Population genomics reveals the origin and asexual evolution of human infective trypanosomes
- University of Glasgow, United Kingdom
- Wellcome Trust Genome Campus, United Kingdom
- University of Glasgow, Scotland
- Université Jean Lorougnon GUEDE, Côte d'Ivoire
- Campus International de Baillarguet, France
- Centre International de Recherche-Développement de l’Elevage en zone Subhumide, Burkina Faso
- UFR Sciences et Techniques, Burkina Faso
- Programme National de Lutte contre la Trypanosomiase Humaine Africaine, Guinea
Figures
Figure 1 with 3 supplements
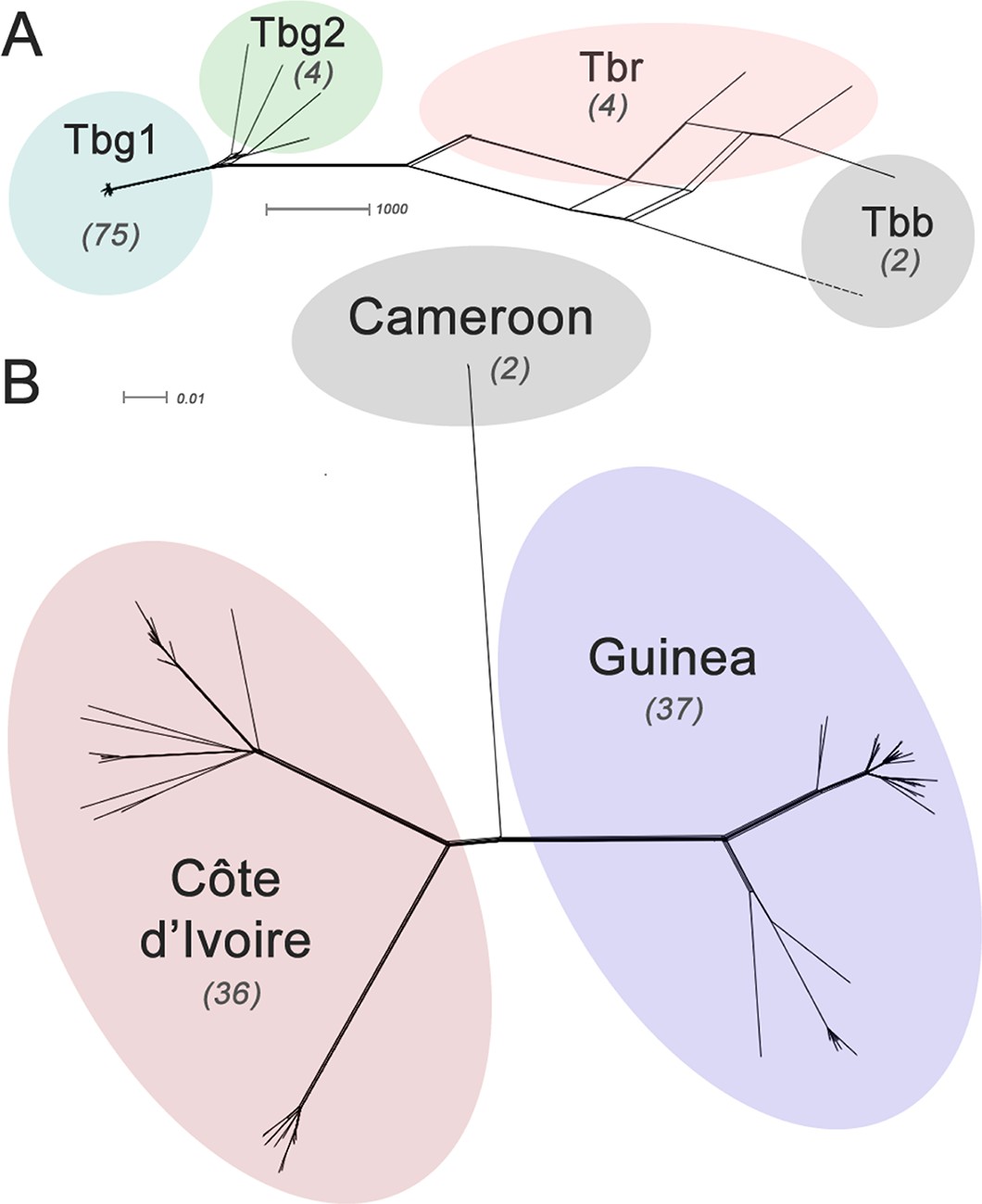
Phylogenetic network analysis.
