Epigenomic landscapes of retinal rods and cones
- Johns Hopkins University School of Medicine, United States
- The Salk Institute for Biological Studies, United States
- Howard Hughes Medical Institute, The Salk Institute for Biological Studies, United States
- Janelia Research Campus, Howard Hughes Medical Institute, United States
- University of California San Diego, United States
- The University of Western Australia, Australia
- Johns Hopkins University, United States
Figures
Figure 1 with 2 supplements
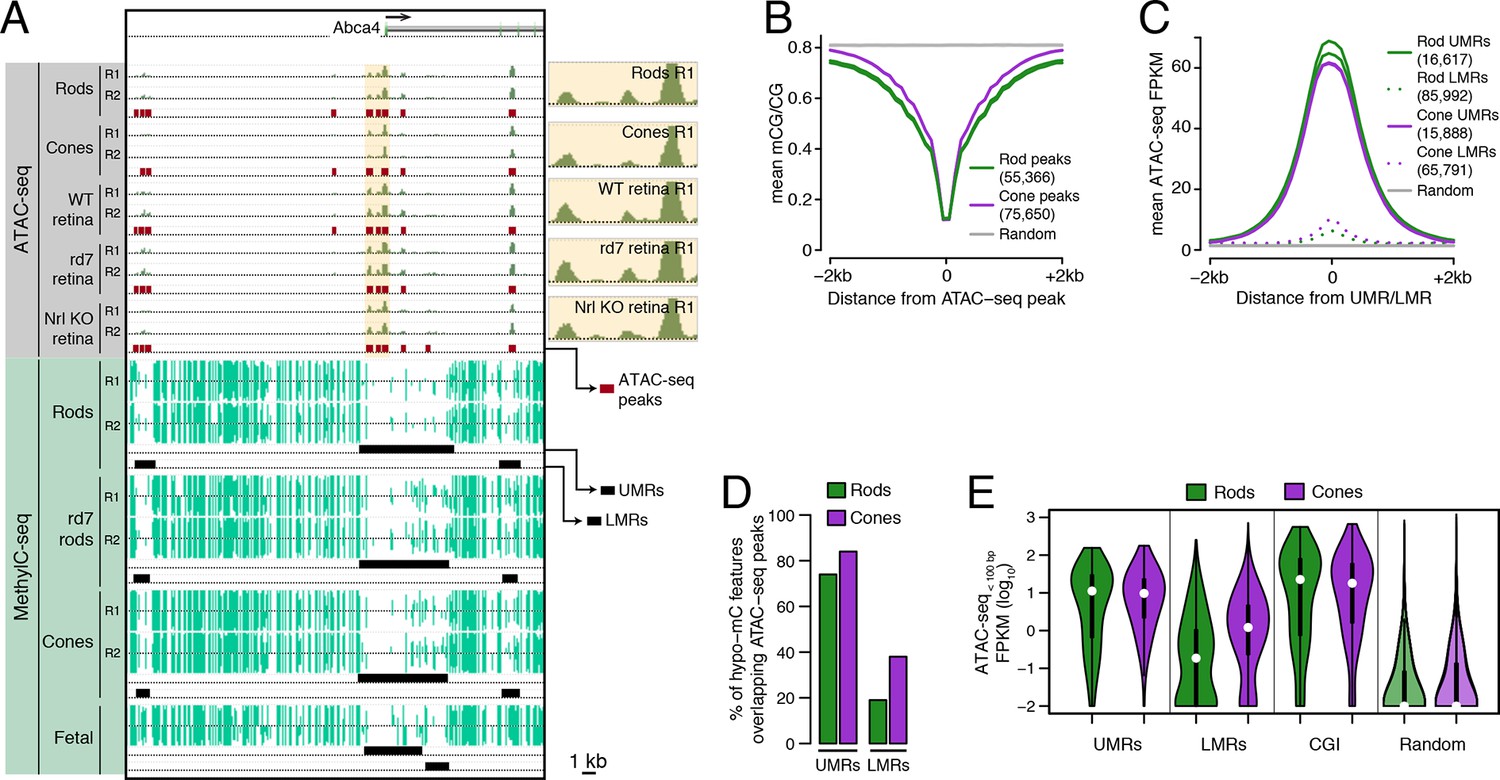
Relationship of DNA methylation and accessible chromatin in retinal rods and cones.
