Functionally important residues from graph analysis of coevolved dynamic couplings
- UCL School of Pharmacy, United Kingdom
- Department of Computer Science, Brunel University London, United Kingdom
- Centre for Host-Microbiome Interactions, Faculty of Dentistry, Oral & Craniofacial Sciences, King’s College London, United Kingdom
- Research Service, Louis Stokes Cleveland Department of Veterans Affairs Medical Center, United States
- Department of Molecular Biology and Microbiology, Case Western Reserve University School of Medicine, United States
- Department of Medicine, Case Western Reserve University School of Medicine, United States
- Departments of Pharmacology, Biochemistry, and Proteomics and Bioinformatics Case Western Reserve University School of Medicine, United States
- CWRU-Cleveland VAMC Center for Antimicrobial Resistance and Epidemiology (Case VA CARES), United States
- University of Tabuk (PFSCBR), Saudi Arabia
- UCL Center for Advanced Research Computing, University College London, United Kingdom
eLife Assessment
This article reports the analysis of coevolutionary patterns and dynamical information for identifying functionally relevant sites. These findings are considered important due to the broad utility of the unified framework and network analysis capable of revealing communities of key residues that go beyond the residue-pair concept. The data are solid and the results are clearly presented.
https://doi.org/10.7554/eLife.105005.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Solid: Methods, data and analyses broadly support the claims with only minor weaknesses
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The relationship between protein dynamics and function is essential for understanding biological processes and developing effective therapeutics. Functional sites within proteins are critical for activities such as substrate binding, catalysis, and structural changes. Existing computational methods for the predictions of functional residues are trained on sequence, structural, and experimental data, but they do not explicitly model the influence of evolution on protein dynamics. This overlooked contribution is essential as it is known that evolution can fine-tune protein dynamics through compensatory mutations either to improve the proteins’ performance or diversify its function while maintaining the same structural scaffold. To model this critical contribution, we introduce DyNoPy, a computational method that combines residue coevolution analysis with molecular dynamics simulations, revealing hidden correlations between functional sites. DyNoPy constructs a graph model of residue–residue interactions, identifies communities of key residue groups, and annotates critical sites based on their roles. By leveraging the concept of coevolved dynamical couplings—residue pairs with critical dynamical interactions that have been preserved during evolution—DyNoPy offers a powerful method for predicting and analysing protein evolution and dynamics. We demonstrate the effectiveness of DyNoPy on SHV-1 and PDC-3, chromosomally encoded β-lactamases linked to antibiotic resistance, highlighting its potential to inform drug design and address pressing healthcare challenges.
Introduction
Quantifying the contribution of individual residues or residue groups to protein function is important to estimate the pathogenic effect of mutations (Stenson et al., 2017). Identifying the functional roles of individual residues has primarily been done through mutagenesis experiments (Matreyek et al., 2018). Bioinformatics methods have complemented these approaches through analysis of multiple sequence alignments (MSA) of homologous proteins and structural data (Poelwijk et al., 2016; Høie et al., 2022; Blaabjerg et al., 2023; Dunham and Beltrao, 2021; Hopf et al., 2017; Radivojac et al., 2013). Among these methods, computational techniques that can decode inter-residue evolutionary relationships from MSAs have paved the way for machine learning (ML)-based strategies that can predict protein structure (Jumper et al., 2021; Lin et al., 2023; Baek et al., 2021; Marks et al., 2012), stability (Broom et al., 2020), and function (Hopf et al., 2017) and extend the scope of computational protein design (Ding et al., 2024; Wu et al., 2019; Russ et al., 2020). A most recent approach has combined experimental data from three proteins, NUDT15, PTEN, and CYP2C9, on the stability and function with sequence and structural features to train an ML model to predict functional sites (Cagiada et al., 2023).
Functional sites are often regulated by both local and global interactions. Changes in these interactions are instrumental for functional events like substrate binding, catalysis, and conformational changes (Wodak et al., 2019). The development of physical models of protein dynamics and the increase in available computational power has stimulated the adoption of computational techniques (Campitelli et al., 2020; Rodrigues et al., 2021) to investigate the conformational dynamics of proteins, an essential component of the many biological functions (Henzler-Wildman and Kern, 2007; James and Tawfik, 2003). Different models have been proposed to describe the interactions between residues during simulations and network models have been particularly popular, including methods on single structures and molecular dynamics (MD) simulations data built by analysing the response to external forces on residue networks (Nevin Gerek et al., 2013), estimating the prevalence of non-covalent energy interaction networks in homologous proteins (Yehorova et al., 2024), or analysing linear or non-linear correlation in atomic fluctuations (Lange and Grubmüller, 2006; Osuna, 2021). These techniques have demonstrated their usefulness in extracting allosteric networks from structural data with applications in enzyme design (Osuna, 2021).
However, none of these techniques incorporate information on residue evolution into the computational approach, while it has been established that evolution through compensatory mutations in dynamic regions, like hinges and loops, can fine-tune protein structural dynamics and introduce promiscuity, thereby diversifying biological function. Assuming that protein functional dynamics is conserved during evolution, significant information on dynamic regions and substrate recognition sites should be recoverable using inter-residue coevolution scores extracted from MSAs (Granata et al., 2017; Liu and Bahar, 2012). Coevolution analysis and MD simulations have independently (Parente et al., 2015) and synergistically been combined in the past to identify important residues for function (Ponzoni et al., 2015; Sutto et al., 2015; Estabrook et al., 2005; Chen et al., 2011; Wang et al., 2013). Yet a method that combines hidden information on dynamics from evolution with direct information on local and global dynamics from conformational ensembles from MD is not yet available.
Here, we present DyNoPy, a computational method that can extract hidden information on functional sites from the combination of pairwise residue coevolution data and powerful descriptors of dynamics extracted from the analysis of MD ensembles. The method can detect coevolved dynamic couplings, that is, residue pairs with critical dynamical interactions that have been preserved during evolution. These pairs are extracted from a graph model of residue–residue interactions. Communities of important residue groups are detected, and critical sites are identified by their eigenvector centrality in the graph (Figure 1). We demonstrate the power of this approach on SHV-1 and PDC-3 β-lactamases of major clinical importance (Olehnovics et al., 2021; Chen et al., 2024). DyNoPy successfully detects residue couplings that align with previous studies, guide in the explanations of mutation sites with previously unexplained mechanisms, and provide predictions on plausible important sites for the emergence of clinically relevant variants.
Figure 1
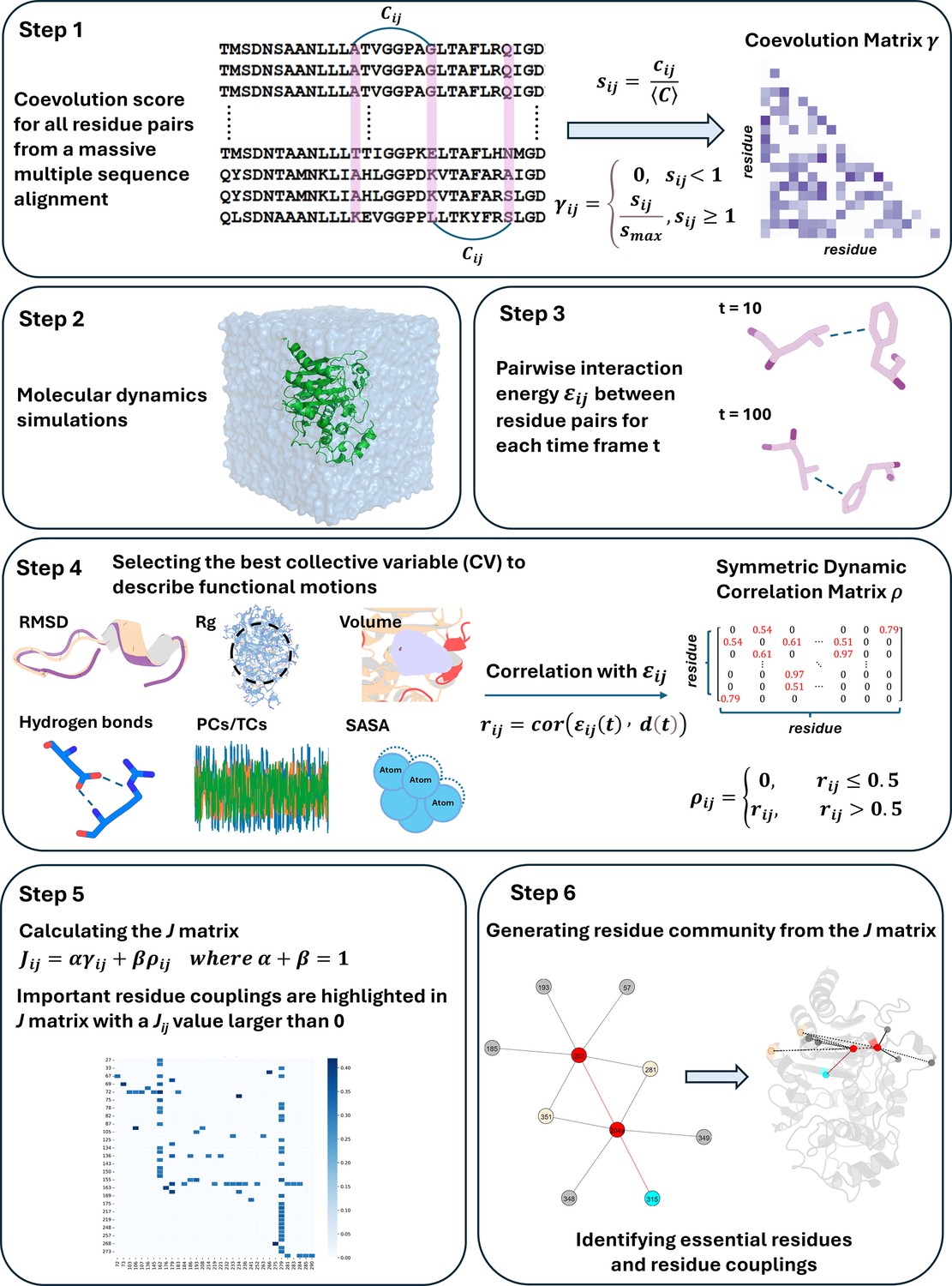
Overview of the DyNoPy workflow.
Results and discussion
β-Lactamases are a group of enzymes capable of hydrolysing β-lactams, conferring resistance to β-lactam antibiotics (Poole, 2004). These enzymes are evolving rapidly as single amino acid substitutions are sufficient to drive their evolution and increase their catalytic spectrum and inhibitor resistance profile (Bush, 2018). The widespread dissemination of β-lactamases across different bacterial species and their extensive emergence highlight their global impact on antibiotic resistance (Bush, 2013). The rapid evolution of β-lactamases and their clinical significance (Bush, 2018) makes them an ideal target for evaluating the robustness of DyNoPy.