SplitsTree phylogenetic networks were constructed using (A) each isolate for the collection of T.b. brucei (Tbb), T.b. rhodesiense (Tbr), T.b. gambiense Group 1 (Tbg1) and T.b. gambiense Group 2 (Tbg2) and (B) for just T.b. gambiense Group 1 isolates. The number of samples in each group is indicated in parenthesis.
-
Figure 1—source data 1
Number of SNP loci with respect to different sub-species.
- https://doi.org/10.7554/eLife.11473.004
-
Figure 1—source data 2
Testing Hardy-Weinberg Equilibrium (HWE) across the T.b. gambiense Group 1 genome.
- https://doi.org/10.7554/eLife.11473.005
-
Figure 1—source data 3
FIS by sub-population.
- https://doi.org/10.7554/eLife.11473.006
-
Figure 1—source data 4
Number and type of T.b. gambiense Group 1 SNPs.
- https://doi.org/10.7554/eLife.11473.007
Figure 1—figure supplement 1
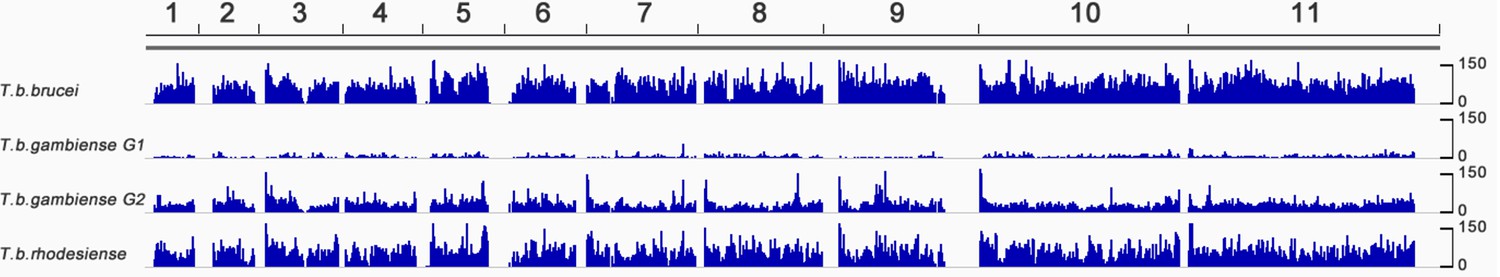
Genome-wide SNP density map for each sub-species.
The densities of SNP loci were assessed among isolates of (a) T.b. brucei (n=2), (b) T.b. gambiense Group 1 (n=75), (c) T.b. gambiense Group 2 (n=4) and T.b. rhodesiense (n=4) over each of the 11 chromosomes (horizontal scale). The number of SNPs per 10Kb window is plotted in blue, with the scale indicating SNP density between 0 and 150 loci per window (vertical scale). T.b. gambiense Group 1 isolates have a SNP density, which is approximately ten-fold lower than other sub-species, however they are evenly distributed throughout the genome.
Figure 1—figure supplement 2

Weir and Cockerham’s fis.
The number of SNP loci exhibiting different levels of fis (Weir and Cockerham’s fis (Goudet, 1995)) was plotted, showing a distribution around -1, indicating strict asexuality.
