Punctuated evolution and transitional hybrid network in an ancestral cell cycle of fungi
- Duke University, United States
- Stanford University, United States
Figures
Figure 1
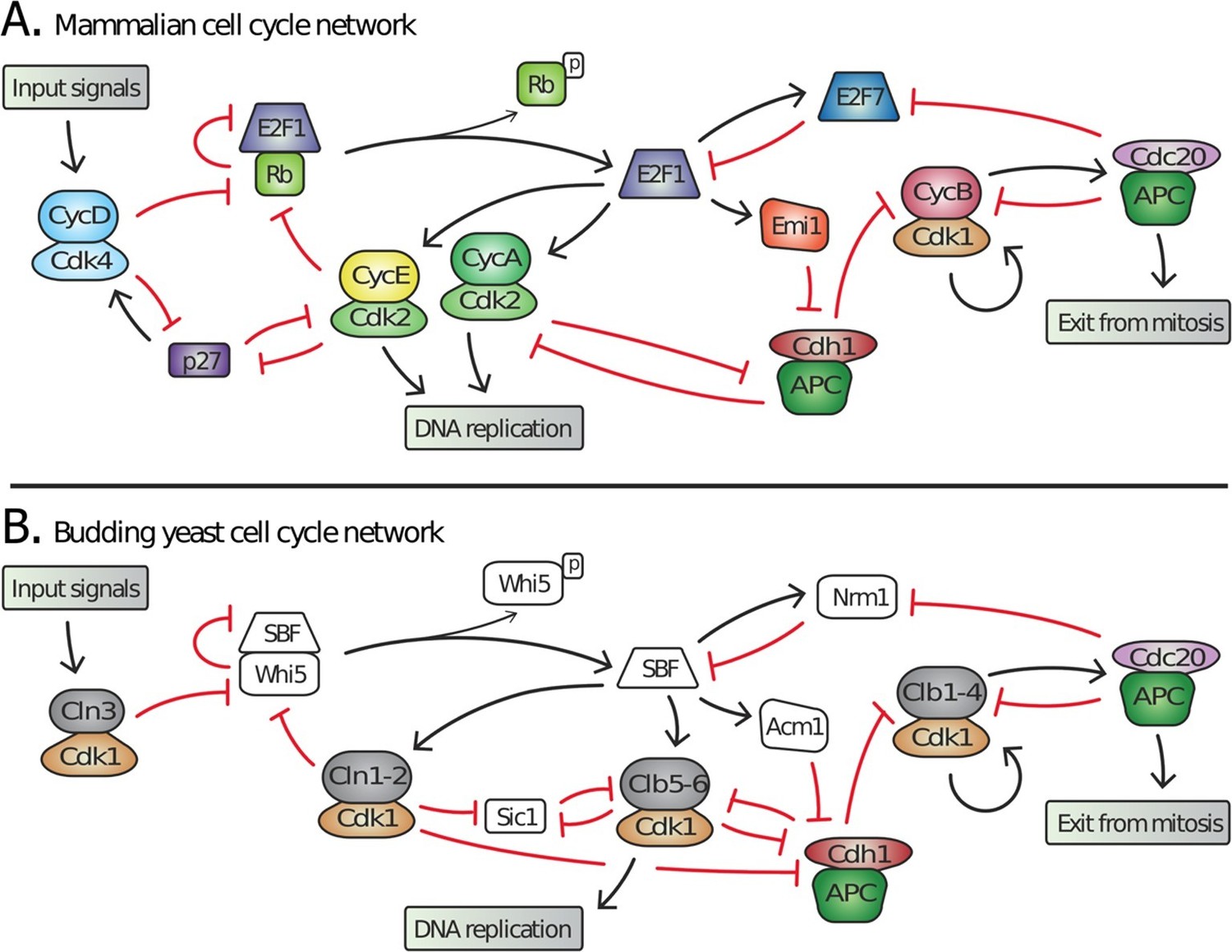
Topology of G1/S regulatory network in mammals and budding yeast is conserved, yet many regulators exhibit no detectable sequence homology.
Schematic diagram illustrating the extensive similarities between (A) animal and (B) budding yeast G1/S cell cycle control networks. Similar coloring denotes members of a similar family or sub-family. Fungal components colored white denote proteins with no identifiable animal orthologs.
Figure 2 with 6 supplements

Animal and plant G1/S regulatory network components were present in the last eukaryotic common ancestor.
Distribution of cell cycle regulators across the eukaryotic species tree (Adl et al., 2012). Animals (Metazoa) and yeasts (Fungi) are sister groups (Opisthokonta), and are distantly related to plants (Charophyta), which are members of the Archaeplastida. Check marks indicate the presence of at least one member of a protein family in at least one sequenced species from the corresponding group. We developed profile-HMMs to detect cell division cycle regulators in eukaryotic genomes. For each cell cycle regulatory family (e.g., Cyclins), we used molecular phylogeny to classify eukaryotic sequences into sub-families (e.g., Cyclin B, Cyclin A, Cyclin E/D). See Figure 2—figure supplement 1 for complete list of regulatory families in all eukaryotic species, and Figure 2—figure supplement 2 (Cyclin), Figure 2—figure supplement 3 (E2F/DP), Figure 2—figure supplement 4 (pRb), Figure 2—figure supplement 5 (Cdc20-family), and Figure 2—figure supplement 6 (CDK) for final phylogenies.
-
Figure 2—source data 1
Reduced set of eukaryotic cell cycle cyclins for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 2—figure supplement 2.
- https://doi.org/10.7554/eLife.09492.005
-
Figure 2—source data 2
Complete set of eukaryotic E2F/DP transcription factors for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 2—figure supplement 3.
- https://doi.org/10.7554/eLife.09492.006
-
Figure 2—source data 3
Complete set of eukaryotic Rb inhibitors for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 2—figure supplement 4.
- https://doi.org/10.7554/eLife.09492.007
-
Figure 2—source data 4
Reduced set of eukaryotic Cdc20-family APC regulators for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 2—figure supplement 5.
- https://doi.org/10.7554/eLife.09492.008
-
Figure 2—source data 5
Reduced set of eukaryotic cyclin-dependent kinases for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 2—figure supplement 6.
- https://doi.org/10.7554/eLife.09492.009
Figure 2—figure supplement 1
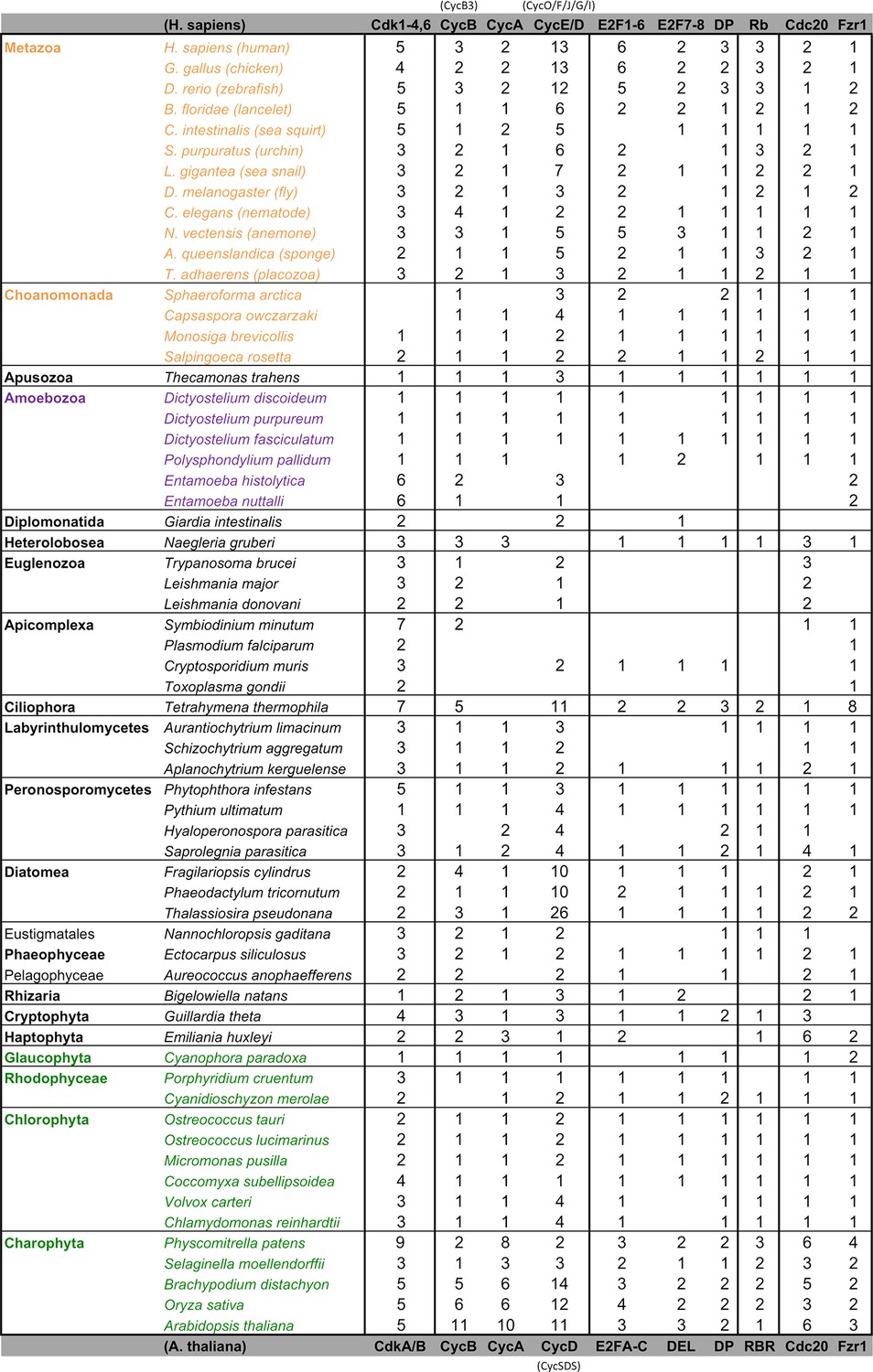
Comparative genomic data of G1/S regulators across eukaryotes.
Each entry lists the number of sub-family members (column) for each eukaryotic genome (row). Grey rows list the sub-family gene names in H. sapiens and A. thaliana. Additional cyclin sub-family members are listed in parentheses. Protein sequences listed in this table, which were used to create molecular phylogenies, can be found in Figure 2—source data 1 through Figure 2—source data 5.
Figure 2—figure supplement 2

Reduced phylogeny of eukaryotic cell cycle cyclins.
