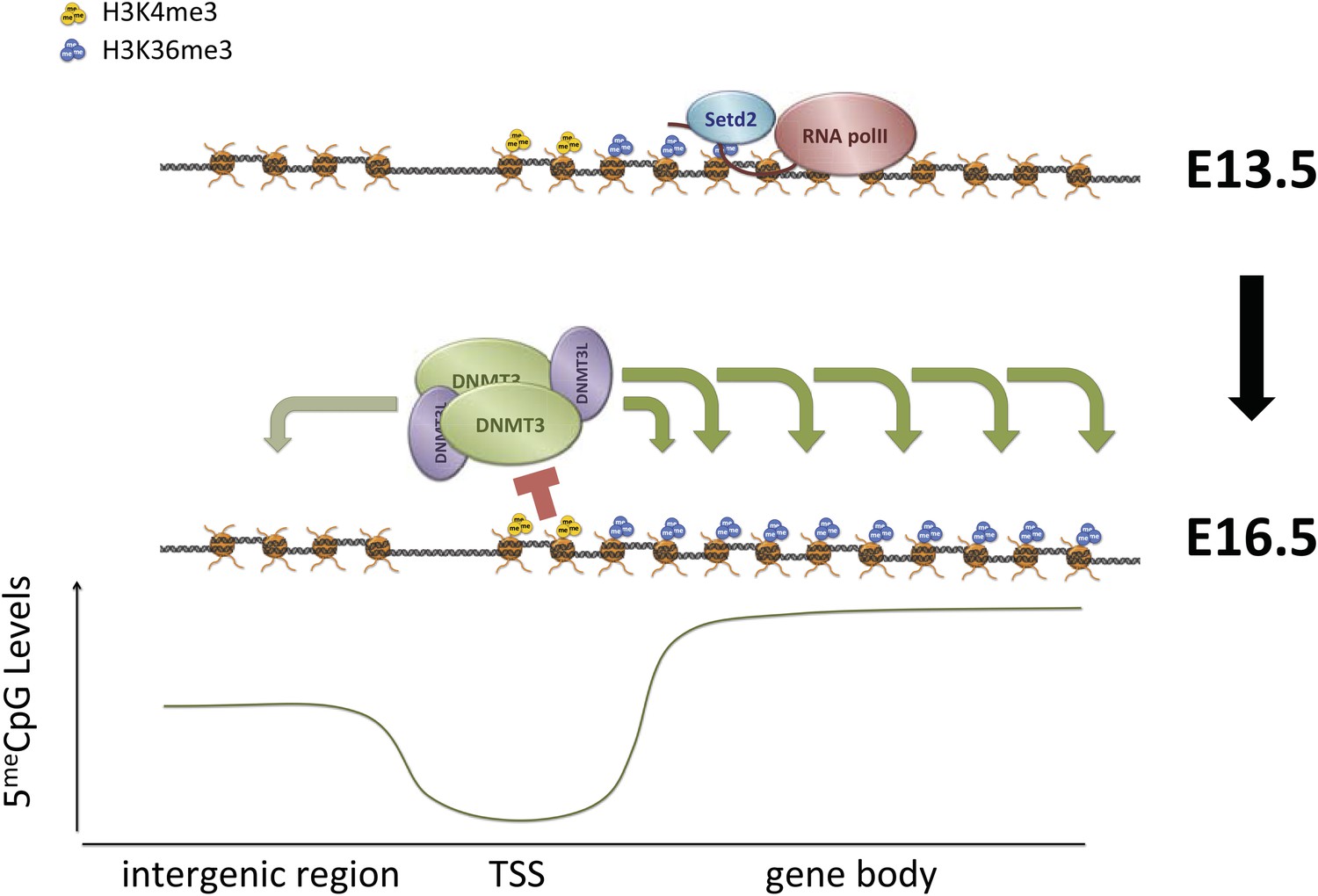
Proposed model for de novo DNA methylation establishment.
Model proposed for the targeting of DNMT3 during events of de novo 5meC establishment after genome-wide erasure of DNA methylation. Our model suggests that the presence of transcription-dependent histone modifications, such as H3K4me3 and H3K36me3, determines the activity of DNMT3b in vivo.