Figure 1—figure supplement 3
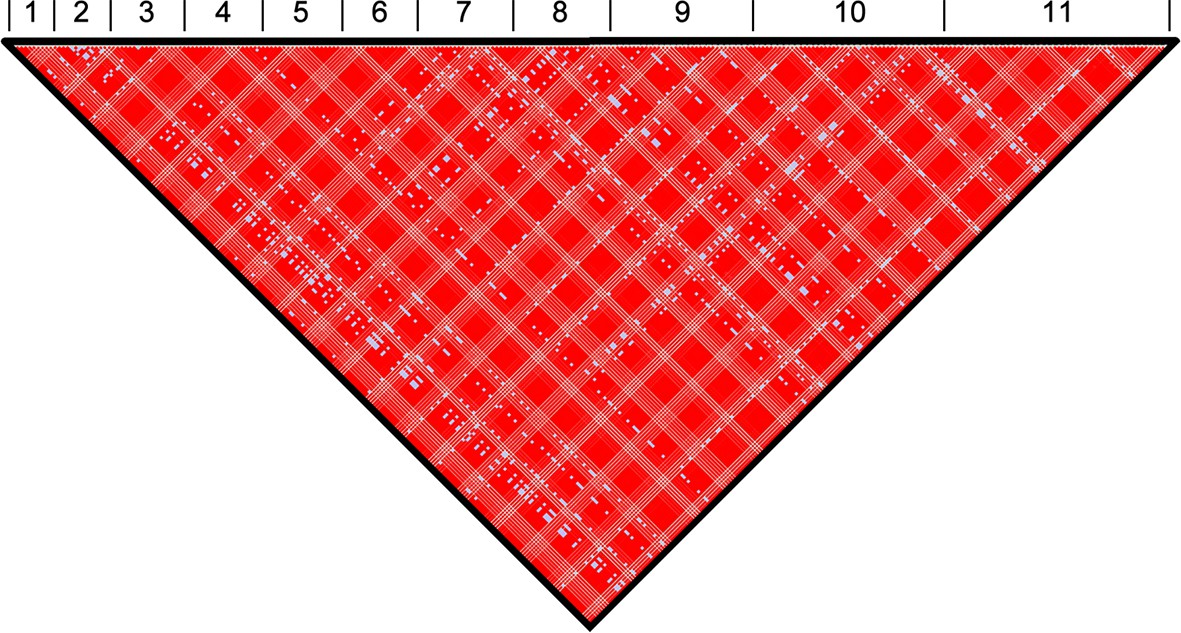
Genome-wide linkage disequilibrium (LD) among T.b. gambiense Group 1 parasites.
Normalised LD was assessed across all eleven chromosomes. The position of each SNP used in this analysis is illustrated. The black triangle illustrates the single linkage group identified in the analysis, which spans the entire genome. Numbers 1–11 represent the 11 chromosomes. Red (D'>1, LOD≥2), Pink (D'<1, LOD≥2), Blue (D'=1, LOD<2) and White (D'<1, LOD<2).
Figure 2 with 2 supplements
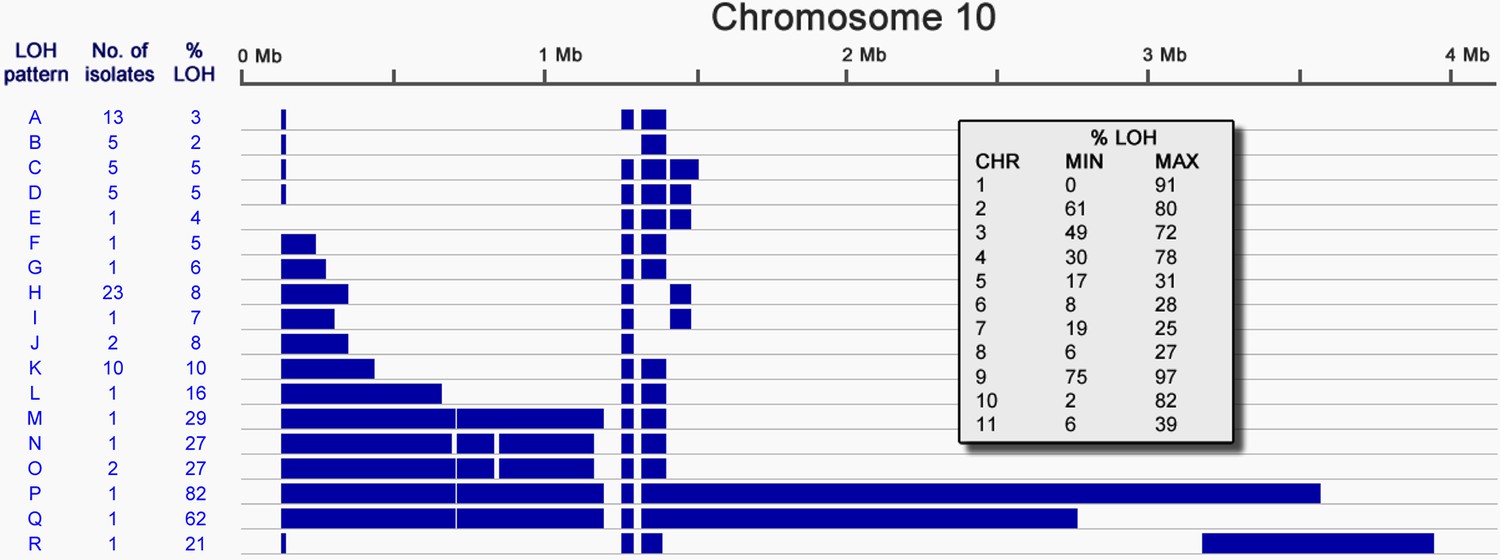
Loss of heterozygosity on chromosome 10.