The cell division cycle (CDC) cyclin family consists of several sub-families with a well-characterized cyclin box: (1) CycA (H. sapiens, A. thaliana), (2) CycB and CLB (H. sapiens, A. thaliana, S. cerevisiae, S. pombe), (3) CycD (H. sapiens, A. thaliana), (4) CycE (H. sapiens), and (5) CLN (S. cerevisiae, S. pombe). We combined CycA, CycB, CycD, CycE, CLB, and CLN sequences from H. sapiens (10 cyclins), A. thaliana (13 cyclins), S. cerevisiae (9 cyclins), and S. pombe (5 cyclins) to create a eukaryotic CDC cyclin profile-HMM (pCYC.hmm) following the procedure outlined in the Methods. Our HMM profile was sensitive enough to discover known, but uncharacterized cyclin sub-families (CycO, CycF, CycG, CycI, CycJ, CycSDS) as bona fide CDC cyclins. A domain threshold of E-20 was used to identify potential CDC cyclin homologs. We first made a phylogeny of all cyclins to classify them. This dataset was then manually pruned to remove long-branches and problematic lineages. Our reduced CDC cyclin dataset (Figure 2—source data 1) has a total of 499 sequences. Columns with the top 10% Zorro score (496 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (23,163 sampled trees, meandiff= 0.01, maxdiff= 0.5)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 2—figure supplement 3
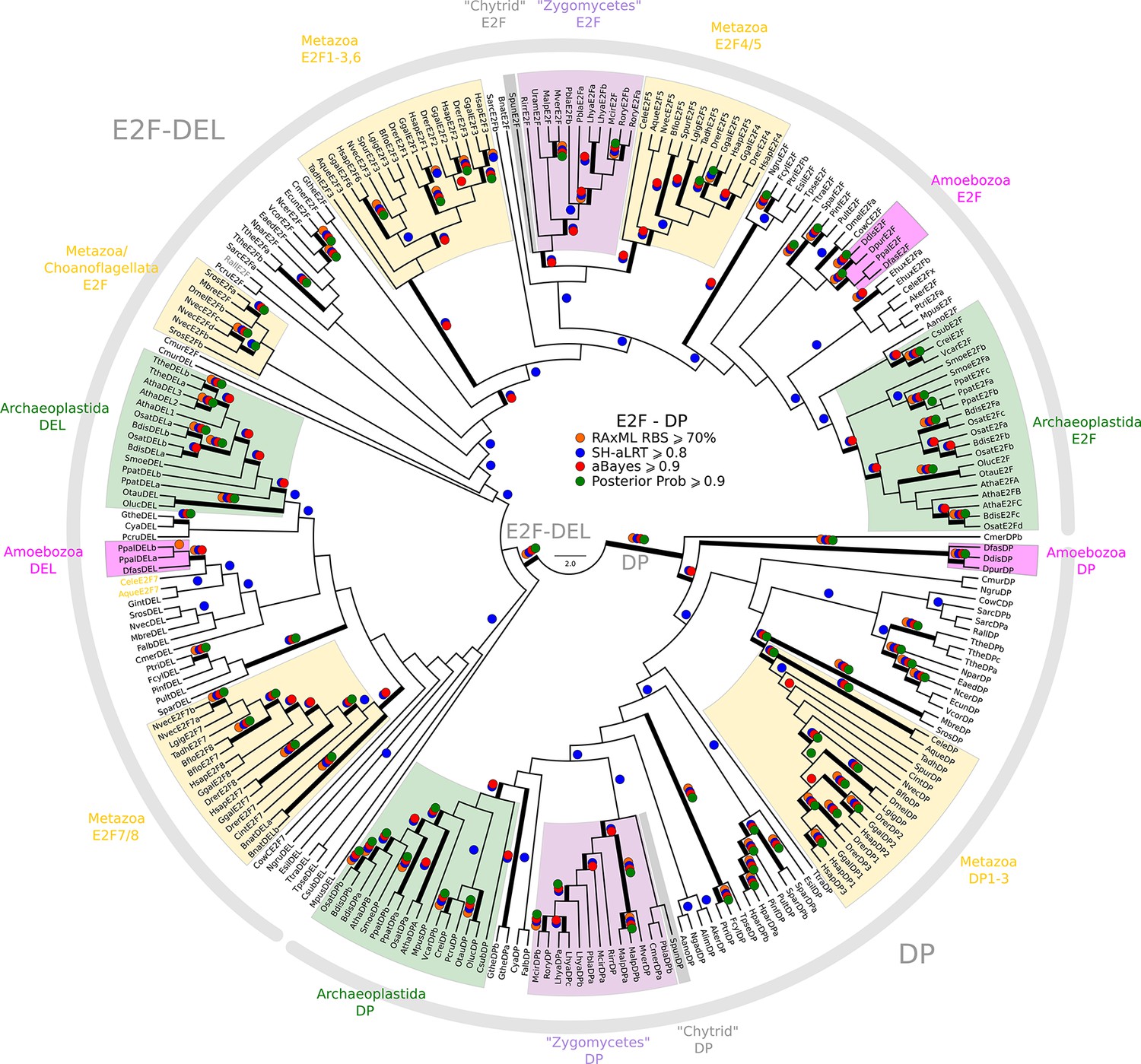
Phylogeny of eukaryotic E2F/DP transcription factors.
E2F-DP is a winged helix-turn-helix DNA-binding domain that is conserved across eukaryotes (van den Heuvel and Dyson, 2008). There are three sub-families within the E2F-DP family: (1) the E2F subfamily, (2) the E2F7-8/DEL subfamily, and (3) the DP subfamily. The E2F family consists of E2F1-6 (H. sapiens) and E2FA-C (A. thaliana). The E2F7-8/DEL family consists of E2F7-8 (H. sapiens) and DEL1-3 (A. thaliana). The DP family consists of DP1-3 (H. sapiens) and DPA-B (A. thaliana). The members of E2F form heterodimers with DP, whereas the DEL family has two DNA-binding domains and does not require DP to bind DNA. We used the E2F_TDP.hmm profile from PFAM to uncover members of the E2F/DP family across eukaryotes. A domain threshold of E-10 was used to identify potential E2F/DP homologs. Our E2F/DP dataset (Figure 2—source data 2) has 248 sequences. Columns with the top 8% Zorro score (284 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (53,009 sampled trees, meandiff=0.0064, maxdiff=0.18)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 2—figure supplement 4
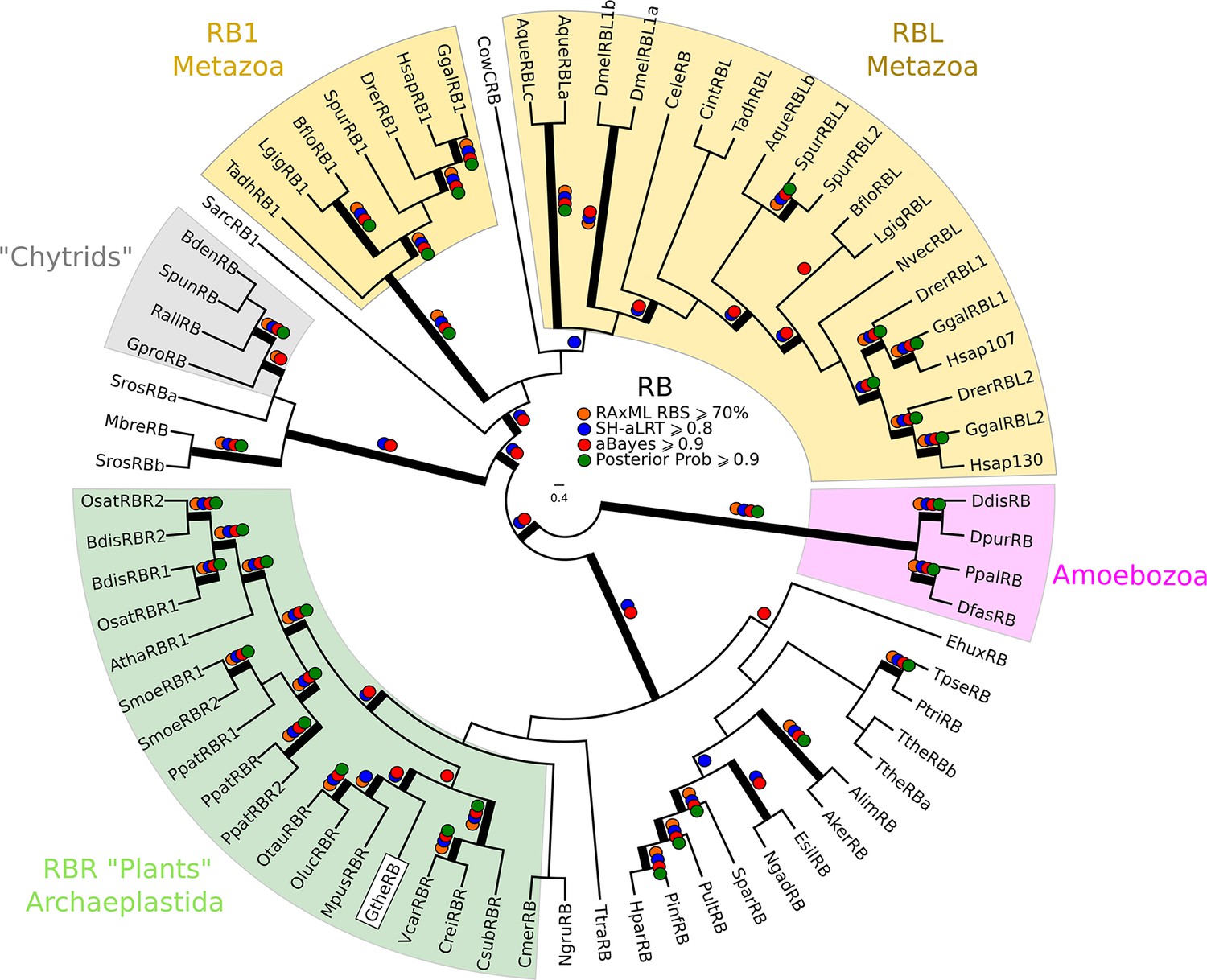
Phylogeny of eukaryotic Rb inhibitors.