In this study, we applied DyNoPy to two model enzymes from different β-lactamase families: class A β-lactamase SHV-1 (a chromosomally encoded enzyme in Klebsiella pneumoniae) and class C β-lactamase PDC-3 (a chromosomally encoded enzyme in Pseudomonas aeruginosa) (Olehnovics et al., 2021; Chen et al., 2024; Figure 2). Both class A and class C β-lactamases comprise an α/β domain and an α helical domain, with the active site situated in between (Matagne et al., 1998; Philippon et al., 2022). Moreover, both enzymes target the carbonyl carbon of the β-lactams using a highly conserved serine residue (Palzkill, 2018; Jacoby, 2009). Despite these similarities, the structures of class A and class C β-lactamases are remarkably different (Figure 2—figure supplements 1 and 2). In class A β-lactamases, the active site is surrounded by three loops: the α3-α4 loop (residues 101–111), the Ω-loop (residues 164–179), and the hinge region (residues 213–218) (Galdadas et al., 2021). The Ω-loop is particularly critical as it positions N170 to hydrogen bond with the E166 via a conserved water molecule, which is essential for initiating the deacylation step (Kuzin et al., 1999). Compared to class A β-lactamases, the active site of class C β-lactamases is wider, conferring a broader substrate binding capability (Medeiros, 1997; Figure 2—figure supplement 3). The active site of class C β-lactamases can be divided into two parts: the R1 site and the R2 site (Jacoby, 2009). The R1 region is surrounded by the extended Ω-loop (residues 183–226), while the R2 site is enclosed by the R2-loop (residues 280–310) (Chen et al., 2024). The Ω-loop in class C β-lactamases is significantly longer than that in class A, enhancing the active site ability to accommodate diverse substrates and contributing to the extended spectrum profile of some class C enzymes (Chen et al., 2024).
Figure 2 with 3 supplements see all
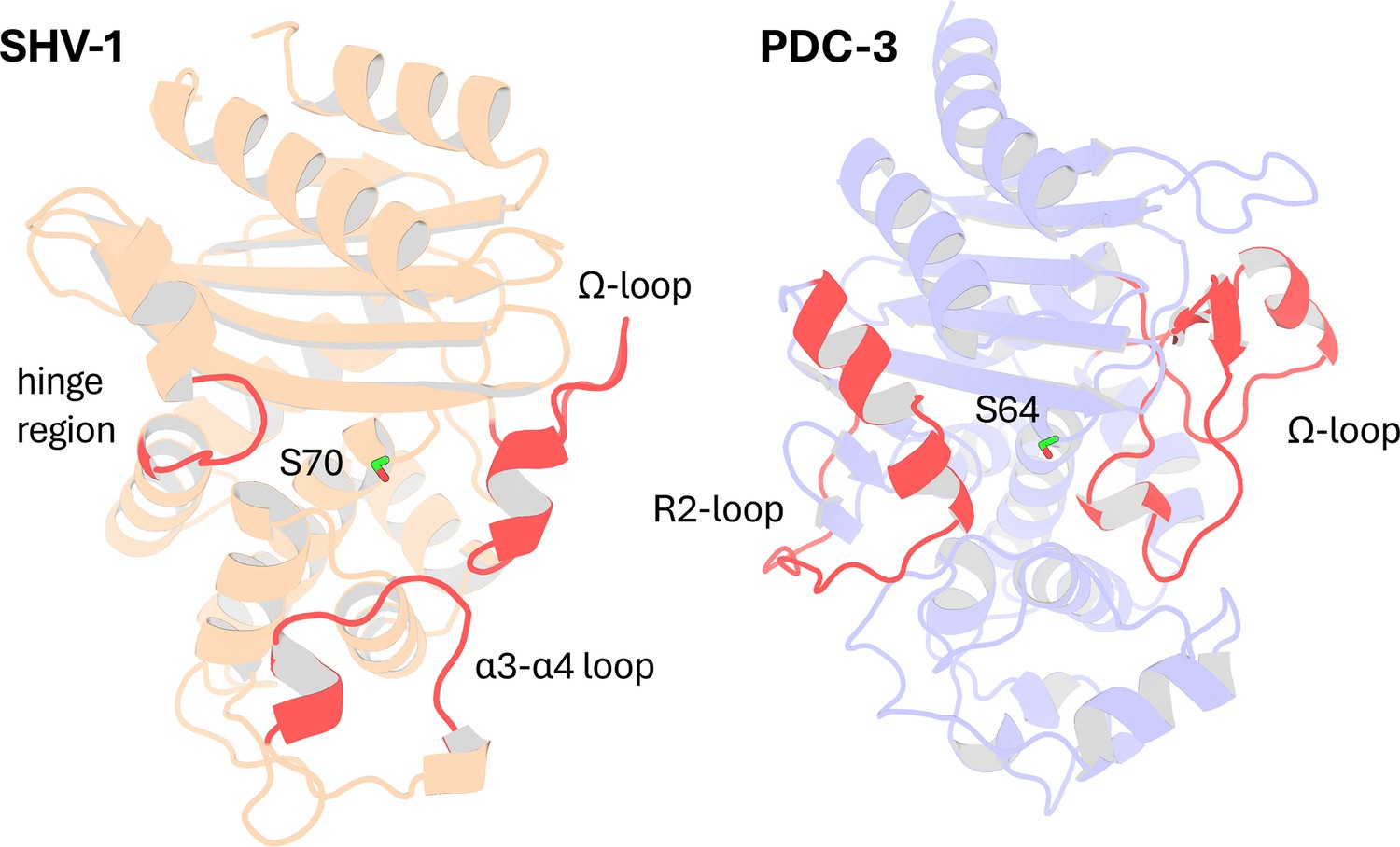
Structural Comparison of SHV-1 (PDB ID: 3N4I) and PDC-3 (PDB ID: 4HEF) β-lactamases.
Catalytic serine S70 (SHV-1) and S64 (PDC3) are highlighted using stick representation. Important loops surrounding the active site are highlighted in red. In SHV-1, highlighted loops are the α3-α4 loop (residues 101–111), the Ω-loop (residues 164–179), and the hinge region (residues 213–218). In PDC-3, highlighted loops are the Ω-loop (residues 183–226) and the R2-loop (residues 280–310).
SHV-1 is a very well-characterized enzyme with a wealth of information on mutations and their corresponding effects on protein function. In contrast, the information available on PDC-3 remains limited. Essential catalytic residues in SHV-1 are S70, K73, S130, E166, N170, K234, G236, and A237 (Ambler et al., 1991), and conserved catalytic residues in PDC-3 include S64, K67, Y150, N152, K315, T316, and G317. Highly conserved stretches of 3–9 hydrophobic residues, annotated as hydrophobic nodes, exist in class A β-lactamases and have been proven to be essential for protein stability (Galdadas et al., 2018). Residues defined as belonging to hydrophobic nodes within SHV-1 are listed in Supplementary file 1a.
In SHV-1, the predominant extended spectrum β-lactamase (ESBL) substitutions occur at L35, G238, and E240, while R43, E64, D104, A146, G156, D179, R202, and R205 appear in ESBLs with lower frequency (Liakopoulos et al., 2016). Mutations at M69, S130, A187, T235, and R244 are known to induce inhibitor resistance in the enzyme (Pagan-Rodriguez et al., 2004). In PDC-3, substitutions primarily occur on the Ω-loop, enhancing its flexibility to accommodate the bulky side chains of antibiotics, while deletions are more common in the R2-loop (Jacoby, 2009). The predominant Ω-loop mutations isolated from clinics are found at positions V211, G214, E219, and Y221 (Barnes et al., 2018).
Emergence of highly conserved dynamic couplings
DyNoPy builds a pairwise model of conserved dynamic couplings detected by combining coevolution scores and information on functional motions into a score Jij (see ‘Methods’ and Figure 1). To this end, a dynamic descriptor should be selected. When the descriptor is associated with functional conformational changes, it is expected that functionally relevant couplings will report higher scores. Dynamic descriptors can be selected from commonly used geometrical collective variables (CVs) for the analysis of MD trajectories (see ‘Methods’). As expected, the average J matrix score varies across the different CVs, with some of them showing no signal of dynamic coupling (Figure 3A).
Figure 3 with 1 supplement see all
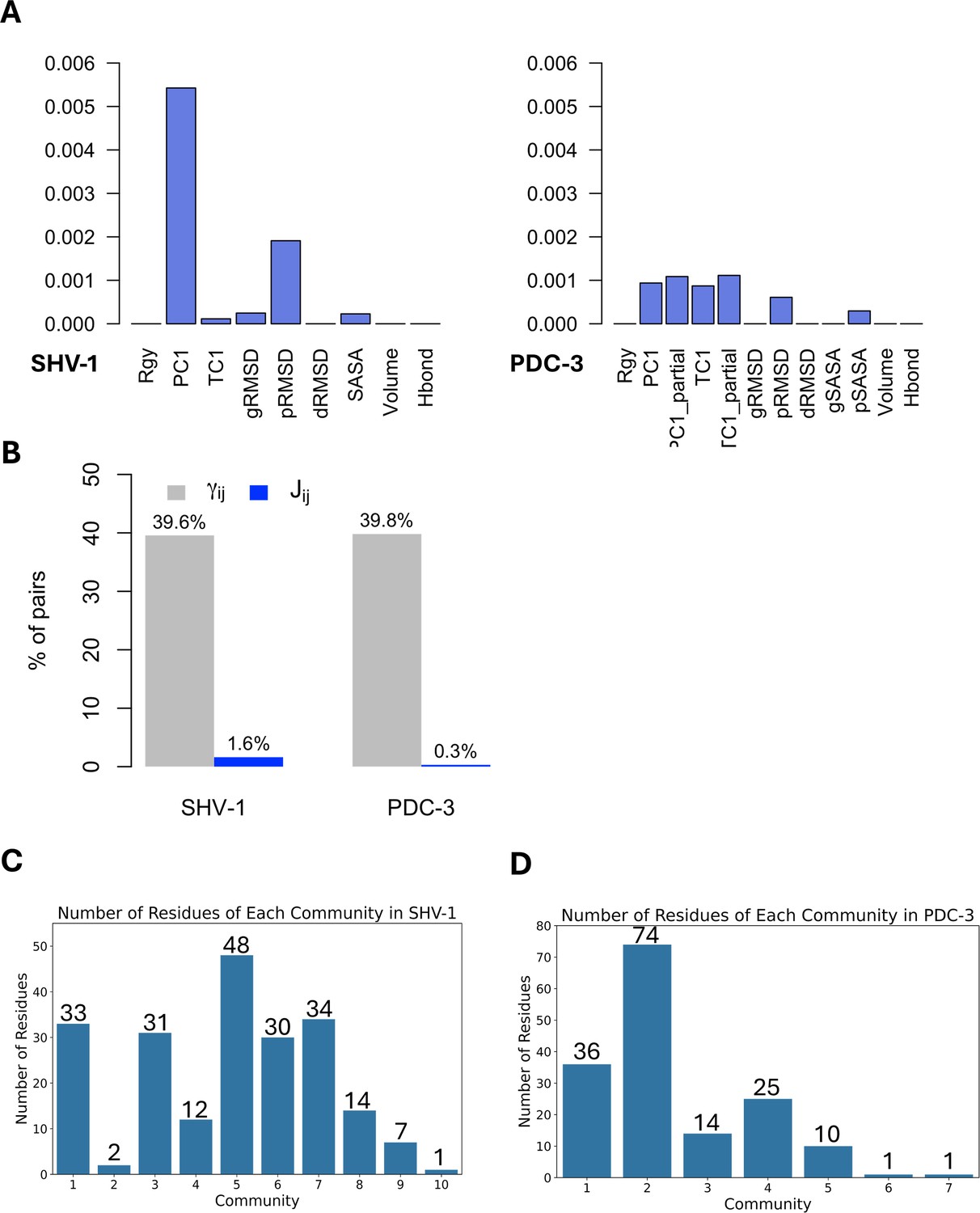
Features and residues in each community.