Loss of heterozygosity regions (blue) spanning Chromosome 10 show 18 different patterns (A-R). The number of isolates possessing each pattern and the percentage of the chromosome affected are indicated. The table (inset) shows the extent of LOH (min and max) for each chromosome as a percentage of chromosome length.
Figure 2—figure supplement 1
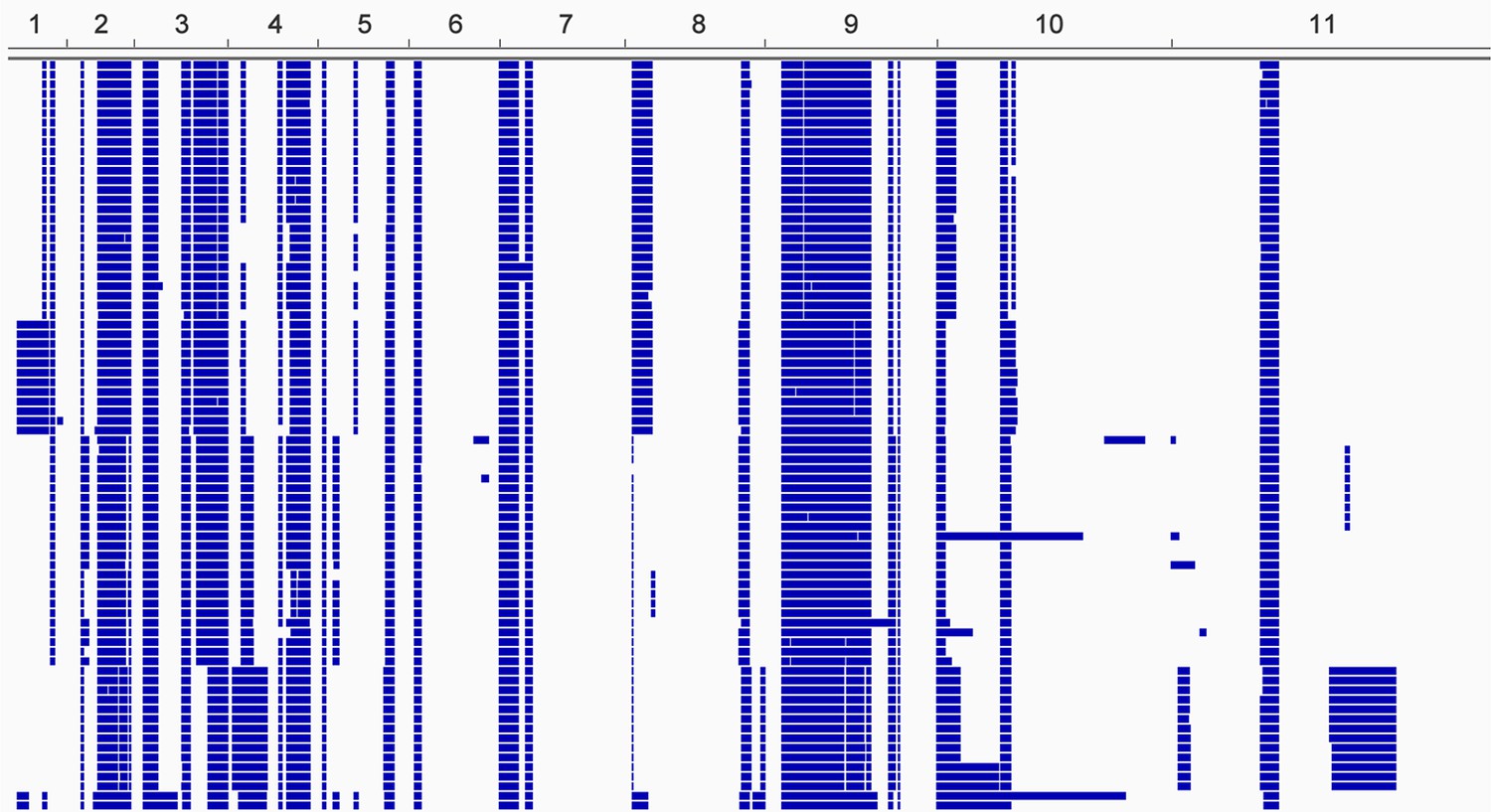
Loss of heterozygosity across the T.b. gambiense Group 1 genome.