H. sapiens has Rb1, RBL1 (p107), and RBL2 (p130), and A. thaliana has RBR1. The model fungi S. cerevisiae and S. pombe do not have any obvious retinoblastoma pocket proteins. We needed more eukaryotic sequences to create a robust HMM profile (pRb.hmm) for the pRb family. Based on the pRb sequences collected in Hallmann, (2009), we built a profile-HMM using putative pRb homologs from H. sapiens, G. gallus, C. intestinalis, D. melanogaster, C. elegans, N. vectensis, T. adhaerens (metazoa); B. dendrobatidis (fungi); D. discoideum, D. purpureum, T. pseudonana, P. tricornutum, N. gruberi, E. huxleyi (protists); C. merolae, O. tauri, O. lucimarinus, M. pusilla, V. carteri, C. reinhartdii, P. patens, S. moellendorfii, A. thaliana (plants). A domain threshold of E-20 was used to identify pRB homologs. Our pRB dataset (Figure 2—source data 3) has 72 sequences. Columns with the top 15% Zorro score (566 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (23,219 sampled trees, meandiff=0.0035, maxdiff=0.067)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 2—figure supplement 5
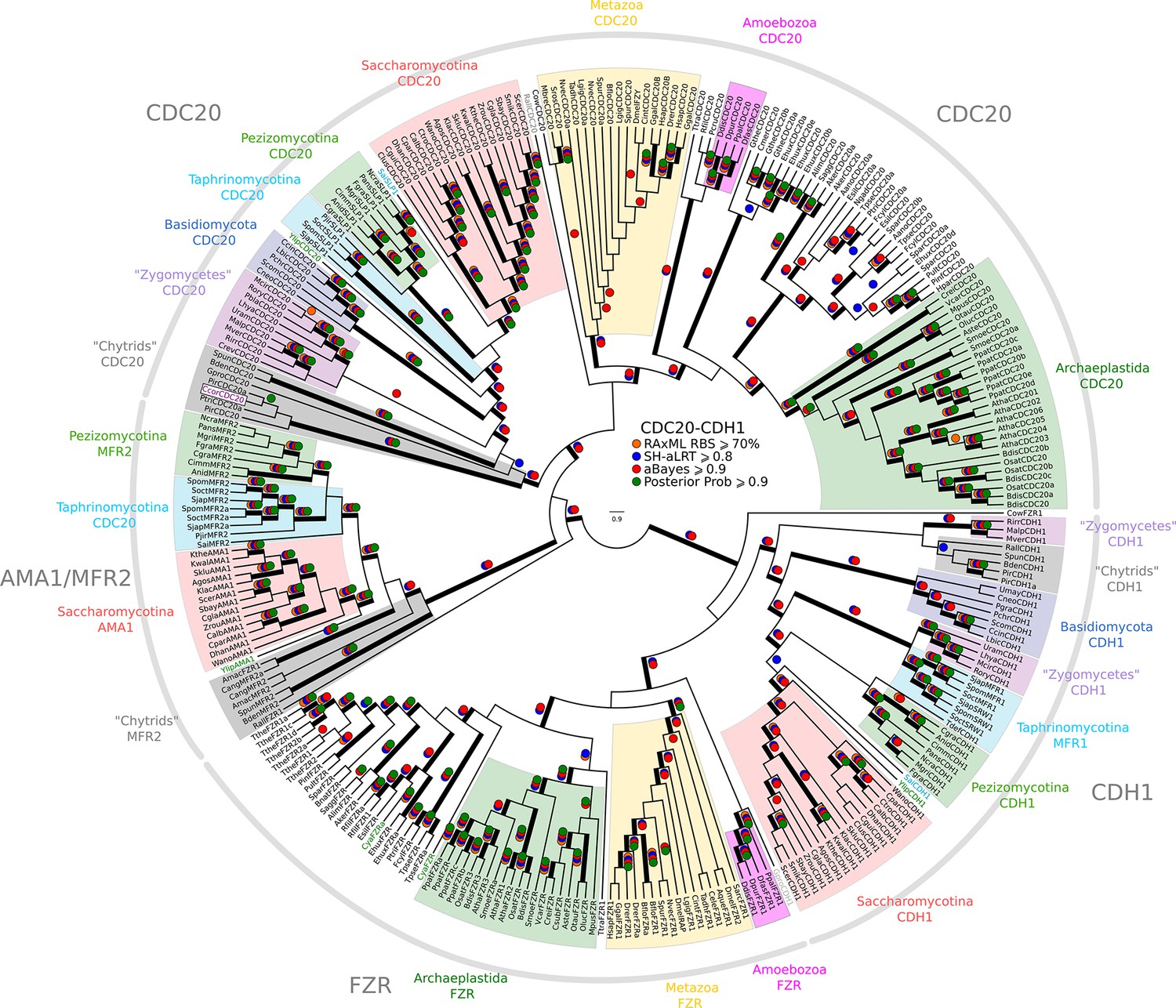
Reduced phylogeny of eukaryotic Cdc20-family APC regulators.
We combined CDC20 and CDH1/FZR1 sequences from H. sapiens (3 members), A. thaliana (9 members), and S. cerevisiae (3 members) to create a eukaryotic CDC20-family APC regulator profile-HMM (pCDC20.hmm) following the procedure outlined in the Methods. A domain threshold of E-50 was used to identify CDC20 homologs. Our pCDC20 dataset has 350 sequences. This dataset was manually pruned to remove long-branches and problematic lineages. Our reduced CDC20 dataset (Figure 2—source data 4) has a total of 289 sequences. Columns with the top 20% Zorro score (608 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (13,638 sampled trees, meandiff=0.015, maxdiff=0.37)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 2—figure supplement 6
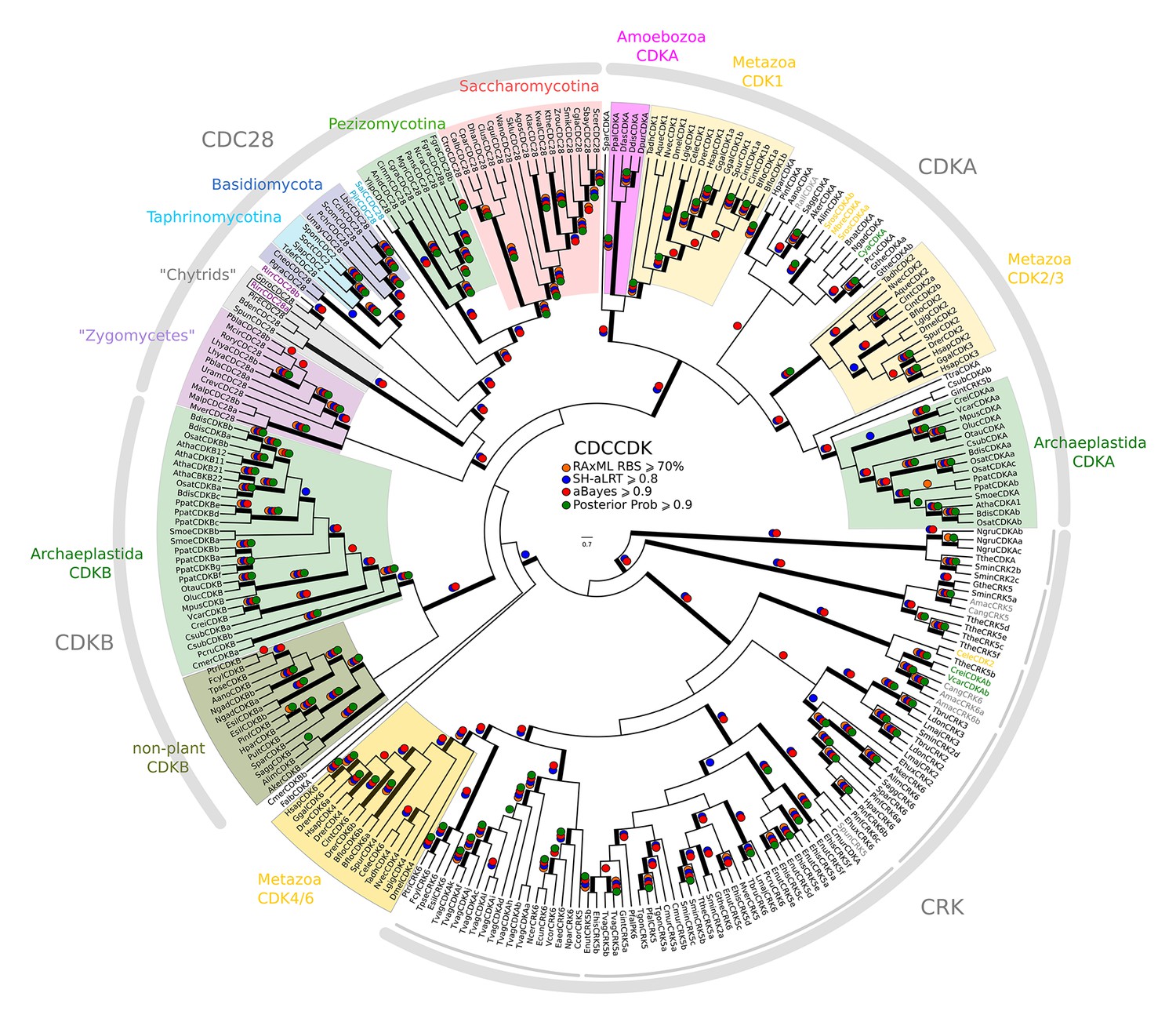
Reduced phylogeny of eukaryotic cyclin-dependent kinases.
To create a profile-HMM (pCDCCDK.hmm) for eukaryotic cell cycle CDK, we combined Cdk1-3, Cdk4, Cdk6 sequences from H. sapiens, CdkA and CdkB from A. thaliana, Cdc28 from S. cerevisiae, and Cdc2 from S. pombe. A domain threshold of E-20 was used to identify potential CDK homologs, and we only kept cell cycle CDKs (i.e. Cdk1-3, Cdk4,6, Cdc28, CdkA, CdkB). Our reduced CDK dataset (Figure 2—source data 5) has 272 sequences. Columns with the top 15% Zorro score (473 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (28,193 sampled trees, meandiff=0.015, maxdiff=0.53)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 3 with 4 supplements

Fungal ancestor evolved novel G1/S regulators, which eventually replaced ancestral cyclins, transcription factors, and inhibitors in Dikarya.
Basal fungi and 'Zygomycota' contain hybrid networks comprised of both ancestral and fungal specific cell cycle regulators. Check marks indicate the presence of at least one member of a protein family in at least one sequenced species from the group; see Figure 3—figure supplement 1 for a complete list of homologs in all fungal species. Cells are omitted (rather than left unchecked) when a family is completely absent from a clade. For each fungal regulatory family (e.g., SBF/MBF), we used molecular phylogeny to classify eukaryotic sequences into sub-families (e.g., SBF/MBF, APSES, Xbp1, Bqt4). See Materials and methods for details and Figure 3—figure supplement 2 (SBF/MBF only), Figure 3—figure supplement 3 (SBF/MBF+APSES), and Figure 3—figure supplement 4 (Whi5/Nrm1) for final phylogenies.
-
Figure 3—source data 1
Complete set of fungal SBF/MBF transcription factors for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 3—figure supplement 2.
- https://doi.org/10.7554/eLife.09492.017
-
Figure 3—source data 2
Complete set of fungal SBF/MBF and APSES transcription factors for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 3—figure supplement 3.
- https://doi.org/10.7554/eLife.09492.018
-
Figure 3—source data 3
Complete set of fungal Whi5/Nrm1 inhibitors for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 3—figure supplement 4.