(A) Average J matrix score varies across different collective variables (CVs). Left: SHV-1; right: PDC-3. (B) Number of non-zero couplings detected by scaled coevolution scores () and J values calculated by DyNoPy (). (C) Number of residues of each community in SHV-1. (D) Number of residues of each community in PDC-3. A reasonable residue community should contain at least three residues.
SHV-1 and PDC-3 exhibit distinct dynamics, requiring a different choice of the CV that best captures the functional dynamics. For SHV-1, the global first principal component (PC1) proved to be the most effective feature, identifying 571 residue pairs with a Jij value greater than 0. Conversely, PDC-3 requires selection of more localized features that can extract the Ω-loop dynamics from the overall protein motion. Among the dynamic descriptors, the partial first time-lagged component (TC1_partial) performed best for PDC-3, detecting 216 residue pairs with a Jij value greater than 0. Consequently, PC1 and TC1_partial were selected to build the J matrix for SHV-1 and PDC-3, respectively. The performance of all 12 CVs for each protein was assessed and listed in Supplementary file 1b.
The importance of dynamical information is evident when coevolution couplings () and conserved dynamic couplings (Jij) are compared: the number of non-zero couplings decreases from 40% to <2% of total residue pairs in the protein (Figure 3B) when information from the dynamics descriptor is added. Thus, the inclusion of protein dynamics in coevolution studies acts as an effective filter that rules out residue pairs that do not have significant correlations with functional motions. Moreover, when relying only on , all the residues in SHV-1 and PDC-3 are included within four identified communities (Supplementary file 1c), suggesting that coevolution scores () alone do not effectively discriminate residues relevant for protein functions. Furthermore, it would be hard to distinguish critical core residues for each community using only as the eigenvector centrality (EVC) values for the residues do not show remarkable differences (Figure 3—figure supplement 1A and B). This means that detailed dynamic investigation of the top residues is needed to determine which pairs should be picked up and further analysed. On the other hand, it is much easier to identify essential residues based on J scores calculated as clear outliers with significantly higher EVC values could be seen for almost all communities (Figure 3—figure supplement 1C and D; Parente et al., 2015; Negre et al., 2018). In conclusion, the lack of specificity in the statistically based coevolution analysis supports the choice of incorporating a score for the correlation between residue interactions and dynamic behaviours that enables deconvolution of community information.
DyNoPy reveals critical residues and predicts evolutionary pathways in SHV-1
DyNoPy identified eight meaningful communities, each consisting of at least three strongly coupled residues within SHV-1 (Figure 3C). All crucial catalytic residues and critical substitution sites previously mentioned participating in one of these communities with the exceptions of R43, R202, and S130. Residues previously known to have critical role in function or conferring ESBLs/IRBLs phenotype are either directly coupled to protein dynamics or act as a central hub. The hubs interact with residues with either a role in catalysis or structural stability through their membership of hydrophobic nodes (Olehnovics et al., 2021). Furthermore, DyNoPy identified key positions (L162 and N136) within some communities that are known to undergo substitutions, conferring an ESBL phenotype in other class A β-lactamases. These substitutions have not yet emerged in the SHV family, providing insightful predictions about the potential future evolution of the enzyme. Detailed discussions of communities with secondary importance for protein function (communities 3, 8, and 9) is provided in the Appendix (Appendix 2—figure 1).
DyNoPy predicts mutation hotspots in SHV-1
DyNoPy detects critical mutation sites (L162 and N136) that are known to extend the range of substrates in other class A β-lactamases but have not yet emerged as variants in the SHV family. These sites have not been modified in the SHV family because of their plausible central role within the communities as they are mediating couplings with key functional residues essential for catalytic activity and structural stability, indicating their critical role in protein function and the potential lower mutation rate. These findings provide insightful predictions about the potential future evolution of the enzyme, as well as plausible explanations for why these mutations have not yet appeared.
L162, positioned at the start of the Ω-loop and adjacent to the crucial catalytic residue E166, is assigned as the core residue for community 1 (Figure 4A). While it remains conserved in the SHV family, variants of L162 have been isolated in other class A β-lactamase and are known to expand the enzyme catalytic spectrum. Single amino acid substitution at L162 can intensify antibiotic resistance in BEL-1 (Pozzi et al., 2016), a class A ESBL clinical variant, exhibiting robust resistance to ticarcillin and ceftazidime (Bogaerts et al., 2007). BEL-2 diverges from BEL-1 by single amino acid substitution (L162F), which alters the kinetic properties of the enzyme significantly and increases its affinity towards expanded-spectrum cephalosporins (Poirel et al., 2010). The relationship between L162 and protein catalytic functions can be explained using DyNoPy model as there are couplings with catalytic important residues M69, K73, E166, and K234. Moreover, the BEL case has confirmed that L162F mutation significantly destabilizes the overall protein structure, highlighting the crucial role of L162 in maintaining protein stability (Pozzi et al., 2016). DyNoPy accurately identifies the centrality of L162 by reporting its connections with 28 backbone residues, including 9 hydrophobic node residues critical for protein stability. Among these, five hydrophobic residues are part of the α2 node: V75, L76, G78, V80, and L81, highlighting the contribution of L162 to the stability of the α2 helix (Olehnovics et al., 2021).
Figure 4
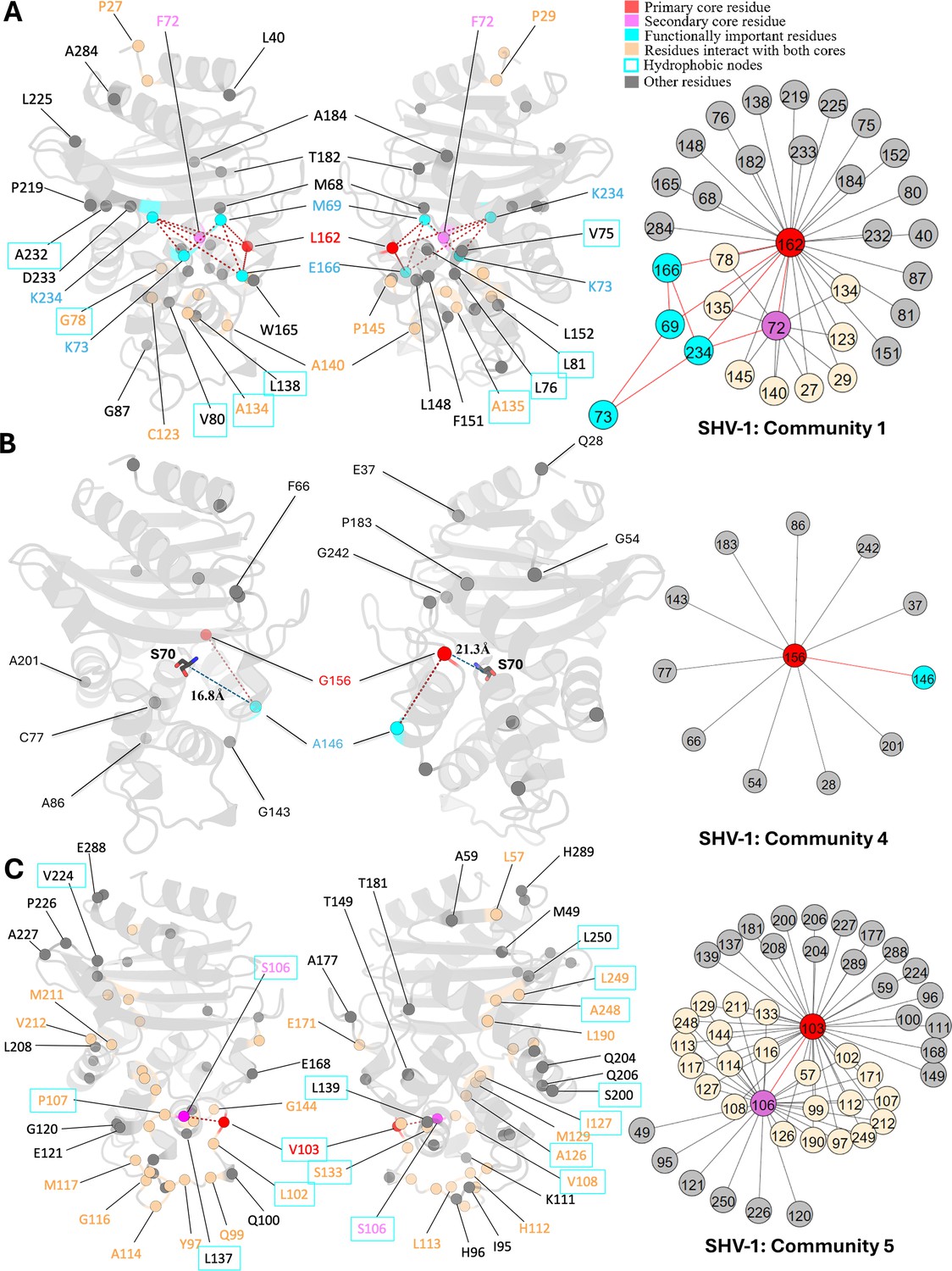
Communities 1, 4, and 5 of SHV-1 β-lactamase.
All the residues are depicted as spheres on the protein structure. The core residue for each community is highlighted in red, while purple is used to emphasize the secondary core residue. Residues that interact with both cores are coloured in light yellow. Functional important residues are marked in cyan. Hydrophobic nodes are enclosed with cyan boxes. (A) Community 1 of SHV-1, comprising 33 residues with L162 being the primary core residue. (B) Community 4 of SHV-1, containing 12 residues and is centred by G156. G156 and A146 are two functional important residues distant from the active site. G156 is 21.3 Å away from the catalytic S70. A146 is 16.8 Å away from S70. (C) Community 5 of SHV-1, embracing 48 residues and showing a strong correlation between V103 and S106.