Figure 7—figure supplement 1
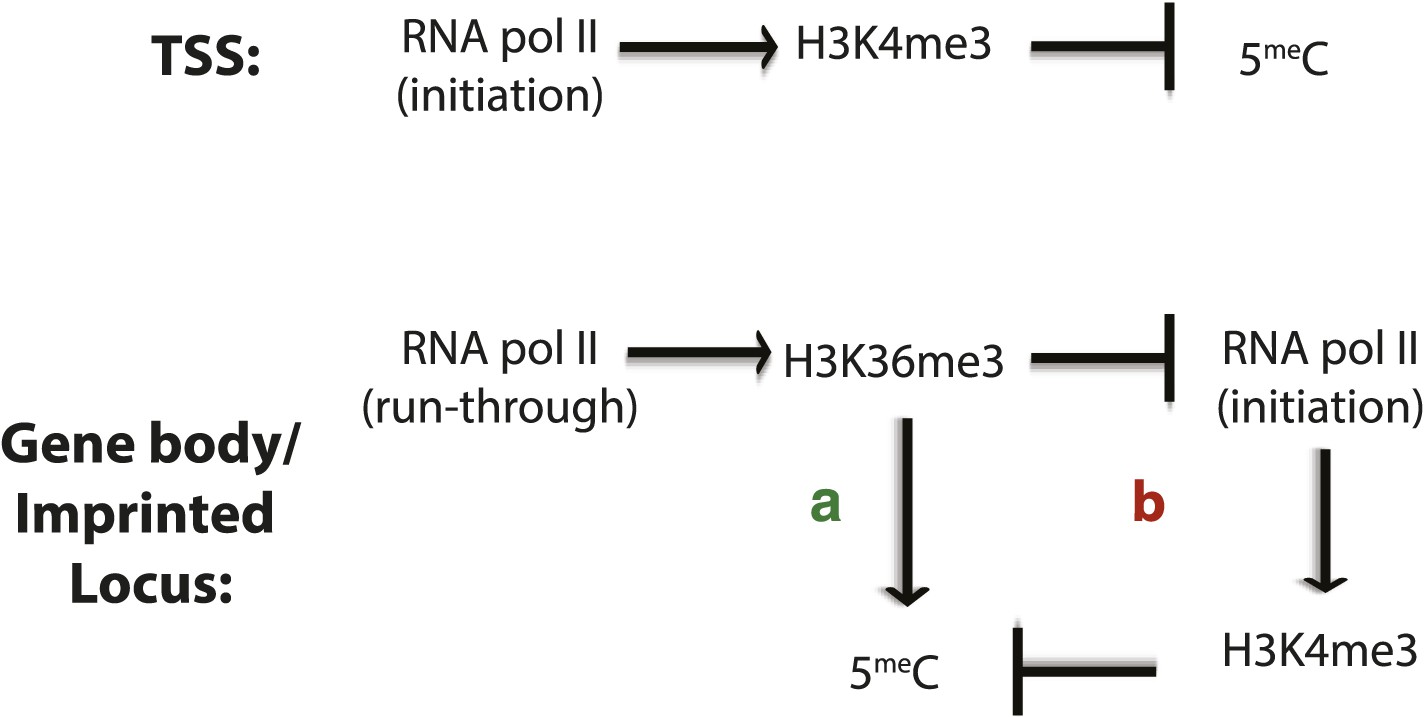
Factors affecting DNA methylation deposition.
(Top) DNA methylation is negatively affected by H3K4me3 at the TSS. (Bottom) H3K36me3 promotes DNA methylation by (A) direct recruitment of DNMT3b and by (B) preventing the methylation of H3K4, which antagonizes 5meC deposition.
Additional files
-
Supplementary file 1
(A) Yeast Whole Genome Bisulfite Sequencing Data. (B) Yeast MNase Sequencing Stats. (C) Yeast mRNA Sequencing Stats. (D) Yeast ChIP Sequencing Stats. (E) Yeast Whole Genome Bisulfite Sequencing Data for mutant strains. (F) Yeast Whole Genome Bisulfite Sequencing in mouse. (G) ChIP Sequencing Stats in mouse.
- https://doi.org/10.7554/eLife.06205.022
-
Supplementary file 2
(A) Yeast dinucleotide context methylation. (B) Yeast mutant strains dinucleotide context methylation. (C) Mouse germ cells dinucleotide context methylation.
- https://doi.org/10.7554/eLife.06205.023
-
Supplementary file 3
(A) Nucleosome called in a DNMT3b-expressing strain. (B) Nucleosome called in a non DNMT3b-expressing strain (EV). (C) Differential nucleosomes between DNMT3b-expressing and non-expressing strain.
- https://doi.org/10.7554/eLife.06205.024
-
Supplementary file 4
(A) Yeast mRNA differential expression using Deseq. (B) Upregulated genes in DNMT3b-expressing strain vs EV. (C) Downregulated genes in DNMT3b-expressing strain vs EV. (D) Gene Ontology (GO) term analysis for upregulated genes in DNMT3b-expressing strain vs EV. (E) GO term analysis for downregulated genes in DNMT3b-expressing strain vs EV. (F) RPKM values of yeast verified ORF in DNMT3b-expressing strain.
- https://doi.org/10.7554/eLife.06205.025
-
Supplementary file 5
Correletion coefficients of DNMT3b occupancy and 5meC levels predictions.
- https://doi.org/10.7554/eLife.06205.026
-
Supplementary file 6
(A) Plasmids used in this study. (B) Yeast strains used in this study. (C) Oligonucleotides used in this study. (D) Antibodies used in this study.
- https://doi.org/10.7554/eLife.06205.027
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse
eLife 4:e06205.
https://doi.org/10.7554/eLife.06205




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}