(A) Browser image showing accessible chromatin (top) and DNA methylation (bottom) near Abca4, a photoreceptor gene expressed by both rods and cones. Enlarged images of ATAC-seq signals in the highlighted area are shown for one replicate of each cell or tissue type. For ATAC-seq, <100 bp ATAC-seq reads are shown. For DNA methylation, mCG/CG is shown. Methylated CG positions are indicated by upward (plus strand) and downward (minus strand) ticks, with the height of each tick representing the fraction of methylation at the site ranging from 0 to 1. Bars below the raw data show locations identified as ATAC-seq peaks, UMRs, and LMRs. Fetal, fetal E13 cerebral cortex from Lister et al., 2013. Biological replicates (R1, R2). (B) Line plot showing lower mean CG methylation at rod and cone ATAC-seq peaks relative to size-matched random genomic regions (repeated 10 times). (C) Line plot showing higher mean ATAC-seq signals at rod and cone UMRs and LMRs relative to size-matched random genomic regions (repeated 10 times). (D) Barplot showing that the percentage of cone LMRs that overlap ATAC-seq peaks (38%) is two-fold higher than the percentage of rod LMRs that overlap ATAC-seq peaks (19%). (E) Violin plot showing that rod LMRs have a bimodal distribution of <100 bp ATAC-seq signals. The median (white dot) and interquartile range (black bar) are indicated. CGI: CpG islands; Random: size-matched random genomic regions (repeated 10 times).
Figure 1—figure supplement 1
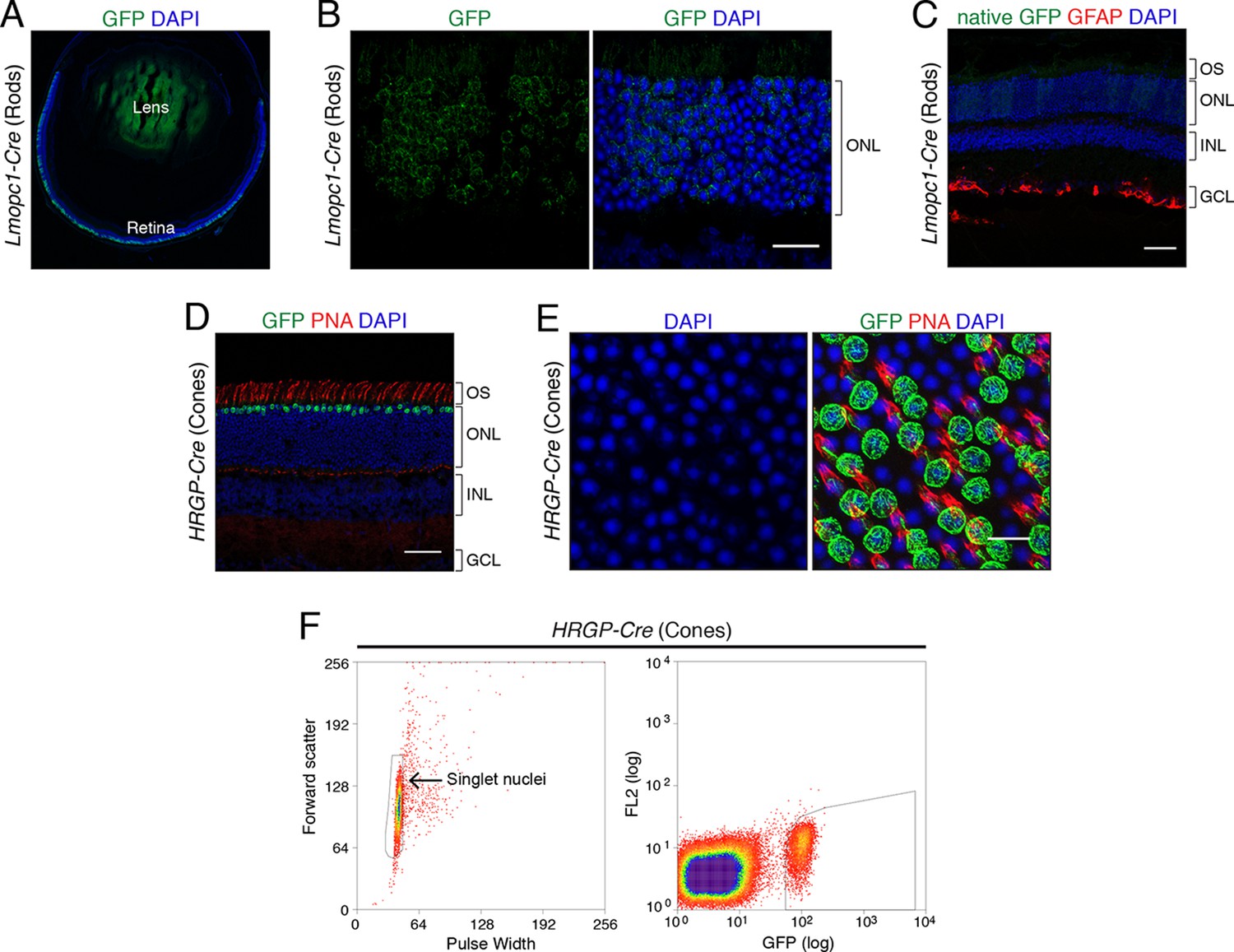
Genetic labeling of mouse rod and cone photoreceptor nuclei.