Just like L162, N136 undergoes advantageous mutations in other class A β-lactamases while remains highly conserved within the SHV family. It is the core residue for community 7 (Figure 5B). This residue forms a hydrogen bond with E166, stabilizing the Ω-loop (Bös and Pleiss, 2008). Although DyNoPy did not detect this direct interaction between N136 and E166, the established relationship between N136 and N170 highlights the role of N136 in influencing E166. N170, an essential catalytic residue located on the Ω-loop, contributes to priming the water molecule for the deacylation step with E166 (Agarwal et al., 2023), and is directly coupled with N136. Due to the essential contribution of N136 in facilitating E166 to maintain its proper orientation, it was previously thought to be intolerant to mutations as substitution of asparagine to alanine at this position would make the enzyme lose its function completely (Cao et al., 2020). However, N136D substitution has emerged as a new clinical variant very recently in PenL, a class A β-lactamase, by increasing its ability in hydrolysing ceftazidime (Cao et al., 2020), suggesting that this site has potential to mutate. This gain of function is mainly triggered by the increased flexibility of the Ω-loop (Cao et al., 2020). DyNoPy correctly detects a dynamical relationship between N136 and the Ω-loop (residues 164–179). Six residues present in the Ω-loop participate within this community, including R164 and D179. These two residues are critical as they are forming the ‘bottleneck’ of the Ω-loop which is essential for the correct position of E166 (Parwana et al., 2024). D179 is also a critical mutation site for SHV-1. Single amino acid substitutions like D179A, D179N, and D179G are enough for the extended spectrum phenotype (Liakopoulos et al., 2016).
Figure 5
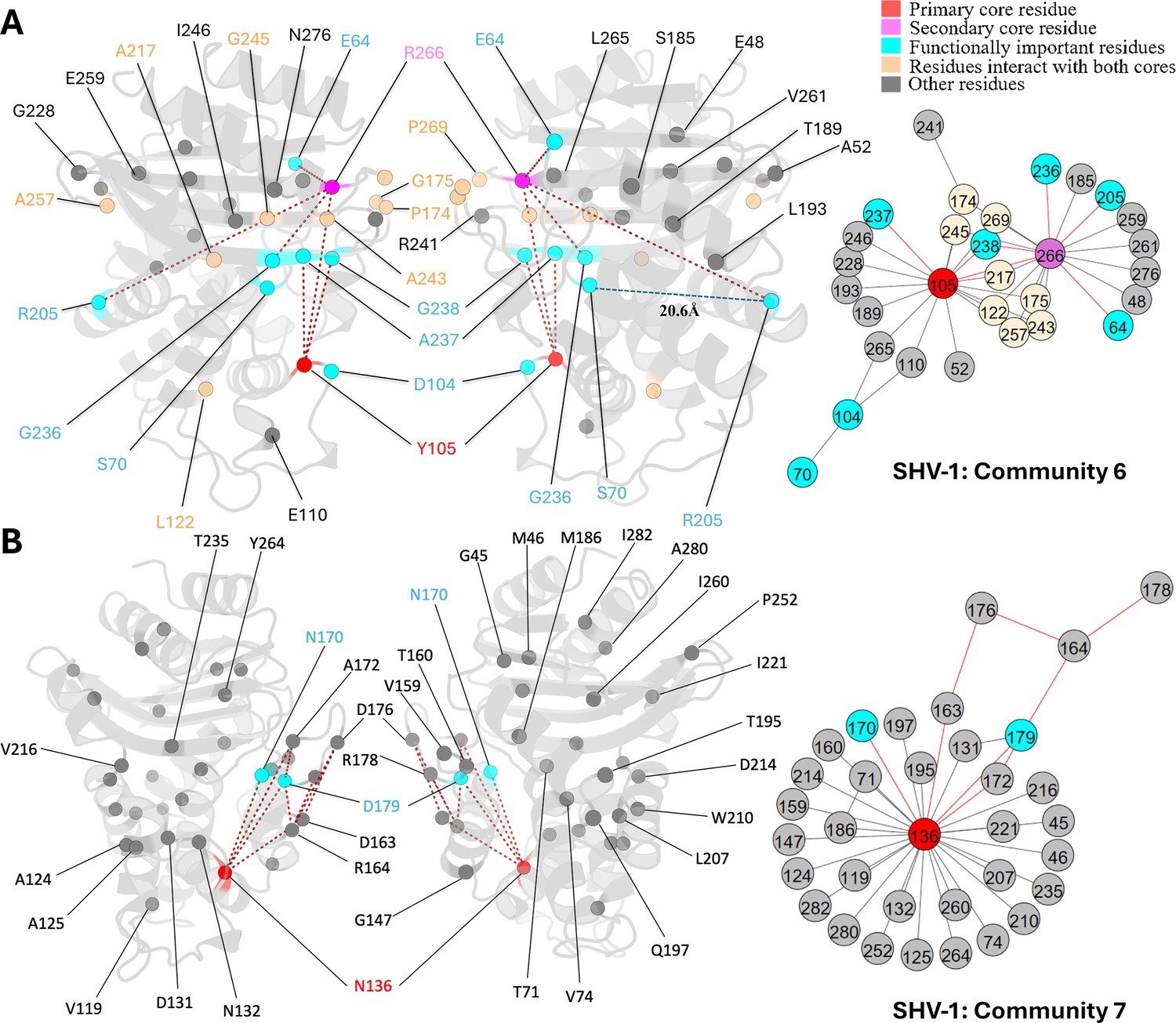
Communities 6 and 7 of SHV-1 β-lactamase.
All the residues are depicted as spheres on the protein structure. The core residue for each community is highlighted in red, while purple is used to emphasize the secondary core residue. Residues that interact with both cores are coloured in light yellow. Functional important residues are marked in cyan. (A) Community 6 of SHV-1, comprising 30 residues with Y105 being the primary core residue. R205 is a functional important residue that is 20.6 Å away from the active site S70. (B) Community 7 of SHV-1, containing 34 residues and is centred by N136.
DyNoPy detects residue couplings essential for protein stability
DyNoPy identifies residue couplings critical for protein functional motions, particularly associated with protein stability. These residue pairs exhibit strong relationships as they are not only directly coupled with each other but also forms various indirect couplings via other residues. As a result, both residues are considered as core residues inside these communities. It is expected that disruption of these couplings through mutation could compromise collective motions essential for enzyme activity.
As the secondary core residues in community 1 (Figure 4A), F72 is showing a strong coupling with the primary core residue L162 and also forms nine indirect couplings with L162, including via the catalytic K234. This network of direct and indirect relationships reveals the importance of F72 and L162 coupling in maintaining protein functional motions. Interestingly, previous studies identified a small hydrophobic cavity formed by L162 and F72, together with L139, and L148, which is essential for the stability of the active site (Pozzi et al., 2016). Notably, DyNoPy successfully recovers the key residues of this local hydrophobic cavity (L162, F72, and L148).
The strong interplay between V103 and S106, which are both residues on the α3-α4 loop, is seen in community 5 (Figure 4C). These residues not only interact with each other directly but are also indirectly coupled via 22 other residues. This community emphasizes the significance of hydrophobic nodes in SHV stability and dynamics. Within the analysed 48 residues, 27 are hydrophobic, out of which 15 residues act as nodes critical for enzyme stabilization. Hydrophobic nodes stabilize their own secondary structures and interconnect to stabilize the overall protein (Galdadas et al., 2021). V103 and S106 themselves are hydrophobic nodes, stabilizing α3 helix and α4 helix, respectively, and are strongly coupled with each other. In CTX-M, another class A enzyme, N106S is a common substitution that results in improved thermodynamic stability and compensate for the loss in stability of the variants (Lu et al., 2022). Interestingly, this residue is already a serine in SHV but still implies its pivotal role in protein stability.
DyNoPy provides valid explanations for mutation sites
During the evolution of β-lactamases, single mutations on specific sites that are distant from the functional sites have been observed to significantly alter protein catalytic functions. Additionally, single mutations on some surface exposed residues can dramatically increase protein stability. Understanding how these distant mutations impact function and stability becomes a major challenge in understanding protein evolutionary pathways. Communities extracted by DyNoPy show these residues linked with functional important residues, providing a rational for these mutation sites with unknown functions.
Mutations of G156 are limited but they lead to ESBL phenotype in the SHV family (Liakopoulos et al., 2016). G156 is the central residue for community 4 (Figure 4B), but it is distant from the active site, over 20 Å away from the catalytic serine S70. Clinical variant SHV-27 has extended resistance ability towards cefotaxime, ceftazidime, and aztreonam (Corkill et al., 2001). It differs from SHV-1 by single amino acid substitution G156D, suggesting that it has directly evolved from SHV-1 (Corkill et al., 2001). Limited research has been done on position G156, and the understanding of how it affects the enzyme catalytic properties given that it is far away from the active site is still unclear. Based on our results, we suggest that this residue is essential for the overall protein function because of its 11 coevolved dynamic couplings with protein dynamics, including A146, another ESBL substitution site.
SHV-38, another ESBL that is capable of hydrolysing carbapenems, harbours a single A146V substitution compared to SHV-1 (Poirel et al., 2003). Like G156, A146 is 16.8 Å away from S70 but shows an ability in altering protein catalytic function. The A146-G156 residue pair shows a strong coevolutionary signal and strong correlation with protein overall dynamics, implying that there may compensatory mutations at these sites with potential to emerge in the SHV family in the future. These two residues are not connected to any catalytic residues but their coupling to functional dynamics can offer plausible explanation to ESBL activity of these two mutations.
Unlike other substitution sites that are adjacent to the active site, R205 is situated more than 20 Å away from catalytic serine S70. Its side chain points outwards from the protein, exposing to the solvent. The R205L substitution often co-occurs with other ESBL mutations and is thought to indirectly contribute to the ESBL phenotype by compensating for stability loss induced by other mutations (Ben Achour et al., 2009). SHV-3 is an ESBL that exhibits significant resistance to cefotaxime and ceftriaxone (Nicolas et al., 1989). Two substitutions in this enzyme, R205L and G238S, extend its resistance profile (Nicolas et al., 1989). Thus, it is promising to see that DyNoPy detected these two mutation sites together within community 6 (Figure 5A).
Y105 and R266 are the core residues for community 6 (Figure 5A). Y105 is situated on the α3-α4 loop positioned at the left side of the binding pocket. It is an important catalytic residue that recognizes and binds to the thiazolidine ring of penicillins or β-lactamase inhibitors (Bethel et al., 2006). There is very limited information on the role of R266, except that it may stabilize the Ω-loop in the SHV family similar to the analogous T266 in TEM (Kuzin et al., 1999). G238 is coupled with an essential catalytic residue Y105, which further links with other catalytic functional residues: S70 and A237, and R266, a residue that is known to stabilize the Ω-loop. This indicates that mutations on G238 would result in an alteration on protein catalytic function, as well as an increased flexibility of the protein, which strongly aligns with previous finding (Nicolas et al., 1989). Its linked mutation site R205 does not show direct coupling with any catalytic residues. Instead, it is directly coupled with R266, which we mentioned as an Ω-loop stabilizer. Thus, it is not surprising that R205 substitution alone is never observed in nature (Neubauer et al., 2020) as it would not give significant evolutionary advantage to the protein.