- https://doi.org/10.7554/eLife.09492.019
Figure 3—figure supplement 1
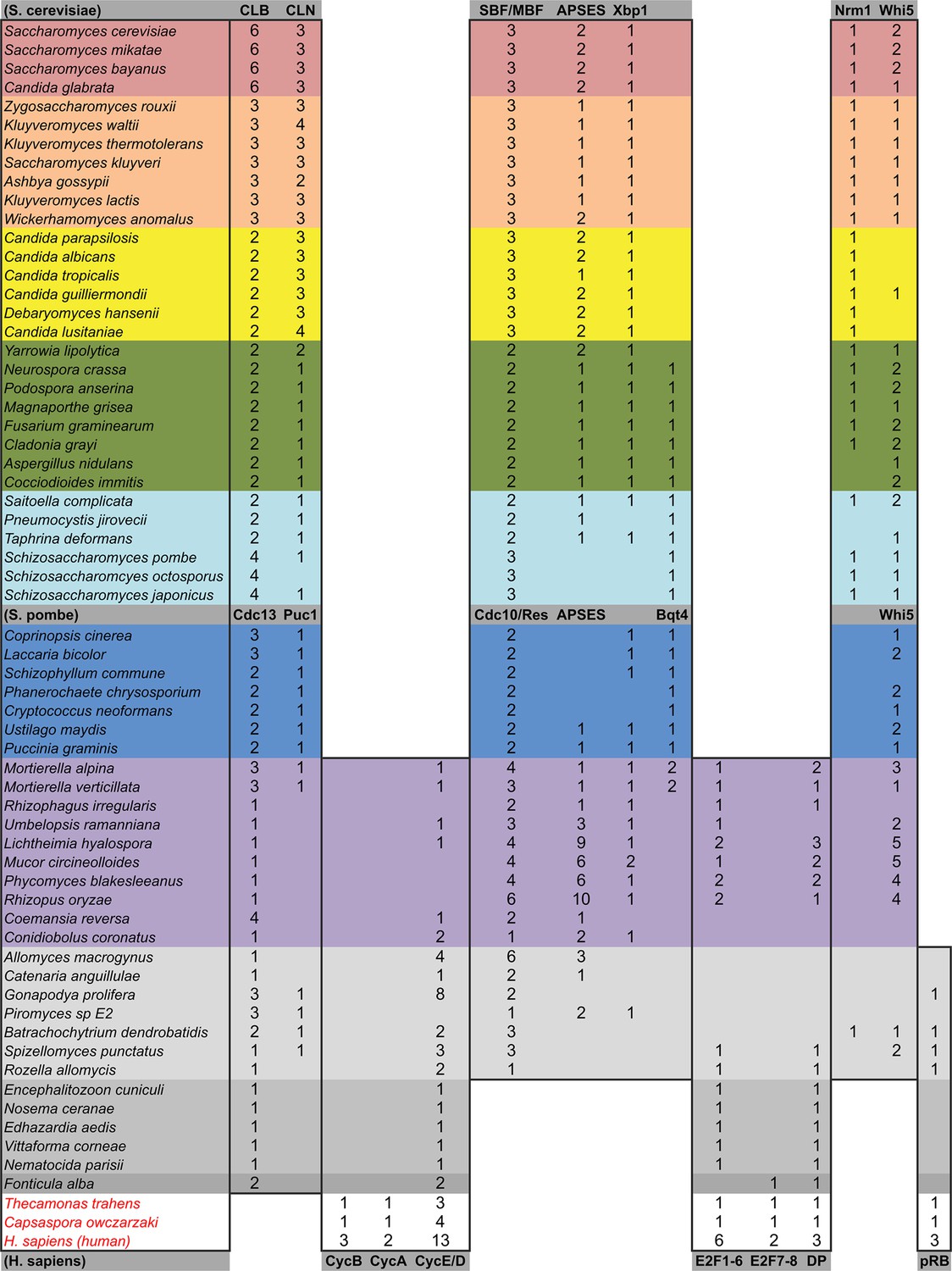
Comparative genomic data of G1/S regulators across fungi.
Grey rows list the sub-family gene names in S. cerevisiae, S. pombe, and H. sapiens. Protein sequences listed in this table, which were used to create new molecular phylogenies not shown in Figure 2, can be found in Figure 3—source data 1 through Figure 3—source data 3.
Figure 3—figure supplement 2
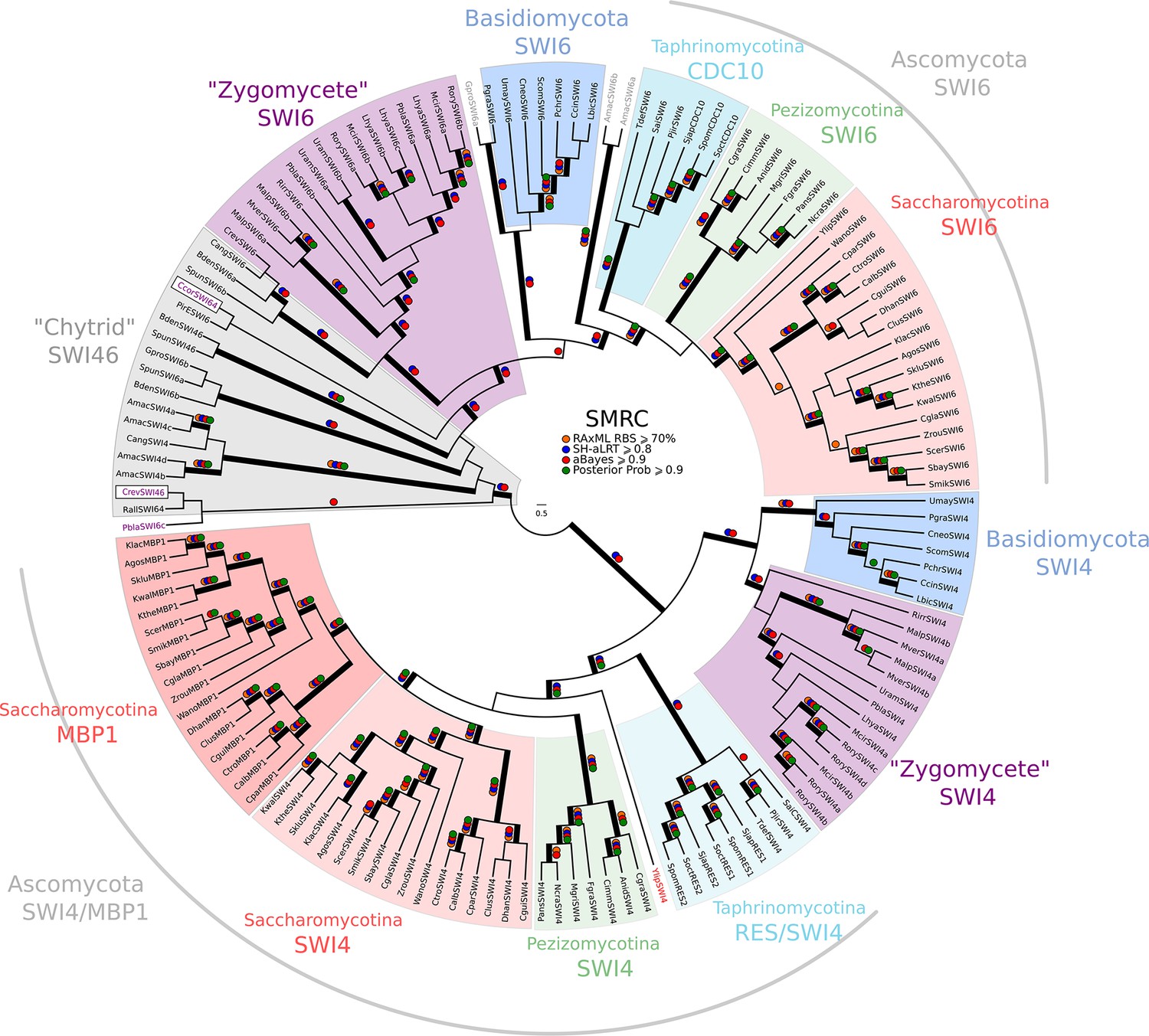
Phylogeny of fungal SBF/MBF transcription factors.
SBF and MBF are transcription factors that regulate G1/S transcription in budding and fission yeast. To detect SMRC (Swi4/6 Mbp1 Res1/2 Cdc10) across fungi, we built a sensitive profile-HMM (pSMRC.hmm) by combining well-characterized SMRC sequences from S. cerevisiae, C. albicans, N. crassa, A. nidulans, and S. pombe. A domain threshold of E-10 was used to identify SMRC homologs. Our SMRC dataset (Figure 3—source data 1) has 147 sequences. Columns with the top 20% Zorro score (709 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (19,457 sampled trees, meandiff=0.0056, maxdiff=0.145)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 3—figure supplement 3
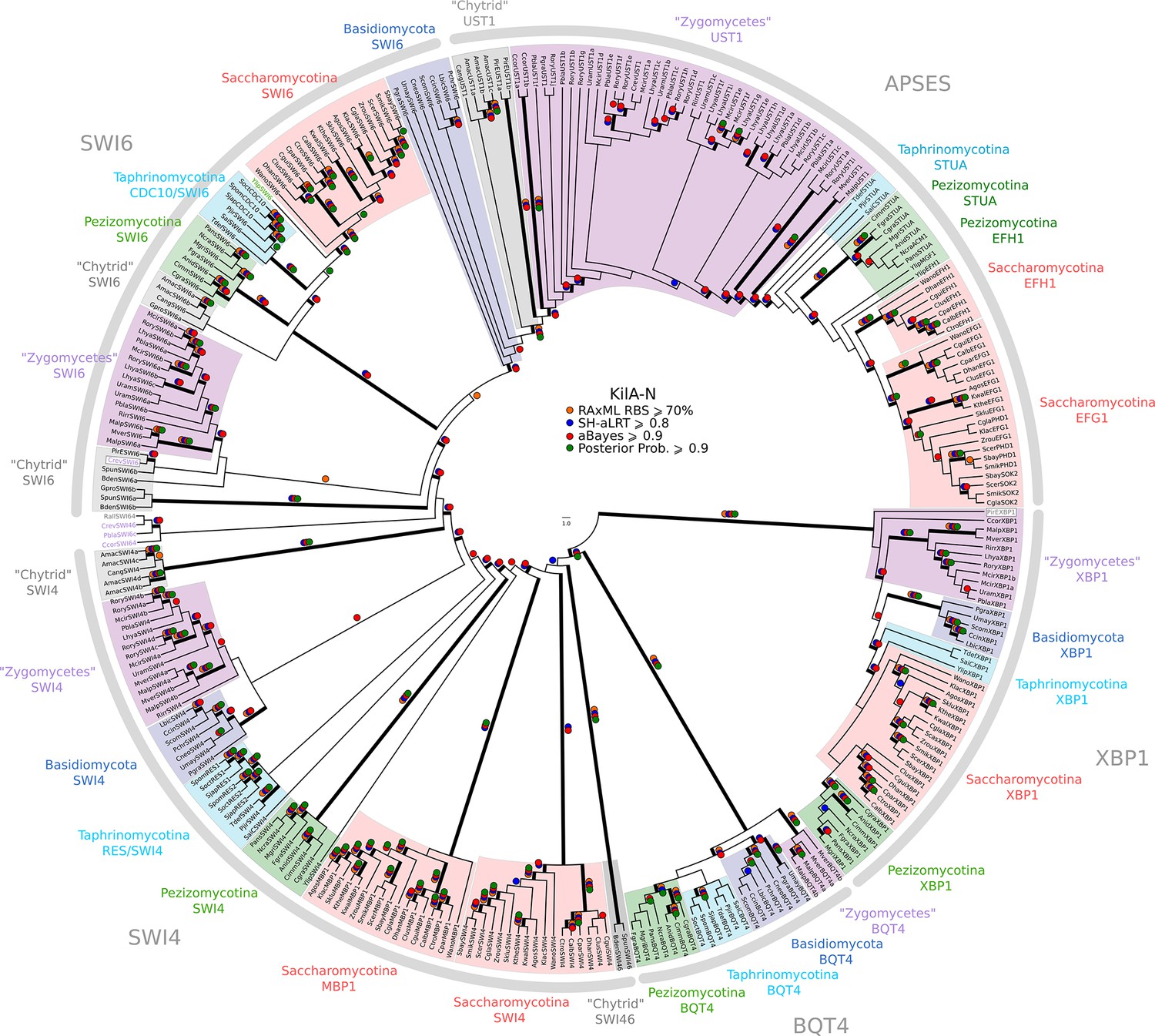
Phylogeny of fungal SBF/MBF and APSES transcription factors.