(A–B) Immunohistochemistry for GFP (green) showing labeling of rod photoreceptors in adult Lmopc1-Cre; R26-CAG-LSL-Sun1-sfGFP-myc retina. Lmopc1-Cre is a rod-specific Cre transgene controlled by the mouse opsin promoter (Le et al., 2006). GFP is restricted predominantly to rods in the outer nuclear layer (ONL). Scale bar (B) 20 µm. (C) Immunohistochemistry for GFAP (red) in adult Lmopc1-Cre; R26-CAG-LSL-Sun1-sfGFP-myc retina. GFAP is normally expressed by retinal astrocytes and is upregulated in reactive Müller glia during retinal stress. Although astrocytes in this retina are strongly GFAP+, there is no detectable GFAP labeling in Müller glia. OS, outer segments; INL, inner nuclear layer; GCL, ganglion cell layer. Native GFP fluorescence is shown (green). Scale bar: 50 µm. (D–E) Immunohistochemistry for GFP (green) and labeling with peanut agglutinin (PNA, red), a marker for cones, in adult HRGP-Cre; R26-CAG-LSL-Sun1-sfGFP-myc mice. HRGP-Cre is a cone-specific Cre transgene controlled by the human red/green pigment promoter (Le et al., 2004). HRGP-Cre predominantly recombines cones, as seen by their location, labeling with PNA, and DAPI staining (Solovei et al., 2009). Scale bars: 50 µm (D) and 10 µm (E). (F) A representative flow cytometry profile of nuclei sorted from HRGP-Cre; R26-CAG-LSL-Sun1-sfGFP-myc retinas. The thresholds used to define singlet nuclei (left) and GFP+ nuclei (right) are outlined in black.
Figure 1—figure supplement 2
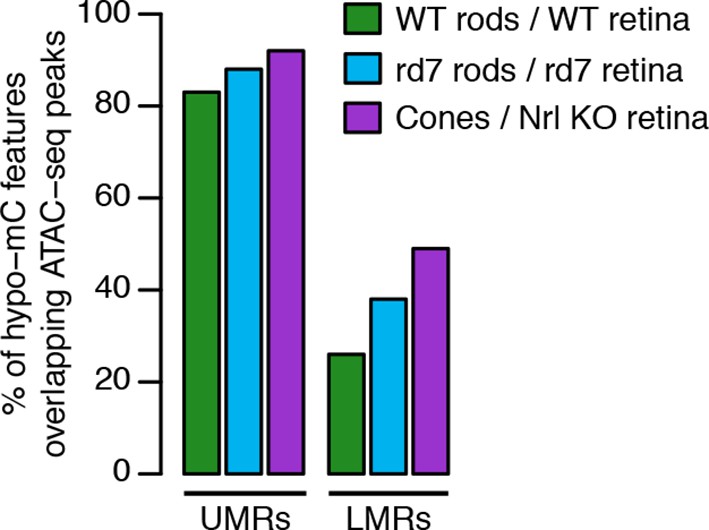
Accessible chromatin in whole retina versus DNA methylation.
Barplot showing the percentage of hypo-methylated features in WT rods, rd7 rods, and cones that overlap with ATAC-seq peaks in whole WT retina, rd7 retina, and Nrl KO retina, respectively.