Insights into the unexplained functional sites of PDC-3
Unlike the extensively studied SHV-1, the functional roles of individual amino acids in PDC-3 remain largely unexplored. This gap in understanding serves as welcome challenge for interpreting the effects of mutations and the dynamic behaviour of PDC-3 from our results. Although several mutation hotspots, such as those on the Ω-loop (Barnes et al., 2018), have been identified, very little is known about the specific contributions of individual amino acids on the functionality of PDC-3.
In PDC-3, mutations have primarily been reported in the Ω-loop. They enhance its flexibility to accommodate the bulky side chains of antibiotics, while deletions are more common in the R2-loop (Jacoby, 2009). DyNoPy detected five communities in total (Figure 3D), with all the four predominant Ω-loop mutations appearing in these communities. Communities 3, 4 and 5 are discussed in the Appendix (Appendix 2—figure 2). Furthermore, DyNoPy also detected several previously unexplored Ω-loop residues.
G214, a known mutation site in PDC-3, is the core residue in community 1. Another two essential mutation sites, E219 and Y221, also participate in this community, directly coupled with G214 (Figure 6A). G214 also has direct couplings with four other Ω-loop residues: A195, A197, G212, and L216. Previous results have demonstrated that substitutions of glycine to alanine or arginine at 214 significantly destabilize the Ω-loop (Chen et al., 2024). The strong correlation between G214 and these Ω-loop residues emphasizes the significant contribution of G214 towards the stability of the Ω-loop, which corroborates with previous results (Chen et al., 2024). Moreover, substitutions such as G214A and G214R and mutations on E219 and Y221 do not affect R2 loop flexibility, resulting in the smaller active site volume among variants (Chen et al., 2024) because none of the residues from the R2 loop are detected in this community, offering plausible explanation to the previously unexplained phenomenon.
Figure 6
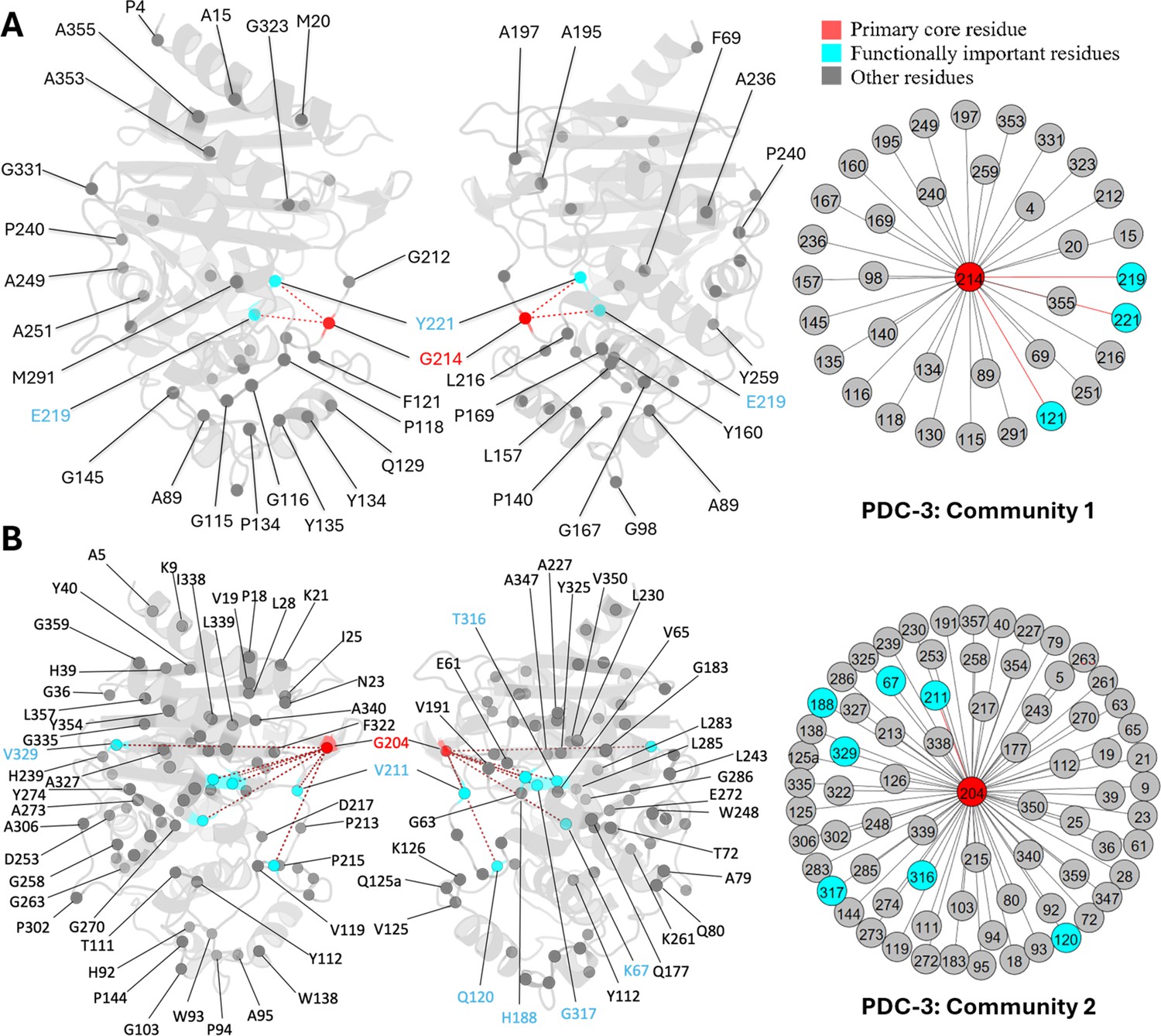
Communities 1 and 2 of PDC-3 β-lactamase.
All the residues are depicted as spheres on the protein structure. The core residue for each community is highlighted in red. Functional important residues are marked in cyan. (A) Community 1 of PDC-3, comprising 36 residues with G214 being the primary core residue. (B) Community 2 of PDC-3, containing 74 residues and is centred by G204.
G204 is the core residue of community 2, coupled with 73 other residues, most of which are distant from the catalytic site, suggesting plausible crucial role in the overall protein stability like L162 in SHV-1 (Figure 6B). G204, a newly emerged mutation site in the PDC family (67), is located on the short β-sheet β5a within the Ω-loop, near the hinge region between β8 and β9 just above the active site. The only known variant of G204 is PDC-466, which was derived from PDC-462 (A89V, Q120K, V211A, N320S), with an addition of G204D (Colque et al., 2021). Coupling of G204 to several catalytically important residues, including K67, K315, and T316 can suggest that mutations at this site can negatively impact catalytic power. This offers a plausible explanation of seeing fewer variants at this site, and mutations at this site could have an impact on the hydrolysing capabilities of PDC variants. This should be confirmed by further experimental studies of variants of G204. Unlike G214, E219, and Y221 mutations which do not influence the dynamics of the R2 loop, substitutions on V211, a member of Ω-loop, have an impact on the dynamics of the R2 loop because of its indirect couplings, through G204 to R2-loop residues (Chen et al., 2024). Two less critical substitution sites, H188 and V329, were also observed in community 2.
Conclusions
DyNoPy offers two distinct advantages over existing computational tools (Yehorova et al., 2024; Osuna, 2021): (a) information on residue–residue coevolution can be directly used to detect the components of protein dynamics that have been preserved during evolution and (b) dynamic descriptors extracted from the MD ensembles can be used to identify the function-specific conserved dynamic couplings. These couplings are then easily modelled as a graph, and network analysis is used to extract epistatic communities and assign roles to residues based on their importance in the graph model. The choice of a relevant descriptor of functional dynamics has an impact on the ability to detect couplings that are involved in functional dynamics.
Here we demonstrated how the choice of relevant global and local descriptors returns a higher number of effective couplings (greater than 0), and in turn leads to interpretable graph models and communities. In other systems, when multiple descriptors can be used to quantify functional conformational change, it is expected that they will differently modulate the effect of coevolution coupling, which will be reflected in a different structure of the associated graph models. This suggests the use of DyNoPy to generate comparative models in proteins with multiple functions associated to distinct dynamical changes.
Mutations of L162 and N136 have not yet emerged in SHV-1, but they are detected by DyNoPy as core residues for communities. These residues are strongly coupled with other functional important residues, which play critical roles in protein stability and catalytic activity. The identification of these couplings shows high consistency with previous studies and highlights the importance of L162 and N136 in SHV-1 functional dynamics. Given their central role in these communities, mutations in L162 and N136 can significantly alter protein function, suggesting their potential for future evolutionary changes. However, their strong relationships with these critical functional residues also suggest that mutation at these sites would need to be balanced to maintain protein function, providing an explanation for why such mutations have not yet emerged in SHV-1 (Soskine and Tawfik, 2010). The ability of DyNoPy in detecting functionally important mutation sites was demonstrated via well-characterized mutation sites including R205 and G238 from SHV-1. Moreover, DyNoPy shows predictive ability on less-studied mutation sites such as G156 and A146, by detecting critical residue couplings that coevolved with functional motions.
Based on the knowledge we have gained from the analysis of SHV-1 functional protein dynamics, we suggest that in PDC-3, mutations at G204 because of its significantly conserved dynamic couplings can lead to new ESBL/IRBL clinical variants. We suggest that DyNoPy can be used as a predictive tool to identify potential functional residues within this enzyme and guide future mutagenesis studies.
In summary, by integrating hidden evolutionary information with direct dynamic interactions, DyNoPy provides a powerful framework for identifying and analysing functional sites in proteins. The tool not only identifies key residues involved in local and global interactions but also improves our ability to predict silent residues with previously unknown roles for future experimental testing. Our application of DyNoPy to broad-spectrum β-lactamases ESBLs and IRBLs demonstrates its potential to address key medical challenges such as antibiotic resistance by providing valid predictions on protein evolution.
Methods
DyNoPy generates a graph representation of the protein structure that captures the couplings between amino acid residues contributing to the functional dynamics of the protein. Residues are represented as graph nodes, and conserved dynamic couplings are recorded as edges. Edge weights quantify the strength of these couplings. The model is built on two assumptions: residue pairs should have (a) coevolved and their (b) time-dependent interactions correlate with a functional conformational change.
Therefore, edge weights () for residue and are calculated as
(1)
where is the scaled coevolution score and is the degree of correlation with the selected functional conformational change. α and β are weights assigned to and that have a sum of 1. The relative weight of the scaled coevolution score (α) is set to 0.5 in this study. When either of the assumptions listed above is not met, is set to zero.