Every T.b. gambiense Group 1 isolate (first column) was analysed for loss of heterozygosity (LOH-see Materials and methods). Blocks of LOH are shown in blue across each of the eleven chromosomes, as indicated.
Figure 2—figure supplement 2
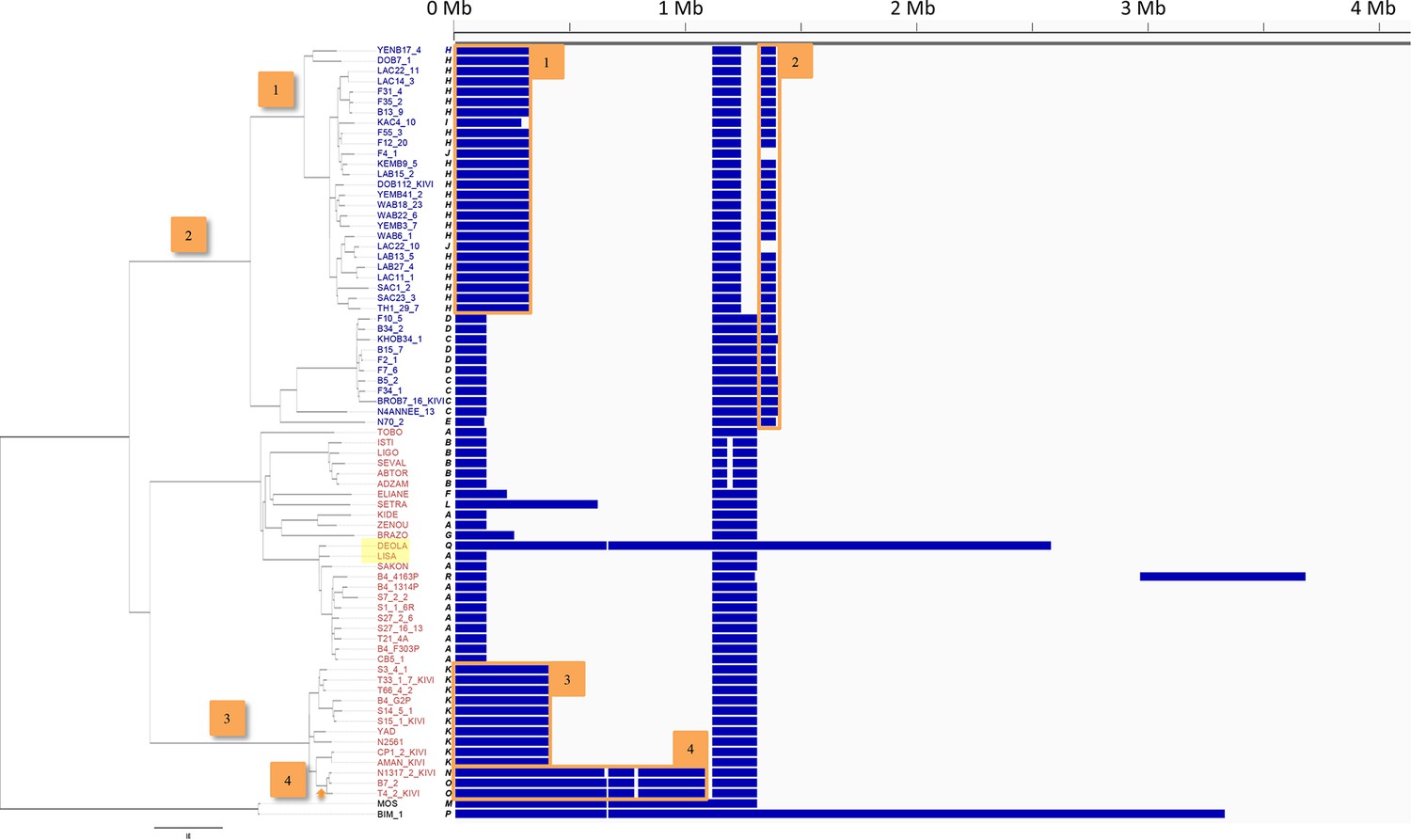
Loss of heterozygosity across chromosome 10.