Figure 2 with 2 supplements
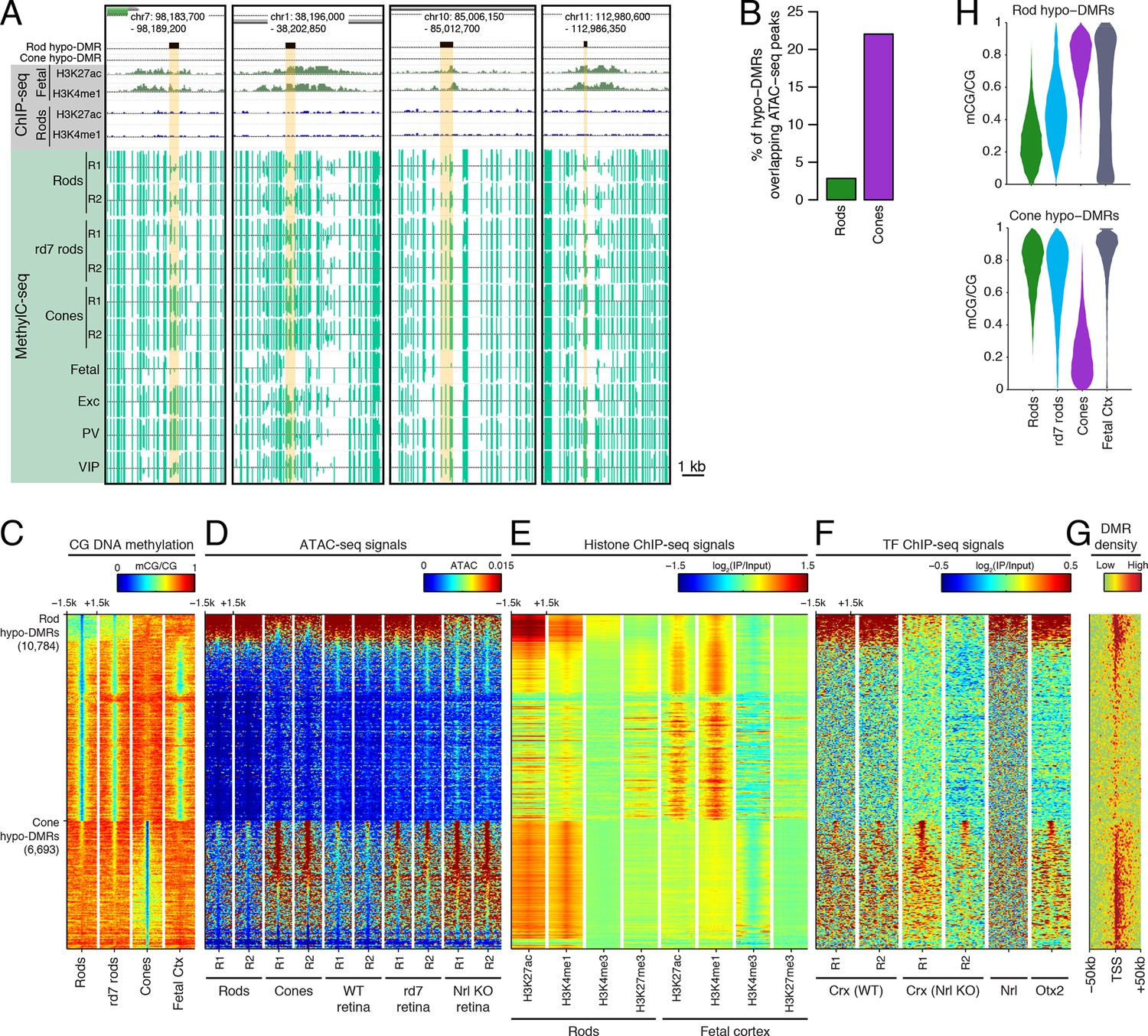
Rod hypo-DMRs show active chromatin marks in early neural development.