SBF/MBF and APSES transcription factors (Asm1, Phd1, Sok2, Efg1, StuA) share a common DNA-binding domain (KilA-N), which is derived from DNA viruses. During our search for SBF and APSES homologs, we consistently detected two additional fungal sub-families with homology to KilA-N: XBP1 (family name taken from S. cerevisiae) and BQT4 (family name taken from S. pombe). To detect APSES, XBP1, and BQT4 homologs, we built profile-HMMs (APSES.hmm, XBP1.hmm, and BQT4.hmm) by combining APSES, XBP1, and BQT4 homologs from S. cerevisiae, C. albicans, N. crassa, A. nidulans, and S. pombe. A domain threshold of E-10 was used to identify APSES, XBP1, and BQT4 homologs. Our final dataset (Figure 3—source data 2) contains all fungal KILA sub-families (SBF/MBF, APSES, XBP1, BQT4) and has a total of 301 sequences. Columns with the top 10% Zorro score (447 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (15,251 sampled trees, meandiff=0.012, maxdiff=0.25)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 3—figure supplement 4
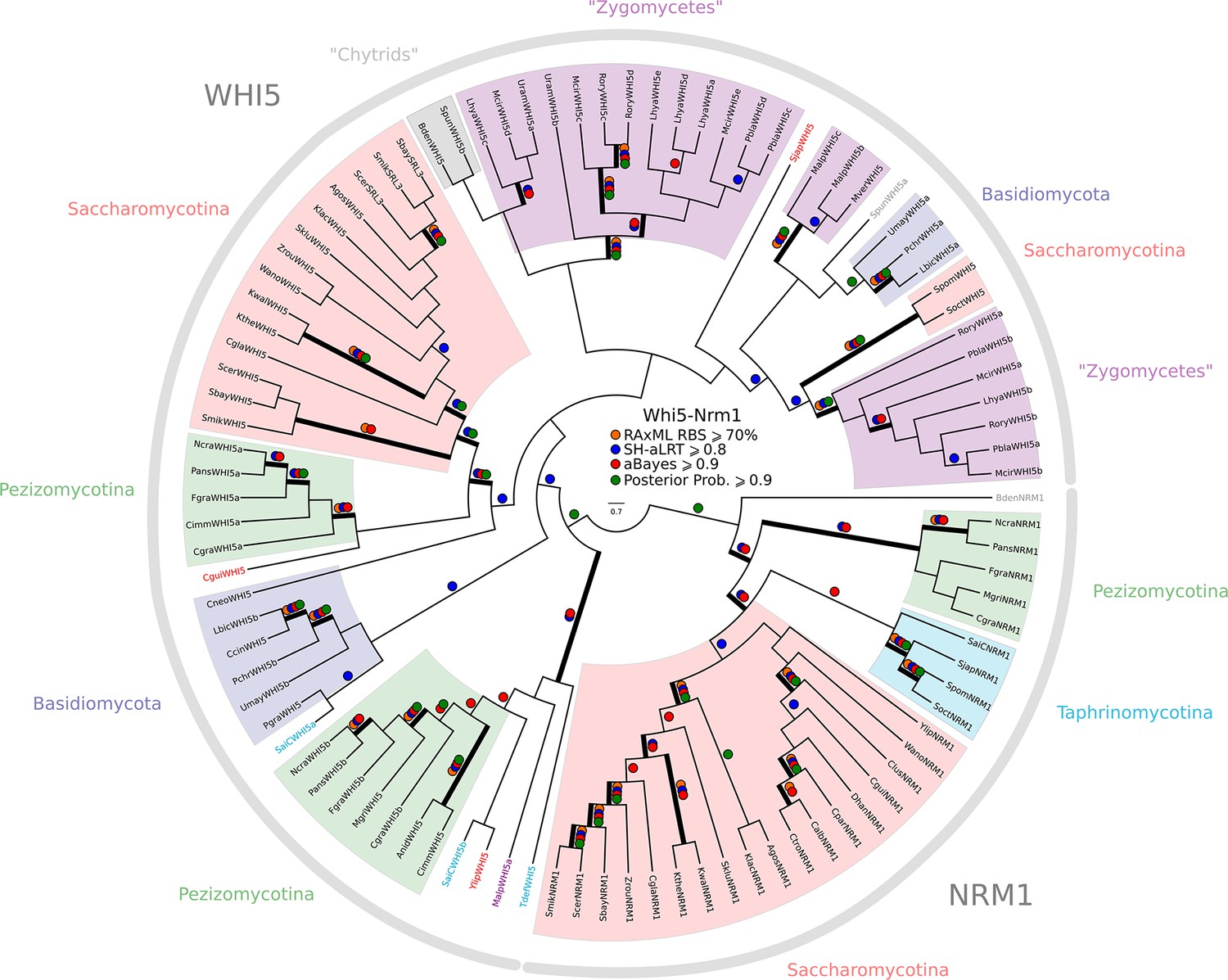
Phylogeny of fungal Whi5/Nrm1 inhibitors.
WHI5 and NRM1 are a yeast-specific protein family that has been identified and functionally characterized across S. cerevisiae, C. albicans, and S. pombe. Both WHI5 and NRM1 are fast evolving proteins. There is a small conserved region of 25 amino-acids (known as the GTB domain) that is responsible for interacting with Swi6/Cdc10 (Travesa et al., 2013). Unfortunately, the Whi5.hmm profile from PFAM is unable to detect an SRL3 paralogue in S. cerevisiae or the NRM1 orthologues in A. gossypii or C. albicans. We built a more sensitive profile-HMM (pWHI5.hmm) by combining WHI5/NRM1 sequences across ascomycetes (including SRL3 from Saccharomyces genomes and NRM1 from Candida genomes). A domain threshold of E-05 was used to identify WHI5 homologs. Our WHI5 dataset (Figure 3—-source data 3) has 98 sequences. Columns with the top 15% Zorro score (260 positions) were used in our alignment. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under the LG model of evolution (aBayes and SH-aLRT metrics with PhyML, RBS with RAxML, Bayesian Posterior Probability with Phylobayes (77,696 sampled trees, meandiff=0.0068, maxdiff=0.11)). Colored dots in branches indicate corresponding branch supports. Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; branch support thresholds are shown in the center of the figure; see Materials and methods.
Figure 4
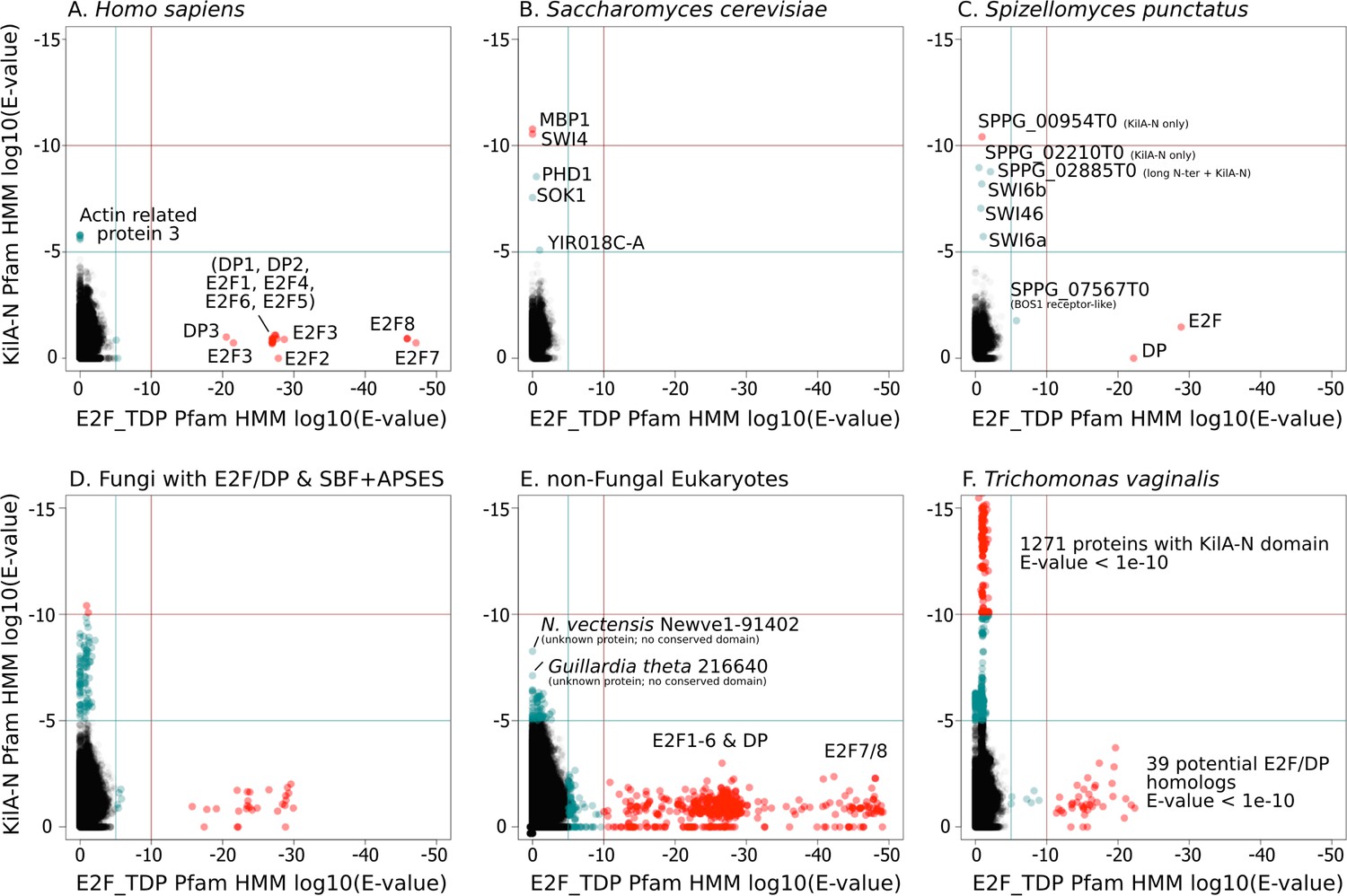
SBF and E2F HMM models detect different sequences.