Scaled coevolution scores
The occurrence of residue–residue coevolution can be estimated and quantified using probabilistic models of correlated mutations from deep MSA. DyNoPy supports generation of the MSA using the HH-Suite package (Remmert et al., 2011) and calculation of scaled coevolution score () using CCMpred (Seemayer et al., 2014) as per the protocol described in Bibik et al., 2024. For SHV-1 and PDC-3, hhblits returned 18,174 sequences (Neff: 11.082) and 27,892 sequences (Neff: 9.951). Sequences were detected from the UniRef30 (v2022_02) database (Mirdita et al., 2017). First, a pairwise residue coevolution matrix (C) is calculated, then these raw scores () are divided by the matrix mean (Equation 2). All scores () smaller than 1 are set to zero, and the remaining values are normalized by the maximum value (Equation 3):
(2)
(3)
Correlation with functional motions
The contribution of a residue pair to a selected functional motion is estimated by how much the change in interaction energy between the two residues over time is correlated with a CV describing the functional motion:
(4)
(5)
where is the pairwise non-bonded interaction energy (see details in Appendix 1) and is the time-dependent value of the CV. Examples of CV and a discussion on the choice of the most relevant CV are presented in the ‘Results’ section. Correlation values smaller than 0.5 are set to 0. In the absence of detectable contributions to the functional dynamics of the system, the couplings extracted by DyNoPy will describe a pure evolutionary model, and the community detection method presented below will be equivalent to a direct decomposition of the residue coevolution network into units.
Graph representation and analysis of conserved dynamic couplings
All pairwise conserved dynamic couplings (Equation 1) are collected into a square matrix J. A graph is built from J using python-igraph v0.11 library (Csárdi and Nepusz, 2006). Nodes represent residues, and edges are drawn between nodes with positive . Edge weights are set to . The relative importance of the residues in this model of protein dynamics is calculated as EVC of the nodes (Newman, 2004). The residues involved in extensive correlated dynamics with other highly connected residues have higher EVC scores. Groups of residues contributing to important collective motions are detected by community analysis of the graph structure. The Girvan–Newman algorithm is used to extract the community structure (Newman, 2006). A meaningful community should contain at least three residues. Applying network analysis on the combined dynamics-coevolution matrix helps us extract higher-order interactions beyond pairwise coupling and detecting critical residues, which show multiple interactions with each other. Moreover, indirect long-range relationships, which would be hard to identify from numerical data, could be detected through community clustering. Community-based analysis offers a more comprehensive understanding of residue relationships and enables the visualization of residue couplings on the protein structure.
Adaptive sampling molecular dynamics simulations
MD simulation data was sourced from our previous studies (Olehnovics et al., 2021; Chen et al., 2024). To summarize, SHV-1 structural coordinates (PDB ID: 3N4I) were obtained from the Protein Data Bank and modified to the wild type by introducing the E104D mutation. Similarly, the PDC-3 structure was derived from PDC-1 (PDB ID: 4HEF) by a T105A substitution. Both enzymes were protonated at pH 7.0 using PropKa from the PlayMolecule platform (Martínez-Rosell et al., 2017). One disulfide bond between C77 and C123 was specified in SHV-1. Both structures were solvated with TIP3P water molecules in a periodic box with a box size of 10 Å. Ions were added to neutralize the overall charge of each system at 150 mM KCl. Amber force field ff14SB was used for all MD simulations (Maier et al., 2015). After an initial minimization of 1000 steps, both the enzymes were equilibrated for 5 ns in the NPT ensemble at 1 atmospheric pressure using the Berendsen barostat (Berendsen et al., 1984). The initial velocities for each simulation were sampled from the Boltzmann distribution at 300 K. Multiple Markov state model (MSM)-based adaptively sampled simulations were performed for both proteins based on the ACEMD engine (Doerr et al., 2016; Harvey et al., 2009). A canonical (NVT) ensemble with a Langevin thermostat (Davidchack et al., 2009) (damping coefficient of 0.1 ps−1) and a hydrogen mass repartitioning scheme were employed to achieve time steps of 4 fs. For SHV-1, each trajectory spanned 60 ns with a time step of 0.1 ns, with a total of 593 trajectories. In the case of PDC-3, 100 trajectories were collected, each containing 3000 frames, lasting 300 ns. To manage the extensive datasets efficiently, trajectories were strategically stridden to ensure that a minimum of 30,000 frames were preserved for each system. The resulting trajectories are summarized in Supplementary file 1d.
Calculation and selection of collective variables
DyNoPy works on the assumption that time-dependent interactions between critical residues, either having significant structural change or not, will correlate with functional conformational motions. Since MD simulation data is high-dimensional, a time-dependent CV is required to extract the most relevant information for the process under study. The usefulness of DyNoPy is dependent on the choice of the CVs. To guide the selection of CVs, we selected 12 distinct features: radius of gyration (Rg), the first principal component (PC1), partial PC1 (PC1_partial), the first time-lagged independent component (TC1), partial TC1 (TC1_partial), global root mean square deviation (gRMSD), partial RMSD (pRMSD), dynamical RMSD (dRMSD), global solvent-accessible surface area (gSASA), partial SASA (pSASA), active site pocket volume, and the number of hydrogen bonds (hbond). A description of the CVs, including the calculation methods and the residues used to calculate the partial variables, is detailed in Appendix 1. CVs were subsequently used as input features for DyNoPy. A good CV should appropriately describe protein functional motions. Thus, a CV that detects the highest number of residue couplings is expected to be the most suitable descriptor. The length of the MD simulations should be appropriate to effectively sample the desired functional process as described by the selected CV.
Appendix 1
Interaction energies (E)
The pairwise non-bonded interaction energies for the upper triangle of the N×N residue matrix are calculated using dyno_pwie.py, which is a wrapper to ccptraj from ambertools. The list of pairs is generated and depending on the number of available threads (nt), ‘n’ sets of pairs are created, and each pair set is assigned to an instance of ccptraj for calculation of pairwise interaction energies. As this is a RAM-dependent operation, based on the size of the MD trajectory, care must be taken to not spawn too many instances of ccptraj as it will slow down each instance of the ccptraj calculation. For each residue pair (), a separate file with coulomb () and van der Waals () interaction energies, as two separate columns, are saved to a h5py compressed file and the total interaction energy ( for each time step can be calculated from this data using Equation A1.
(A1)
Collective variables description
Radius of gyration (Rg)
The radius of gyration (Rg) measures the compactness of the protein structure through the calculation of the root mean square distance of all atoms from the centre of mass. A lower Rg indicates a more compact structure, while a higher Rg suggests a more expanded or unfolded state. This CV was calculated using the MDAnalysis v2.7.0 (Gowers et al., 2016; Michaud-Agrawal et al., 2011), focusing on the whole protein for both enzymes.
The first principal component (PC1)
Principal component analysis identifies the major dynamical feature of a protein by reducing the dimensionality directly from molecular dynamics simulation data. PC1 represents the largest variance on the selected features across the simulation. Global PC1 was computed using PyEMMA v2.5.12 (Scherer et al., 2015), focusing on backbone torsion angles and χ1 angles for all residues, capturing the global dominant structural changes.
The partial PC1 (PC1_partial)
PC1_partial was calculated using the same methodology as PC1 but only focusing on functionally significant residues and essential secondary structures. This approach prioritizes the dynamics within regions critical to the protein’s function, thereby providing a more targeted analysis of pertinent conformational changes. It is particularly well-suited for studying proteins that are predominantly rigid with localized flexible regions.
Time-lagged independent component 1 (TC1)
Time-lagged independent component analysisidentifies slow, independent processes within the protein dynamics. TC1 captures the slowest conformational changes in the protein over time. It was computed globally using backbone torsion angles and χ1 angles for all residues, with PyEMMA v2.5.12 (Scherer et al., 2015).
Partial time-lagged independent component 1 (TC1_partial)
Similarly, TC1_partial was calculated, but focused on specific, functionally important regions. By analysing these key residues and structures, TC1_partial represents the slow conformational changes crucial within essential regions, which are highly corresponding with protein functions.
Global root mean square deviation (gRMSD)
The RMSD measures the average deviation of a protein’s atomic positions from a reference structure, typically the starting conformation. gRMSD provides an overview of the protein’s structural deviation over time. In our case, trajectories were first aligned, and Cα RMSD was calculated using MDAnalysis v2.7.0 (Gowers et al., 2016; Michaud-Agrawal et al., 2011), utilizing the starting conformation of each protein as a reference.
Partial root mean square deviation (pRMSD)
This CV is calculated similarly to gRMSD but focuses on specific residues or regions of interest. pRMSD provides insight into the structural stability of functionally important areas of the protein by focusing on catalytical residues and essential loops.
Dynamical root mean square deviation (dRMSD)
dRMSD is an extension of RMSD that considers the dynamic nature of protein motions by analysing fluctuations over time rather than static deviations. More specifically, instead of calculating RMSD to a fixed reference structure, it focuses on the difference between adjacent frames. This approach allows the analysis of short-timescale fluctuations and provides insight into the dynamic nature of protein motions over time. It was also calculated using MDAnalysis v2.7.0 (Gowers et al., 2016; Michaud-Agrawal et al., 2011) focusing on the Cα and offers a more nuanced understanding of the protein’s conformational landscape.
Global solvent-accessible surface area (gSASA)
SASA quantifies the surface area of the protein accessible to the solvent. gSASA was calculated using GROMACS v2020.1 (Abraham et al., 2015) and provides information about the protein’s overall exposure to the solvent, which is relevant for understanding folding and binding interactions.
Partial solvent-accessible surface area (pSASA)
pSASA focuses on the solvent exposure of specific active site residues, offering insights into how binding sites or functional regions of the protein interact with the solvent.
Active site pocket volume
The volume of the active site pocket was calculated using Mdpocket (Schmidtke et al., 2011). This CV is important for understanding the size and shape changes in the binding site, which can influence ligand binding and protein function. Upon importing the topology file and trajectories, Mdpocket autonomously identifies potential binding pockets within the protein structure. Subsequently, the relevant pocket was manually selected (Figure 2—figure supplement 3)
Number of hydrogen bonds (hbond)
The number of hydrogen bonds was calculated per frame using VMD v1.9.3 (Humphrey et al., 1996), with a distance threshold of 3.5 Å and an angle cut-off of 40°. This CV provides insight into the stability of the protein structure and interactions that are critical for maintaining its conformation and function.
Residues and regions used in partial CVs
For SHV-1, the backbone torsion angles and χ1 angles for catalytic important residues—S70, T71, K73, S130, N132, K234, and T235—and Ω-loop residues (R164-D179) were used as the input feature to calculate the partial CVs. Similarly, for PDC-3, the backbone torsion angles and χ1 angles for catalytic important residues—K67, Y150, N152, K315, T316, and G317—Ω-loop residues (G183-S226), and R2-loop residues (L280-Q310) were utilized to calculate all the partial variables.
Appendix 2
Other SHV-1 communities
Core residues for some of the communities identified by DyNoPy have never been studied in class A β-lactamases before. DyNoPy predicts these residues as essential due to their coevolution trends with many other residues and their critical role in class A β-lactamase dynamics (Appendix 2—figure 1).