Every T.b. gambiense Group 1 isolate was analysed for loss of heterozygosity (LOH). Blocks of LOH across chromosome 10 (horizontal scale) are shown in blue. Eighteen different patterns (A-R) are evident and the phylogenetic tree is shown on the left. Patterns of LOH can be seen to cluster in agreement with the tree, clearly showing that a stable LOH profile may be inherited. A selection of inherited blocks of LOH together with the branch on which they emerge are marked 1–4. In addition, very recent LOH events can be observed, for example the large LOH region in ‘DEOLA’, which is not observed in its close relative ‘LISA’, highlighted in yellow.
Figure 3
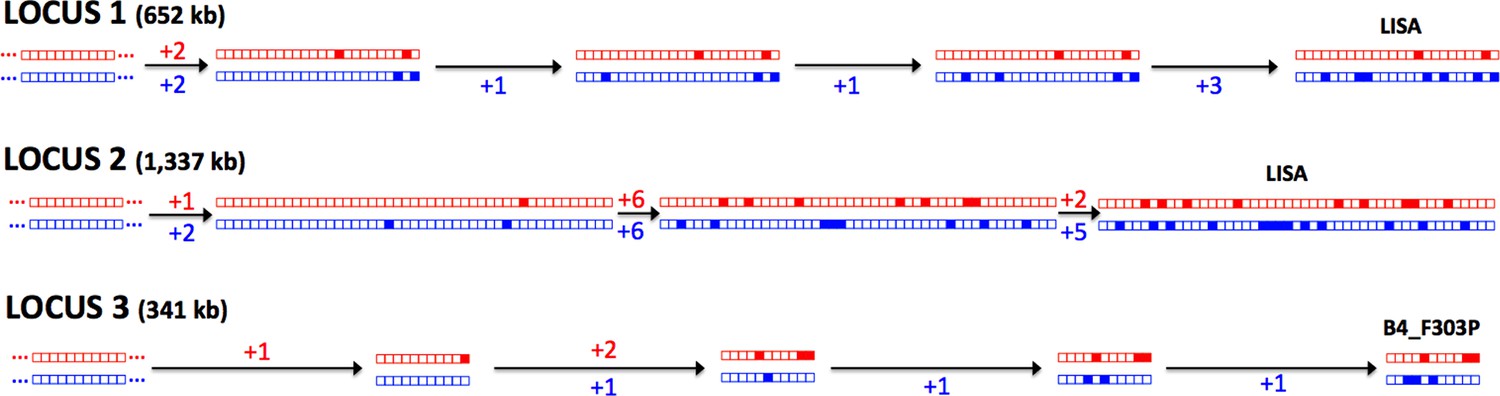
The Meselson effect.
An accumulation of mutations on separate haplotypes, the ‘Meselson Effect’, is illustrated using three regions on chromosome 10. For each region (1, 2 and 3) the two haplotypes are shown for a series of isolates with the accumulating mutations (filled boxes) indicated in red or blue for each haplotype. The sequences of accumulating mutations observed in the isolates ‘Lisa’ and ‘B4_F303P’ are shown. The number of mutations arising between each ancestral haplotype pair is indicated with two distinct lineages apparent at each locus, illustrating the independent evolution of each haplotype.
-
Figure 3—source data 1
Estimated time since the most recent common ancestor.
- https://doi.org/10.7554/eLife.11473.015
Figure 4 with 3 supplements

Haplotype co-evolution of chromosome 8.
(A) Phylogenetic tree of phased haplotype sequences of chromosome 8 shows a complete divergence between A (blue) and B (red) haplotypes of T.b. gambiense Group 1 (identical genotypes removed). The tree is rooted using a Group 2 isolate (green); (B) Co-phylogenetic analysis reveals 100% consensus between the A and B haplotype trees of a subset of T.b. gambiense Group 1 isolates.
-
Figure 4—source data 1
Co-phylogenetic analysis.