We used the Pfam HMMER model of the E2F/DP DNA-binding domain (E2F_TDP.hmm) and SBF DNA-binding domain (KilA-N.hmm). Every protein in the query genome (listed at top) was scored using hmmsearch with E2F/DP HMM (x-axis) and KilA-N HMM (y-axis). All scores below 1E-5 (i.e., marginally significant) are blue and those below 1E-10 (i.e. highly significant) are red. All hits with E-values between 1E-5 and 1E-10 were further validated (or rejected) using an iterative search algorithm (Jackhmmer) against the annotated SwissProt database using the HMMER web server (Finn et al., 2011). We then inspected these sequences manually for key conserved KilA-N residues. (A) Homo sapiens only has E2F/DP, (B) Saccharomyces cerevisiae only has KilA-N (i.e. SBF, MBF, APSES). (C) Spizellomyces punctatus and (D) other basal fungi have both E2F/DP and KilA-N. (E) All the non-fungal eukaryote genomes that we surveyed only have E2F/DP. (F) Trichomonas vaginalis is one of the few eukaryotes outside of fungi that has both E2F/DP and KilA-N. The E2F/DP HMM and KilA-N HMM always have orthogonal hits (i.e. no protein in our dataset significantly hits both HMMs).
Figure 5
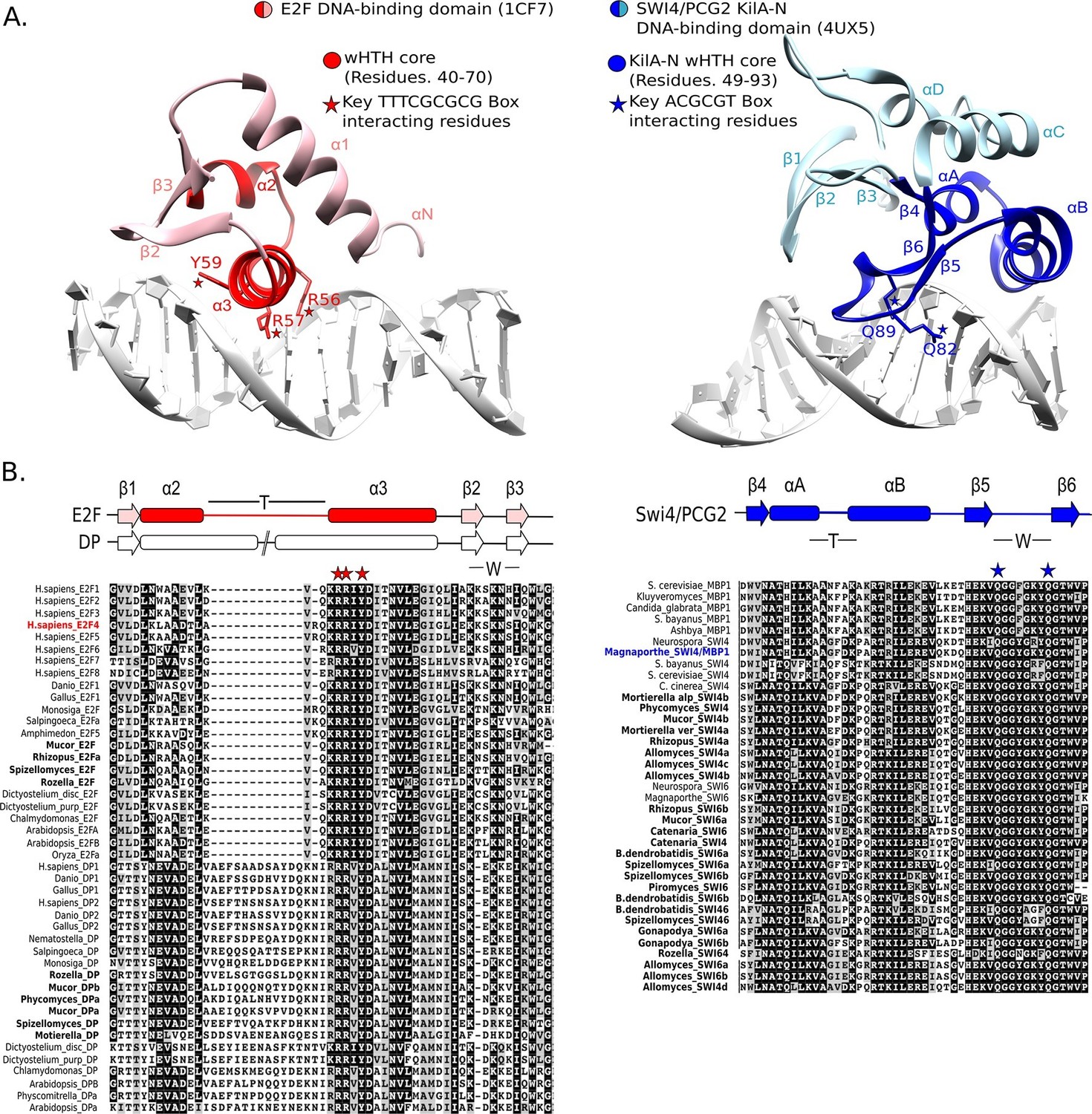
E2F and SBF show incongruences in sequence, structure, and mode of DNA binding.
(A) Although both proteins share a winged helix-turn-helix (wHTH) domain, the E2F/DP and SBF/MBF superfamilies do not exhibit significant sequence identity or structural similarity to suggest a common recent evolutionary origin according to CATH or SCOP databases. Furthermore, each wHTH has a different mechanism of interaction with DNA: the arginine and tyrosine side-chains of recognition helix-3 of E2F (E2F4 from Homo sapiens [Zheng et al., 1999]) interact with specific CG nucleotides, where as the glutamine side-chains of the 'wing' of SBF/MBF (PCG2 from Magnaporthe oryzae [Liu et al., 2015]) interact with specific CG nucleotides. (B) Sequence alignment of the DNA binding domain of representative eukaryotic E2F/DP (left) and fungal SBF/MBF (right). The corresponding secondary structure is above the sequence alignment. Evolutionary conserved residues of sequence aligned DNA binding domains are highlighted in black. Bold sequence names correspond to E2F/DP and SBF/MBF sequences from basal fungi. Colored sequence names correspond to sequences of the structures shown in panel A. PDB IDs for the structures used are shown in parentheses. W = wing; T= turn.
Figure 6
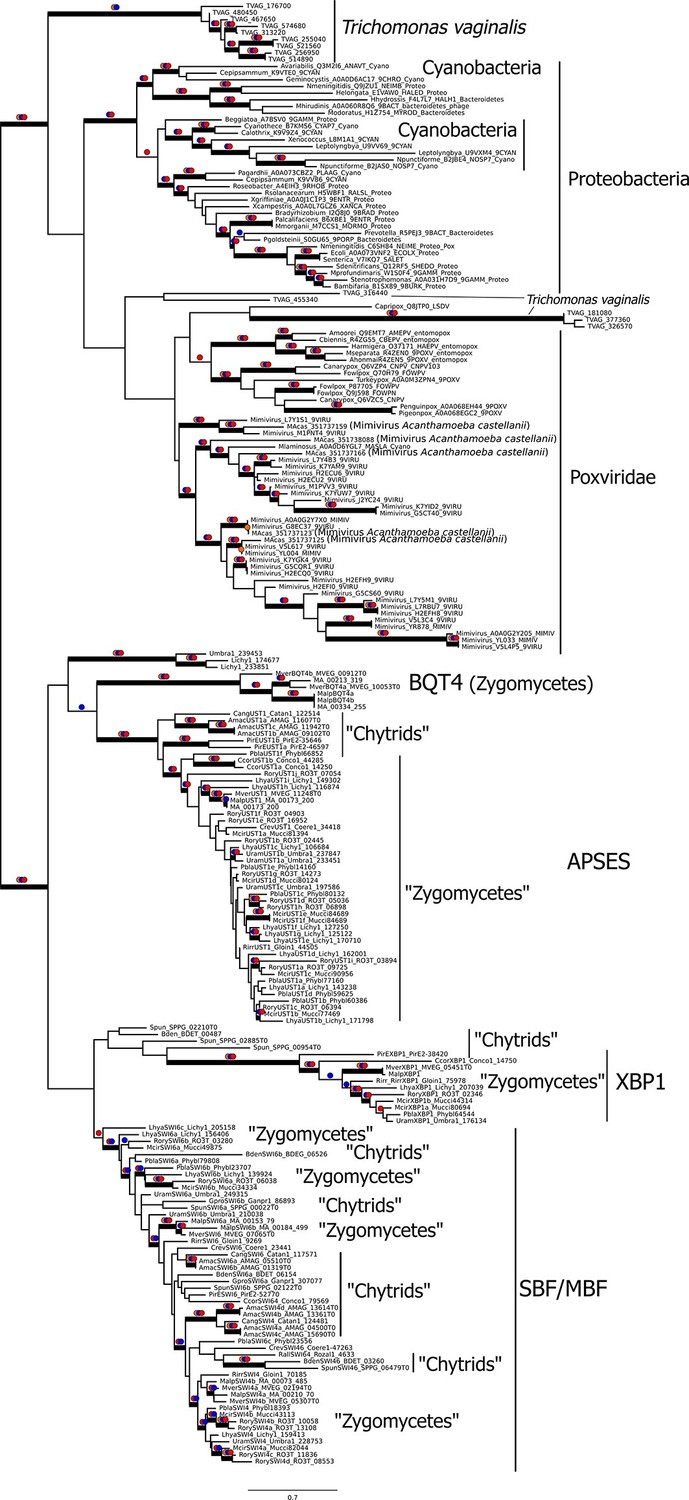
Viral origin of yeast cell cycle transcription factor SBF.
Maximum likelihood unrooted phylogenetic tree depicting relationships of fungal SBF-family proteins, KilA-N domains in prokaryotic and eukaryotic DNA viruses. The original dataset was manually pruned to remove long-branches and problematic lineages. Our reduced KilA-N dataset has a total of 219 sequences (Figure 6—source data 1), 130 positions. Confidence at nodes was assessed with multiple support metrics using different phylogenetic programs under LG model of evolution. Colored dots in branches indicate corresponding branch supports (red dots: PhyML aBayes ≥0.9; blue dots: PhyML SH-aLRT ≥0.80; orange: RAxML RBS ≥70%). Thick branches indicate significant support by at least two metrics, one parametric and one non-parametric; scale bar in substitutions per site; see Materials and methods.