D267 is the core residue in community 3 (Appendix 2—figure 1A). This residue is located on the loop connecting β-sheet β9 and α-helix α12. It is outside the catalytic site and has not undergone essential substitutions, thus remaining unexplored in class A β-lactamases. However, DyNoPy indicates its potential importance due to its relationships with three known essential mutation sites: L35, E240, and A187. L35 and E240 are predominant ESBL mutation sites, while A187 substitution confers an inhibitor-resistant phenotype (Chang et al., 2001). The relationship between D267 and these mutation sites suggests a trend towards coevolution, and interactions between D267 and these mutation sites are important for protein dynamics.
Similarly, I279, located also on α12, is the core residue of community 8 (Appendix 2—figure 1B). This residue forms relationships with R244, a critical inhibitor resistant mutation site (Giakkoupi et al., 1998). Other residues within this community are predominantly positioned on the α1 and α12 helices, near the protein’s terminus, highlighting the significant contribution of I279 to protein integrity and stability.
For community 9, I155 serves as the core residue (Appendix 2—figure 1C). This community is relatively localized and does not encompass any known essential residues. Most of the residues involved in this community are spatially close to each other, suggesting that I155 plays a vital role in the local dynamics, especially surrounding α-helices α7 and α9.
Appendix 2—figure 1
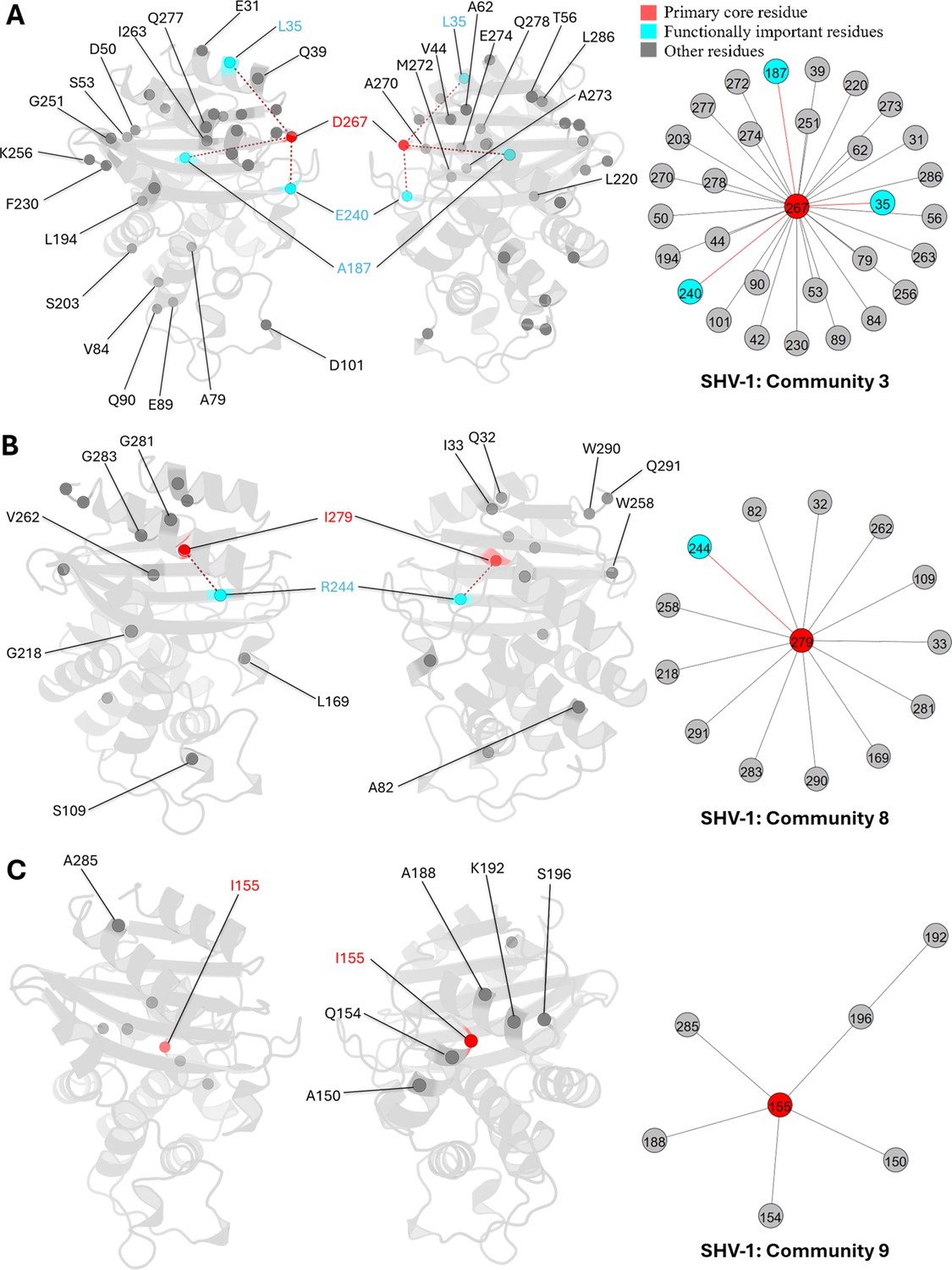
Communities 3, 8, and 9 of SHV-1 β-lactamase.
All the residues are depicted as spheres on the protein structure. The core residue for each community is D267, I279, and I155, respectively. They are highlighted in red. Functional important residues are marked in cyan. (A) Community 3 of SHV-1, comprising 31 residues. (B) Community 8 of SHV-1, containing 14 residues. (C) Community 9 of SHV-1, with 7 residues.
Other PDC-3 communities
The core residues in the other PDC-3 communities have not been extensively studied. However, their detection by DyNoPy suggests a significant trend in co-evolution and highlights their crucial role in protein dynamics (Appendix 2—figure 2). This emphasizes the capability of DyNoPy to predict essential residues in previously unexplored proteins, potentially offering valuable insights for future experimental research.
E49, D206, and R210 are core residues for community 3, a small community containing only 14 residues (Appendix 2—figure 2A). R210 is the primary core residue in this community, linking to six residues, while both E49 and R210 are secondary core residues that show a relationship with four residues. Unlike other communities with widespread interactions, community 3 illustrates a localized relationship among Ω-loop residues, primarily on the short β5a and β5b β-sheets and the short helix α7a. Also, 8 out of 14 residues are Ω-loop residues, with the remainder located on adjacent loops or near the terminals of adjoining secondary structures, all of which are flexible regions. This community indicates that R210 is crucial for maintaining local structural integrity and the stability of the Ω-loop.
G202, the core residue of community 4, appears essential for active site stability by interacting with residues whose side chains point into the active site (Appendix 2—figure 2B). It also stabilizes two α-helices, α2 and α5, located adjacent to the active site. P154 is a special mutation site in the PDC family. P154L occur in PDC-73 and PDC-81, giving the protein a mild increase in resistance to ceftazidime (Berrazeg et al., 2015).
K204a and R207 are the central residues in community 5, each establishing six interactions and thus sharing an equal position of importance within this community. K281 and K351, which interact with both core residues, are highlighted in light yellow (Appendix 2—figure 2C). K204a forms direct interactions with six residues, primarily located on the opposite side of the active site, either on the R2 loop or at the beginning of helix α11. This suggests that K204a plays a crucial role in maintaining the conformation of the R2 site. Additionally, K204a shows direct correlations with the catalytically significant residue K315. Although R207 is spatially close to K204a, it interacts with residues on loops that are distant from the catalytic site.
Appendix 2—figure 2
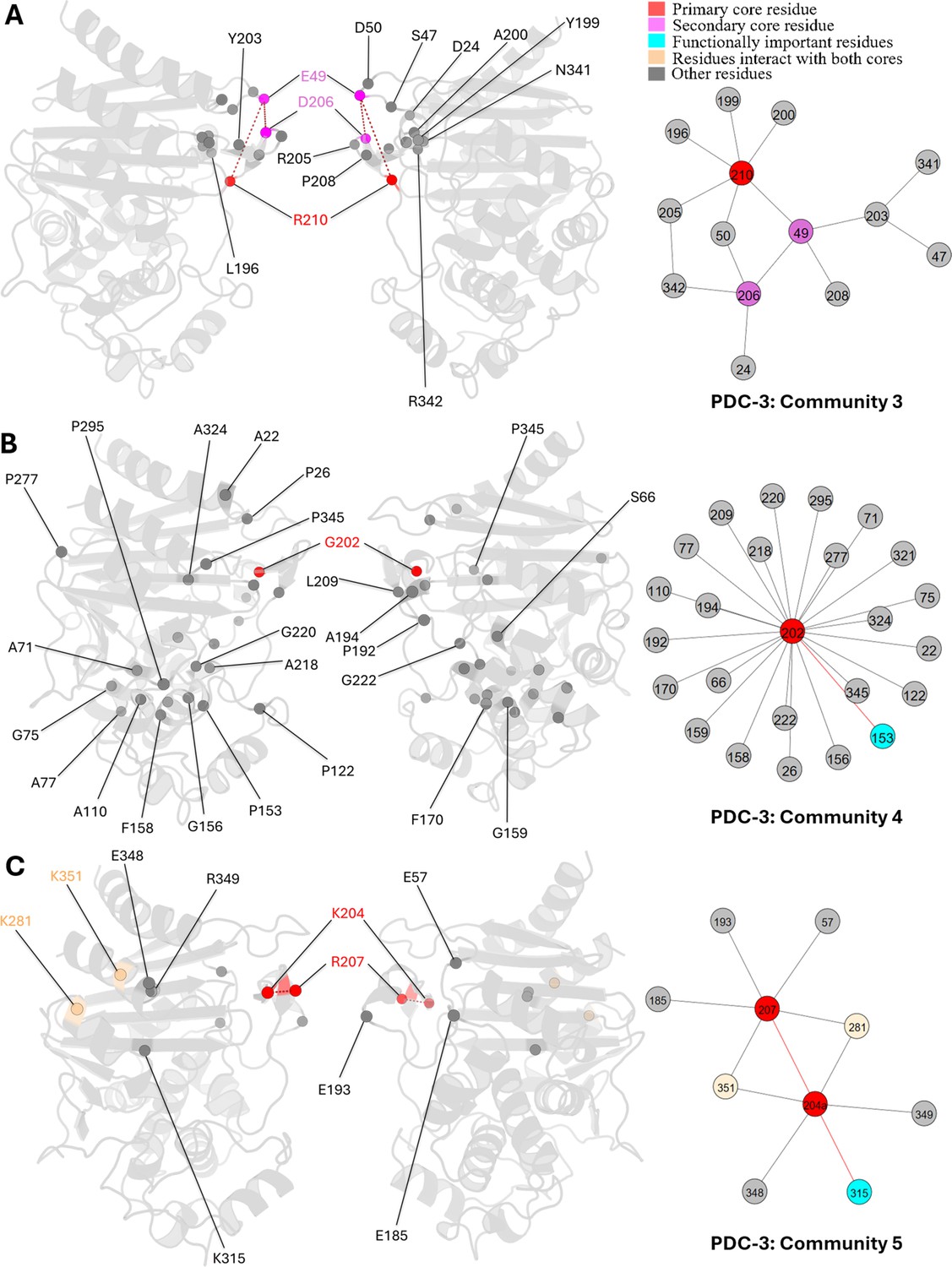
Communities 3, 4, and 5 of PDC-3 β-lactamase.