- https://doi.org/10.7554/eLife.11473.017
Figure 4—figure supplement 1

Phylogenetic trees of phased data showing ‘A’ and ‘B’ haplotypes.
Maximum likelihood phylogenetic trees of phased haplotype sequences demonstrate divergence between A (blue) and B (red) haplotypes of T.b. gambiense Group 1 over each of the 11 chromosomes. This represents all SNP loci identified in each sub-species (n = 230,891). The haplotypes of non-Group 1 isolates are shown in green. Divergence of A and B haplotypes of Group 1 isolates can be observed for every chromosome.
Figure 4—figure supplement 2
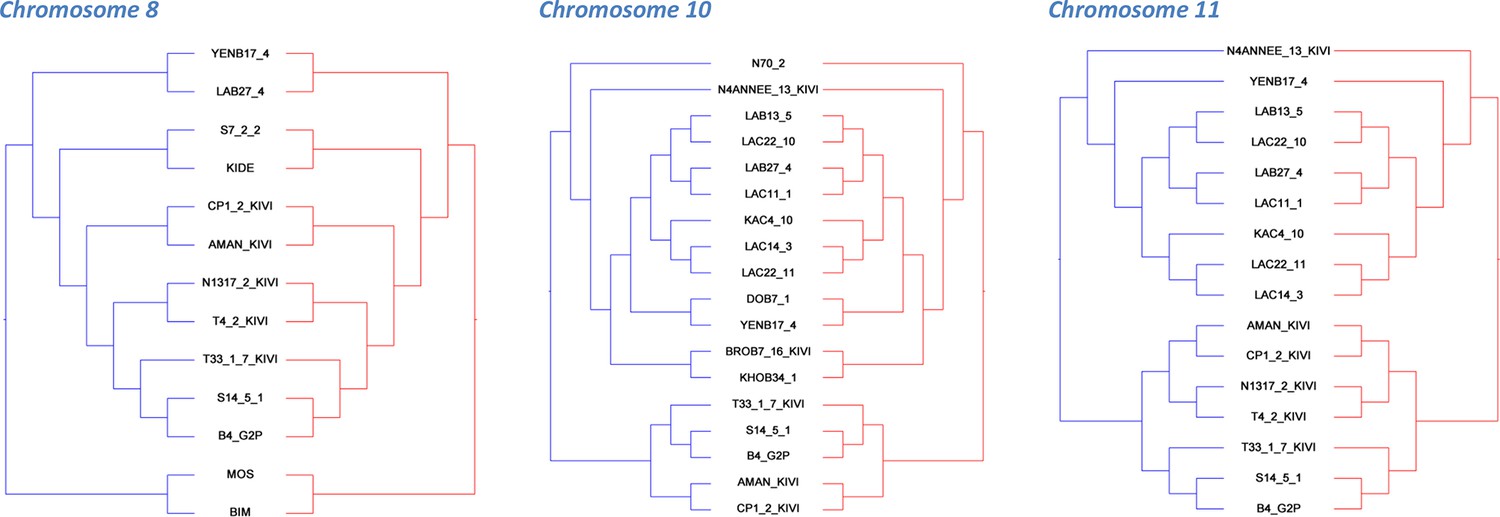
Co-phylogenetic analysis.
For the three chromosomes with more than 1,000 SNP loci (Figure 1—source data 4), subsets of isolates (discriminated by highly supported nodes, see Materials and methods) were used to construct haplotype trees. In each case, the A and B haplotype trees show identical topology, illustrating the co-evolution of partner haplotypes.