(A) Browser images showing examples of rod hypo-DMRs that are enriched for active enhancer histone marks in fetal E14.5 brain (from Shen et al., 2012) but not in adult rods. These rod hypo-DMRs also display low levels of DNA methylation in both WT rods and fetal E13 cerebral cortex (from Lister et al., 2013), but not in cones or in most adult cortical neuron types (Exc, PV, VIP; from Mo et al., 2015). In addition, rd7 rods show higher levels of methylation than WT rods but lower levels than cones. (B) Cone hypo-DMRs show a six-fold higher overlap with ATAC-seq peaks, compared to rod hypo-DMRs with rod ATAC-seq peaks. (C–G) Heatmap showing CG methylation levels in a 3 kb window centered at rod and cone hypo-DMRs (C). At the same genomic regions, the following were plotted: ATAC-seq signal (D), ChIP-seq signals for histone modifications in adult rods (this study) or in E14.5 fetal brain (from Shen et al., 2012) (E), and ChIP-seq signals for retinal TFs (from Corbo et al., 2010; Hao et al., 2012; Samuel et al., 2014) (F). The density of DMRs relative to their closest TSS is shown in (G) for a 100 kb window around the TSS. For (C–G), the rows are ordered by decreasing rank of the absolute signals of rod and cone ATAC-seq data at rod and cone hypo-DMRs, respectively. (H) The fetal cortex shares low CG DNA methylation with rods at a substantial fraction of rod hypo-DMRs (top), but shows high methylation at the majority of cone hypo-DMRs (bottom). Furthermore, methylation levels in rd7 rods are generally intermediate between WT rods and cones, particularly at rod hypo-DMRs.
Figure 2—figure supplement 1
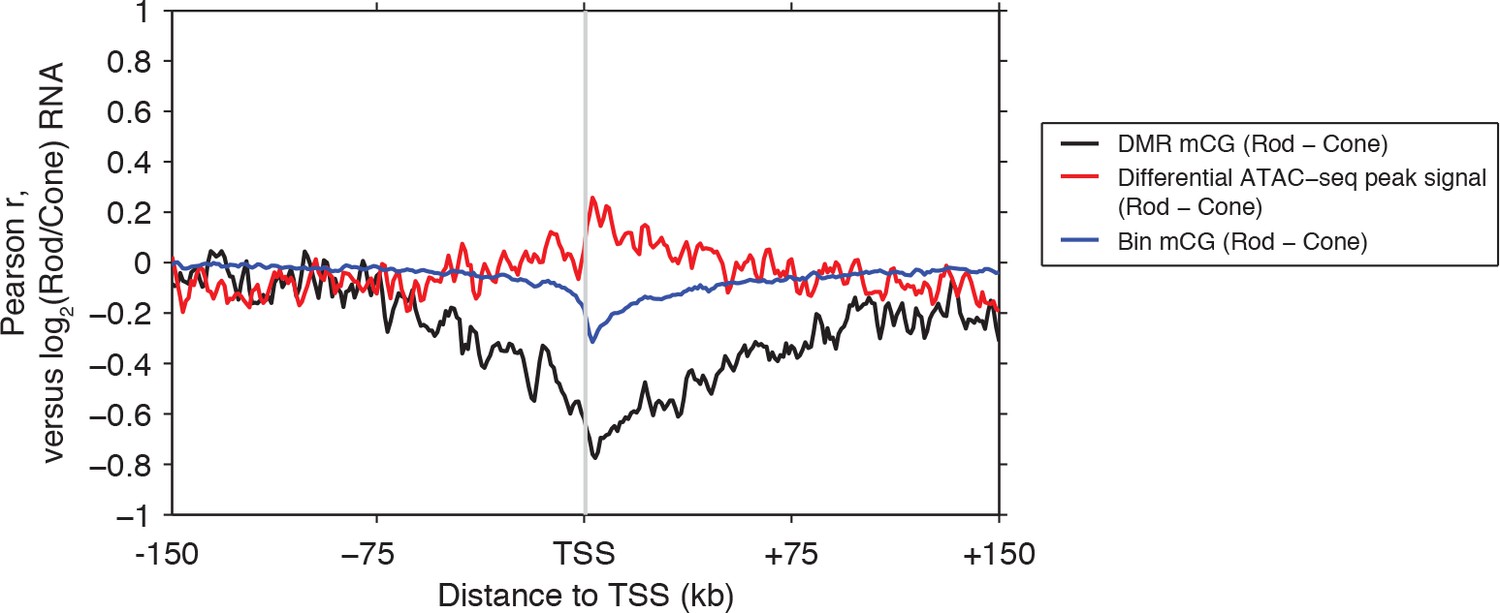
Rod versus cone DNA methylation levels at DMRs are strongly anti-correlated with relative gene expression.