All the residues are depicted as spheres on the protein structure. The core residue for each community is highlighted in red, while purple is used to emphasize the secondary core residue. Residues that interact with both cores are coloured in light yellow. Functional important residues are marked in cyan. (A) Community 3 of PDC-3, comprising 14 residues with R210 being the primary core residue. (B) Community 4 of PDC-3, containing 25 residues and is centred by G202. (C) Community 5 of PDC-3, embracing 10 residues and having two core residues K204 and R207.
Data availability
All files required to run the simulations (topology, coordinates, input), processed trajectories (xtc), corresponding coordinates (pdb), can be downloaded from https://doi.org/10.57760/sciencedb.15876 (PDC-3) and https://doi.org/10.5281/zenodo.13693144 (SHV-1). DyNoPy is available at https://github.com/alepandini/DyNoPy, (copy archived at Pandini, 2024).
-
Science Data BankΩ-Loop mutations control the dynamics of the active site by modulating a network of hydrogen bonds in PDC-3 β-lactamase.https://doi.org/10.57760/sciencedb.15876
-
ZenodoFunctionally Important Residues from Graph Analysis of Co-evolved Dynamical Couplings.https://doi.org/10.5281/zenodo.13693144
References
-
Detailed investigation of catalytically important residues of class A β-lactamaseJournal of Biomolecular Structure & Dynamics 41:2046–2073.https://doi.org/10.1080/07391102.2021.2023645
-
A standard numbering scheme for the class A beta-lactamasesThe Biochemical Journal 276 (Pt 1):269–270.https://doi.org/10.1042/bj2760269
-
Molecular dynamics with coupling to an external bathThe Journal of Chemical Physics 81:3684–3690.https://doi.org/10.1063/1.448118
-
Mutations in β-Lactamase AmpC Increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporinsAntimicrobial Agents and Chemotherapy 59:6248–6255.https://doi.org/10.1128/AAC.00825-15
-
Role of Asp104 in the SHV β-LactamaseAntimicrobial Agents and Chemotherapy 50:4124–4131.https://doi.org/10.1128/AAC.00848-06
-
Emergence and dissemination of BEL-1-producing Pseudomonas aeruginosa isolates in BelgiumAntimicrobial Agents and Chemotherapy 51:1584–1585.https://doi.org/10.1128/AAC.01603-06
-
Conserved water molecules stabilize the Omega-loop in class A beta-lactamasesAntimicrobial Agents and Chemotherapy 52:1072–1079.https://doi.org/10.1128/AAC.01035-07
-
Proliferation and significance of clinically relevant β-lactamasesAnnals of the New York Academy of Sciences 1277:84–90.https://doi.org/10.1111/nyas.12023
-
Past and present perspectives on β-lactamasesAntimicrobial Agents and Chemotherapy 62:e01076-18.https://doi.org/10.1128/AAC.01076-18
-
Discovering functionally important sites in proteinsNature Communications 14:4175.https://doi.org/10.1038/s41467-023-39909-0
-
The role of conformational dynamics and allostery in modulating protein evolutionAnnual Review of Biophysics 49:267–288.https://doi.org/10.1146/annurev-biophys-052118-115517
-
Non-catalytic-region mutations conferring transition of class a β-lactamases into ESBLsFrontiers in Molecular Biosciences 7:598998.https://doi.org/10.3389/fmolb.2020.598998
-
Diversity of SHV and TEM beta-lactamases in Klebsiella pneumoniae: gene evolution in Northern Taiwan and two novel beta-lactamases, SHV-25 and SHV-26Antimicrobial Agents and Chemotherapy 45:2407–2413.https://doi.org/10.1128/AAC.45.9.2407-2413.2001
-
SHV-27, a novel cefotaxime-hydrolysing beta-lactamase, identified in Klebsiella pneumoniae isolates from a Brazilian hospitalThe Journal of Antimicrobial Chemotherapy 47:463–465.https://doi.org/10.1093/jac/47.4.463
-
Langevin thermostat for rigid body dynamicsThe Journal of Chemical Physics 130:234101.https://doi.org/10.1063/1.3149788
-
Protein design using structure-based residue preferencesNature Communications 15:1639.https://doi.org/10.1038/s41467-024-45621-4
-
HTMD: high-throughput molecular dynamics for molecular discoveryJournal of Chemical Theory and Computation 12:1845–1852.https://doi.org/10.1021/acs.jctc.6b00049
-
Exploring amino acid functions in a deep mutational landscapeMolecular Systems Biology 17:e10305.https://doi.org/10.15252/msb.202110305
-
ConferenceMDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics SimulationsPython in Science Conference.https://doi.org/10.25080/Majora-629e541a-00e
-
ACEMD: accelerating biomolecular dynamics in the microsecond time scaleJournal of Chemical Theory and Computation 5:1632–1639.https://doi.org/10.1021/ct9000685
-
Mutation effects predicted from sequence co-variationNature Biotechnology 35:128–135.https://doi.org/10.1038/nbt.3769
-
VMD: visual molecular dynamicsJournal of Molecular Graphics 14:33–38.https://doi.org/10.1016/0263-7855(96)00018-5
-
Conformational diversity and protein evolution--a 60-year-old hypothesis revisitedTrends in Biochemical Sciences 28:361–368.https://doi.org/10.1016/S0968-0004(03)00135-X
-
Generalized correlation for biomolecular dynamicsProteins 62:1053–1061.https://doi.org/10.1002/prot.20784
-
A review of SHV extended-spectrum β-Lactamases: neglected yet ubiquitousFrontiers in Microbiology 7:1374.https://doi.org/10.3389/fmicb.2016.01374
-
Sequence evolution correlates with structural dynamicsMolecular Biology and Evolution 29:2253–2263.https://doi.org/10.1093/molbev/mss097
-
ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SBJournal of Chemical Theory and Computation 11:3696–3713.https://doi.org/10.1021/acs.jctc.5b00255
-
Protein structure prediction from sequence variationNature Biotechnology 30:1072–1080.https://doi.org/10.1038/nbt.2419
-
PlayMolecule proteinprepare: a web application for protein preparation for molecular dynamics simulationsJournal of Chemical Information and Modeling 57:1511–1516.https://doi.org/10.1021/acs.jcim.7b00190
-
Catalytic properties of class A beta-lactamases: efficiency and diversityThe Biochemical Journal 330 (Pt 2):581–598.https://doi.org/10.1042/bj3300581
-
β-Lactamases: quality and resistanceClinical Microbiology and Infection 3:4S2–4S9.https://doi.org/10.1016/S1198-743X(14)65030-8
-
MDAnalysis: a toolkit for the analysis of molecular dynamics simulationsJournal of Computational Chemistry 32:2319–2327.https://doi.org/10.1002/jcc.21787
-
Uniclust databases of clustered and deeply annotated protein sequences and alignmentsNucleic Acids Research 45:D170–D176.https://doi.org/10.1093/nar/gkw1081
-
A genotype-phenotype correlation study of shv β-lactamases offers new insight into SHV resistance profilesAntimicrobial Agents and Chemotherapy 64:e02293-19.https://doi.org/10.1128/AAC.02293-19
-
Structural dynamics flexibility informs function and evolution at a proteome scaleEvolutionary Applications 6:423–433.https://doi.org/10.1111/eva.12052
-
Detecting community structure in networksThe European Physical Journal B - Condensed Matter 38:321–330.https://doi.org/10.1140/epjb/e2004-00124-y
-
The role of hydrophobic nodes in the dynamics of class a β-lactamasesFrontiers in Microbiology 12:720991.https://doi.org/10.3389/fmicb.2021.720991
-
The challenge of predicting distal active site mutations in computational enzyme designWIREs Computational Molecular Science 11:1502.https://doi.org/10.1002/wcms.1502
-
Tazobactam inactivation of SHV-1 and the inhibitor-resistant Ser130 -->Gly SHV-1 beta-lactamase: insights into the mechanism of inhibitionThe Journal of Biological Chemistry 279:19494–19501.https://doi.org/10.1074/jbc.M311669200
-
The structural role of N170 in substrate-assisted deacylation in KPC-2 β-LactamaseAngewandte Chemie 63:e202317315.https://doi.org/10.1002/anie.202317315
-
Class c β-lactamases: molecular characteristicsClinical Microbiology Reviews 35:e0015021.https://doi.org/10.1128/cmr.00150-21
-
The context-dependence of mutations: a linkage of formalismsPLOS Computational Biology 12:e1004771.https://doi.org/10.1371/journal.pcbi.1004771
-
Emergence in Klebsiella pneumoniae of a chromosome-encoded shv β-lactamase that compromises the efficacy of imipenemAntimicrobial Agents and Chemotherapy 47:755–758.https://doi.org/10.1128/AAC.47.2.755-758.2003
-
BEL-2, an extended-spectrum β-lactamase with increased activity toward expanded-spectrum cephalosporins in Pseudomonas aeruginosaAntimicrobial Agents and Chemotherapy 54:533–535.https://doi.org/10.1128/AAC.00859-09
-
Resistance to beta-lactam antibioticsCellular and Molecular Life Sciences 61:2200–2223.https://doi.org/10.1007/s00018-004-4060-9
-
Crystal structure of the Pseudomonas aeruginosa bel-1 extended-spectrum β-lactamase and its complexes with moxalactam and imipenemAntimicrobial Agents and Chemotherapy 60:7189–7199.https://doi.org/10.1128/AAC.00936-16
-
PyEMMA 2: a software package for estimation, validation, and analysis of markov modelsJournal of Chemical Theory and Computation 11:5525–5542.https://doi.org/10.1021/acs.jctc.5b00743
-
Mutational effects and the evolution of new protein functionsNature Reviews. Genetics 11:572–582.https://doi.org/10.1038/nrg2808
Article and author information
Author details
Sarath Chandra Dantu

Funding
Leverhulme Trust (RPG-2017-222)
- James A Garnett
- Alessandro Pandini
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
SCD was supported by Leverhulme Trust grant RPG-2017-222 awarded to AP and JAG. The authors would like to thank Arianna Fornili for insightful suggestions on the design of DyNoPy methodology.
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.105005. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2025, Xu, Dantu et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,728
- views
-
- 126
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 3
- citations for umbrella DOI https://doi.org/10.7554/eLife.105005
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Functionally important residues from graph analysis of coevolved dynamic couplings
eLife 14:RP105005.
https://doi.org/10.7554/eLife.105005.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}