Figure 4—figure supplement 3
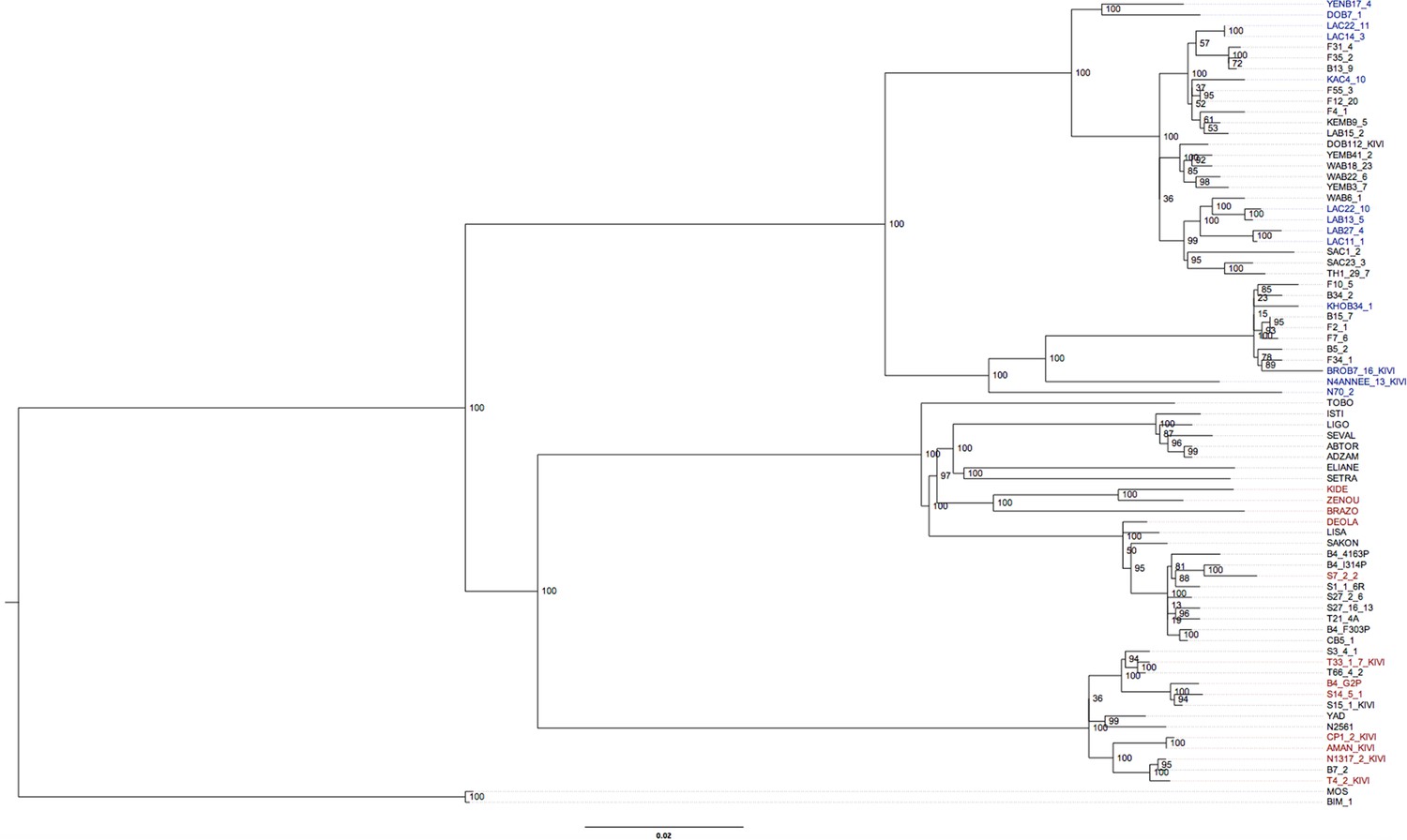
Phylogenetic tree of all T.b. gambiense Group 1 isolates.
A maximum likelihood phylogenetic tree was constructed with all T.b. gambiense Group 1 isolates using the panel of SNPs associated with derived alleles (Figure 1—source data 4). Bootstrap support is shown for each node. The 27 isolates chosen to illustrate co-evolution of partner haplotypes in Figure 4—figure supplement 2 are shown in blue (Guinea) and red (Côte d'Ivoire). The clade used for molecular clock calculations is marked with an asterisk.
Additional files
-
Supplementary file 1
Isolates used in this study.
For each isolate, the year of isolation, host, country and location are given along with the results of the BIIT test (Blood Incubation Infectivity Test), which determines human infectivity. The presence/absence of TgSGP, the T.b. gambiense Group 1 human serum resistance gene and SRA, the T.b. rhodesiense human serum resistance gene are indicated. The majority of samples in this study were T.b. gambiense Group 1, details of which have been previously published (Heitman, 2006; Thorvaldsdottir et al., 2013).
- https://doi.org/10.7554/eLife.11473.021
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Population genomics reveals the origin and asexual evolution of human infective trypanosomes
eLife 5:e11473.
https://doi.org/10.7554/eLife.11473




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}