-
Figure 6—source data 1
Reduced set of KilA-N domains for phylogenetic analysis.
These files contain the protein sequences used to create molecular phylogeny in Figure 5.
- https://doi.org/10.7554/eLife.09492.027
Figure 7
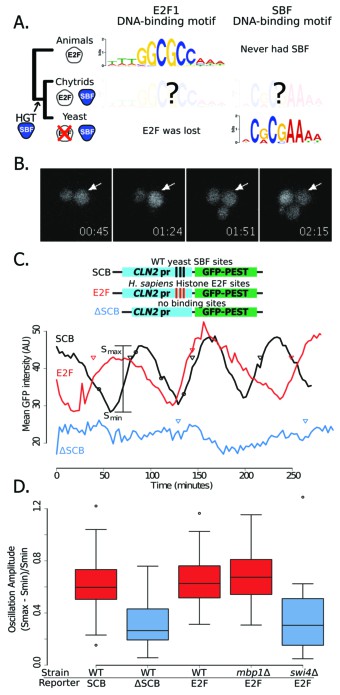
Yeast cell cycle transcription factor SBF can regulate cell cycle-dependent transcription via E2F binding sites in vivo.
(A) Phylogenetic tree of animals, chytrids, yeast labelled with E2F, SBF or both transcription factors (TF) if present in their genomes. The known DNA-binding motifs of animal E2F (E2F1) and yeast SBF (Swi4) were taken from the JASPAR database, where as Chytrid E2F and SBF motifs are unknown. (B) Fluorescence images of cells expressing a destabilized GFP from the SBF-regulated CLN2 promoter. (C) Oscillation of a transcriptional reporter in budding yeast. Characteristic time series of GFP expression from a CLN2 promoter (SCB), a CLN2 promoter where the SBF binding sites were deleted (∆SCB), or a CLN2 promoter where the SBF binding sites were replaced with E2F binding sites from the human gene cluster promoters (E2F). Oscillation amplitudes were quantified by scaling the mean fluorescence intensity difference from peak to trough divided by the trough intensity (Smax - Smin)/Smin. Circles denote time points corresponding to (B). Triangles denote budding events. (D) Distribution of oscillation amplitudes for different genotypes and GFP reporters. swi4∆ and mbp1∆ strains have deletions of the SBF and MBF DNA-binding domain subunits respectively. t-test comparisons within and across red and blue categories yield p-values >0.3 or <0.01 respectively. Boxes contain 25th, median and 75th percentiles, while whiskers extend to 1.5 times this interquartile range.
Figure 8 with 2 supplements
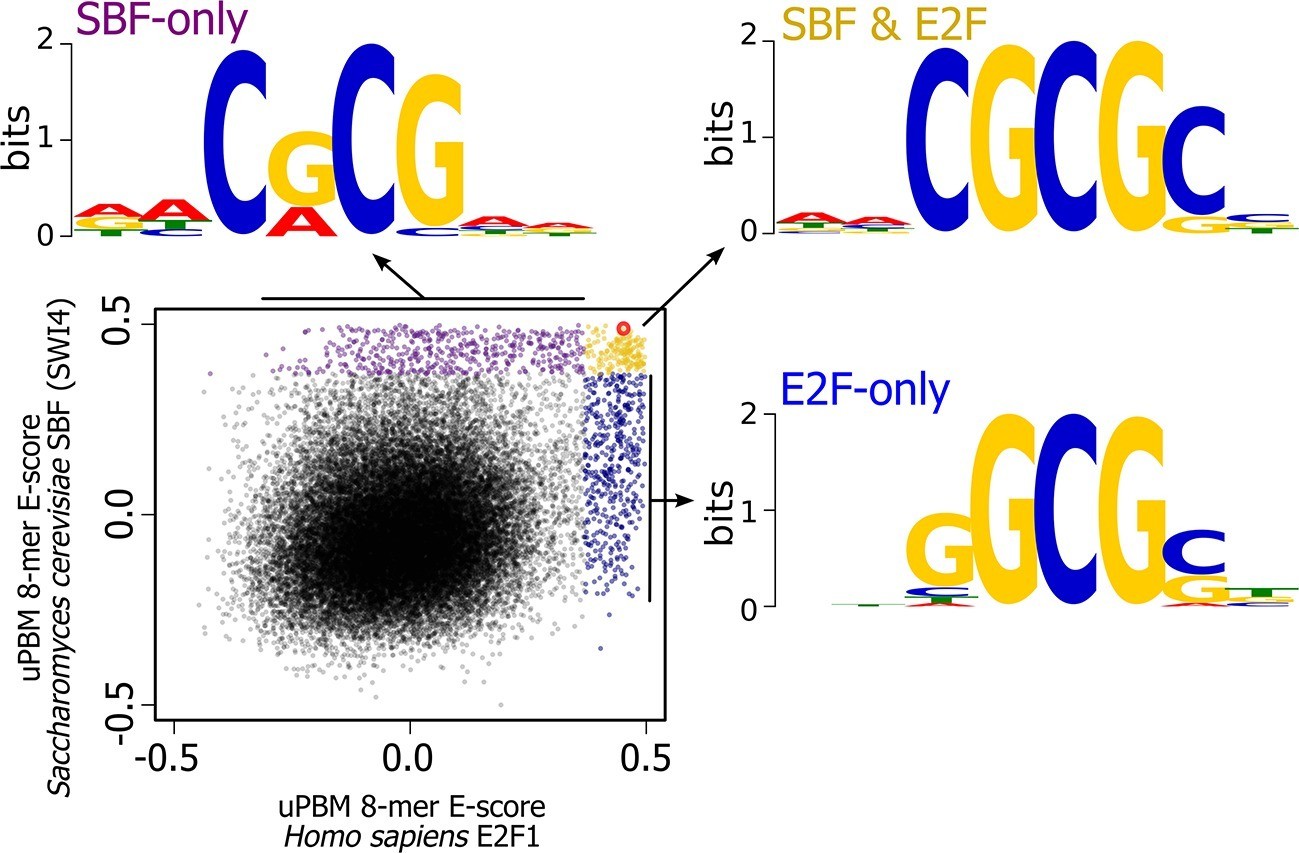
High-throughput DNA binding data for yeast SBF and human E2F shows that SBF and E2F can bind shared and distinct DNA-binding sites.
Plot of in vitro protein binding microarray 8-mer E-scores for Homo sapiens E2F1 (Afek et al., 2014) versus S. cerevisiae SBF protein Swi4 (Badis et al., 2008). All 8-mer motifs colored (E-score > 0.37) are considered significant targets with a false positive discovery rate of 0.001 (Badis et al., 2009). Yellow are common 8-mer motifs bound by both E2F1 and SBF, blue are E2F-only motifs, and purple are SBF-only motifs. The E2F motif from histone cluster promoters used in Figure 7 is circled in red.
Figure 8—figure supplement 1
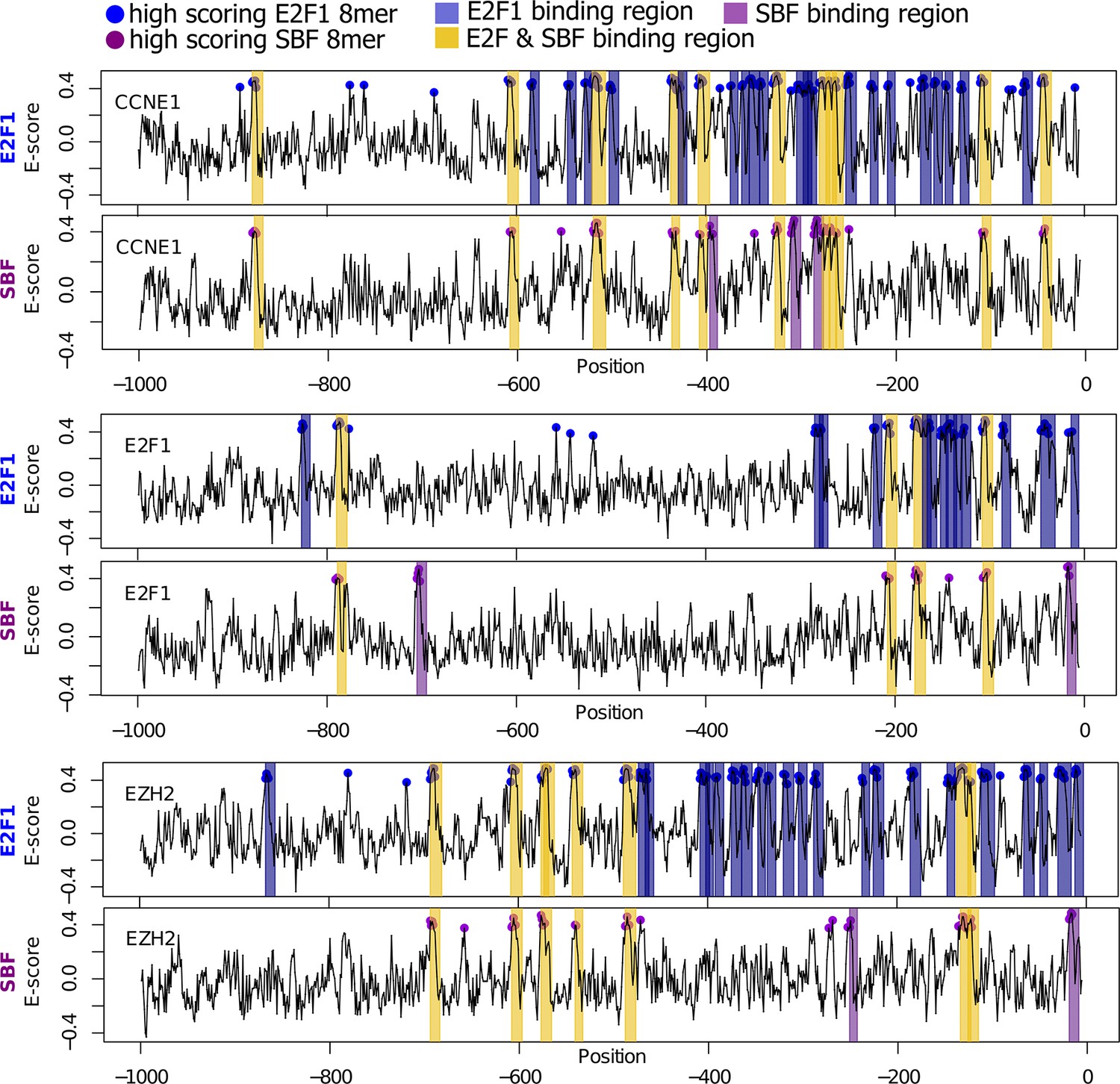
Bioinformatic scan of E2F-regulated human promoters suggests possible regulation by SBF.
E2F1 (top) and SBF (bottom) PBM motifs were used to scan the proximal (1000 bp) promoters of E2F-regulated promoters (CCNE1, E2F1, and EZH2). Promoter regions with a significant hit (8-mer E-score > 0.37) to a E2F or SBF motif have blue or purple dot, respectively. Predicted TF-binding regions were defined as at least 2 consecutive overlapping 8-mers (7 nucleotide overlap) and shaded as blue (E2F binding region) or purple (SBF-binding region). Common or E2F & SBF binding regions were colored yellow and defined as regions that overlap in at least one full 8-mer. Note that the number of E2F binding sites predicted by in vitro PBM data is larger than the actual number of functional in vivo E2F binding sites. This is to be expected because, in the nuclear environment, E2F can be outcompeted at putative DNA binding sites by nucleosomes or other nuclear proteins.
Figure 8—figure supplement 2
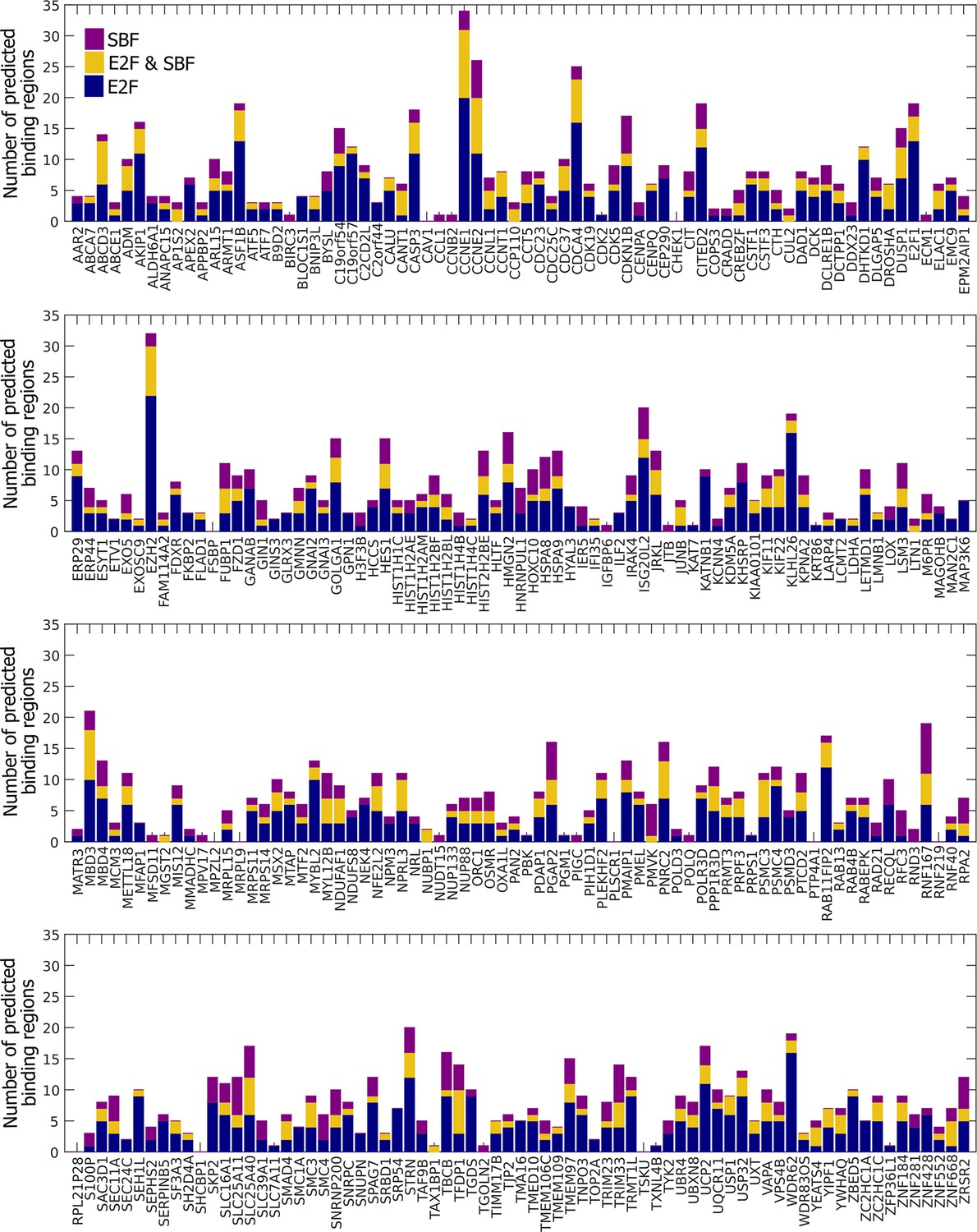
Many E2F-regulated genes in humans could be bound by SBF.
Summary of E2F-only regions (blue), SBF-only regions (purple), and E2F and SBF co-regulated regions (yellow) for a set of 290 E2F-regulated promoters.
Figure 9
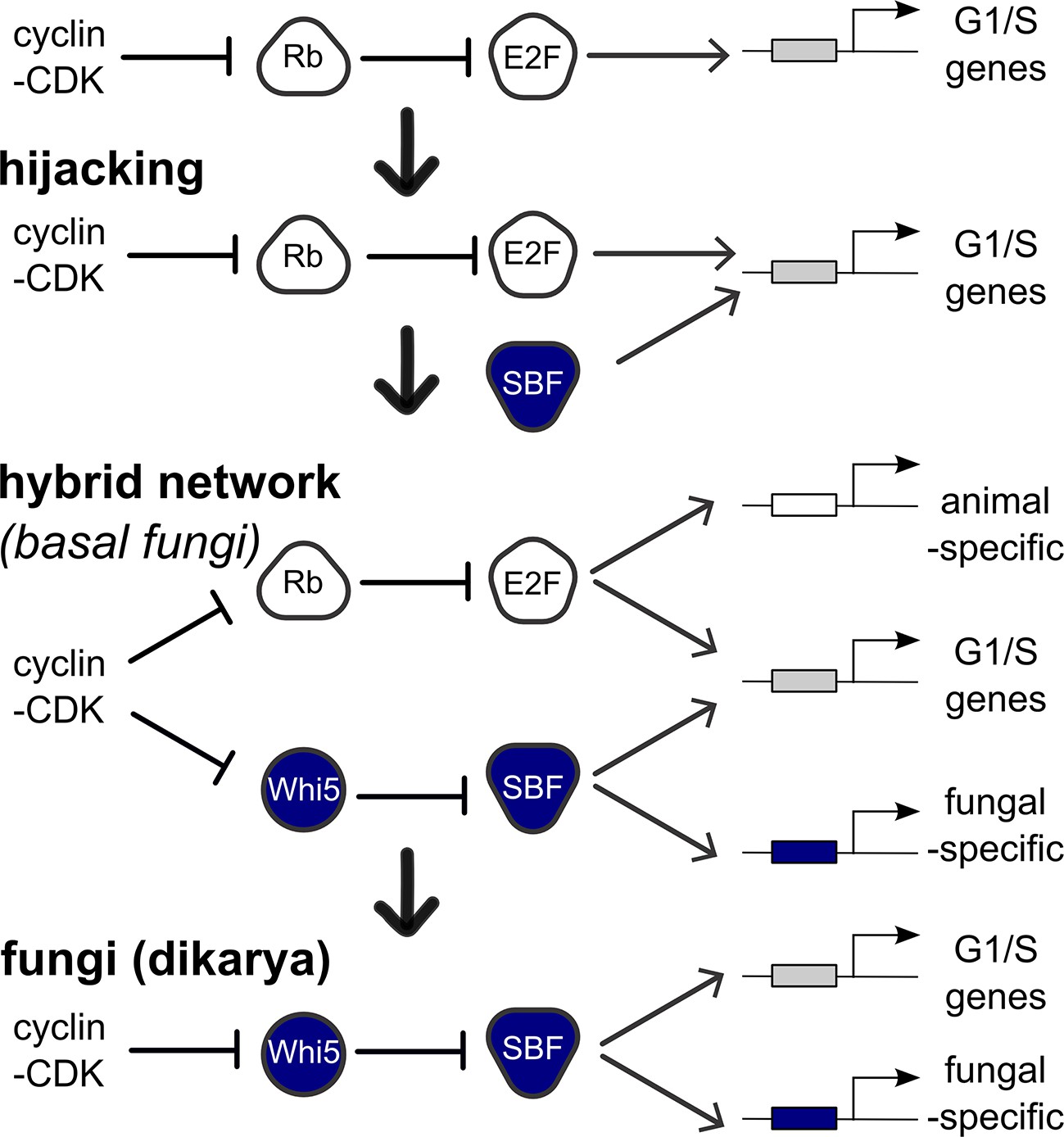
Punctuated evolution of a conserved regulatory network.
Evolution can replace components in an essential pathway by proceeding through a hybrid intermediate. Once established, the hybrid network can evolve dramatically and lose previously essential regulators, while sometimes retaining the original network topology. We hypothesize that SBF may have hijacked the cell cycle of a fungal ancestor by binding cis-regulatory DNA sites of E2F and activating expression of G1/S genes, thus promoting cell cycle entry. Cell cycle hijacking in a fungal ancestor was followed by evolution of Whi5 to inhibit SBF and Whi5 was subsequently entrained to upstream cell cycle control through phospho-regulation by old or new cyclin-CDKs to create a hybrid network with parallel pathways. The hybrid network likely provided redundant control of the G1/S regulatory network, which could explain the eventual loss of E2F and its replacement by the SBF pathway in more derived fungi (Dikarya). Interestingly, zoosporic fungi such as Chytrids have hybrid networks and are transitional species because they exhibit animal-like features of the opisthokont ancestor (centrioles, flagella) and fungal-like features (cell wall, hyphal growth). We hypothesize that E2F and SBF also bind and regulate a subset of animal-specific and fungal-specific G1/S genes, which could help explain the preservation of the hybrid network in Chytrids. Ancestral SBF expanded to create an entire family of transcription factors (APSES) that regulate fungal-specific traits such as sporulation, differentiation, morphogenesis, and virulence.
Additional files
-
Supplementary file 1
(A) List of eukaryotic genomes. We downloaded and analyzed the following annotated genomes using the 'best' filtered protein sets when available. We gratefully acknowledge the Broad Institute, the DOE Joint Genome Institute, Génolevures, PlantGDB, SaccharomycesGD, AshbyaGD, DictyBase, JCV Institute, Sanger Institute, TetrahymenaGD, PythiumGD, AmoebaDB, NannochloroposisGD, OrcAE, TriTryDB, GiardiaDB, TrichDB, CyanophoraDB, and CyanidioschizonDB for making their annotated genomes publicly available. We especially thank D. Armaleo, I. Grigoriev, T. Jeffries, J. Spatafora, S. Baker, J. Collier, and T. Mock for allowing us to use their unpublished data. (B) Plasmids. (C) Strains. All yeast strains were derived from W303 and constructed using standard methods.
- https://doi.org/10.7554/eLife.09492.033
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Punctuated evolution and transitional hybrid network in an ancestral cell cycle of fungi
eLife 5:e09492.
https://doi.org/10.7554/eLife.09492




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}