Relative gene expression (log2(Rod/Cone) RNA TPM) has a stronger magnitude of correlation with DNA methylation levels at DMRs (black line), than with either ATAC-seq signals at differential ATAC-seq peaks (red line) or mean mCG levels in 1 kb genomic bins (blue line).
Figure 2—figure supplement 2
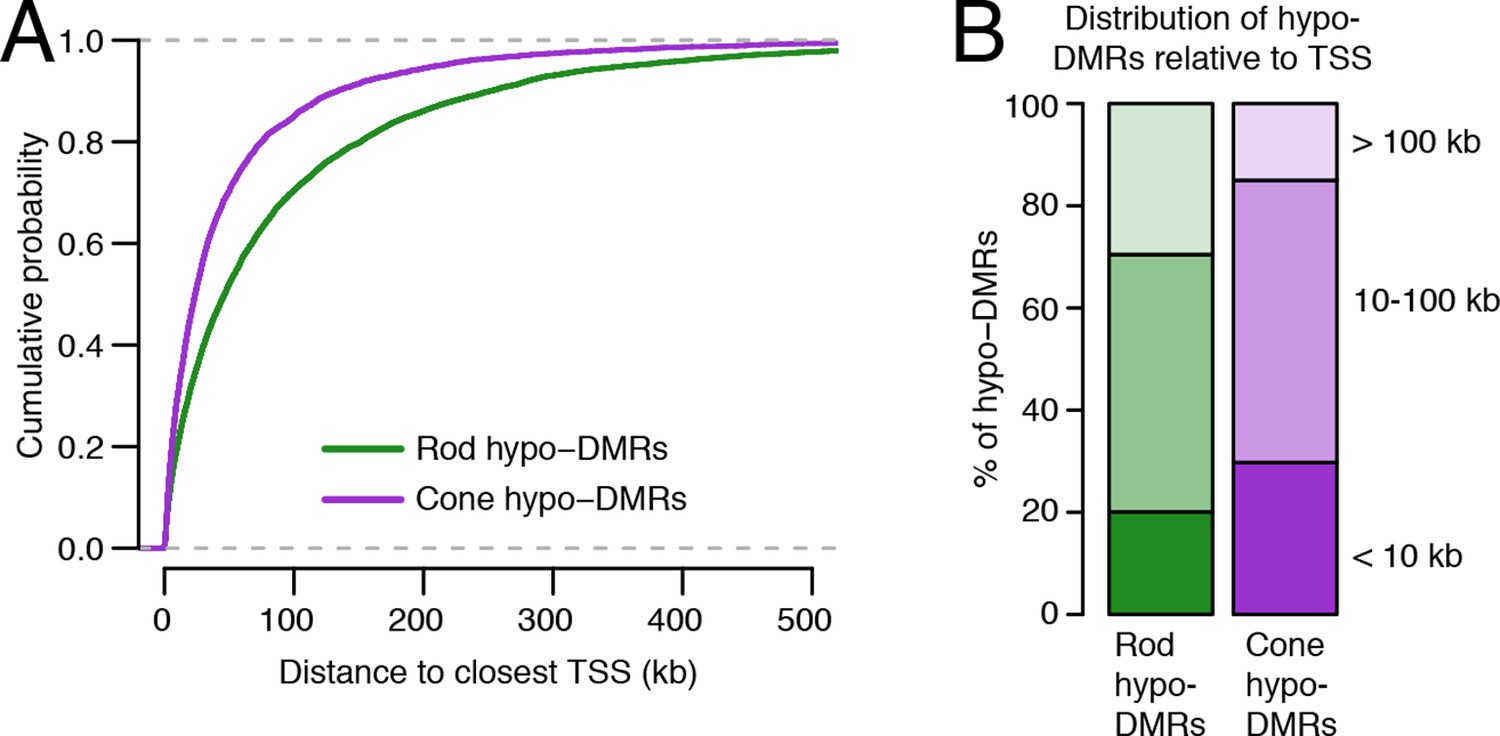
Relationship of rod and cone hypo-DMRs to gene promoters.
(A) Cone hypo-DMRs are distributed closer to the TSS than rod hypo-DMRs. (B) Barplot showing the percentage of rod and cone hypo-DMRs that fall proximal (<10 kb) or distal (10–100 kb and >100 kb) to a TSS.
Figure 3 with 3 supplements
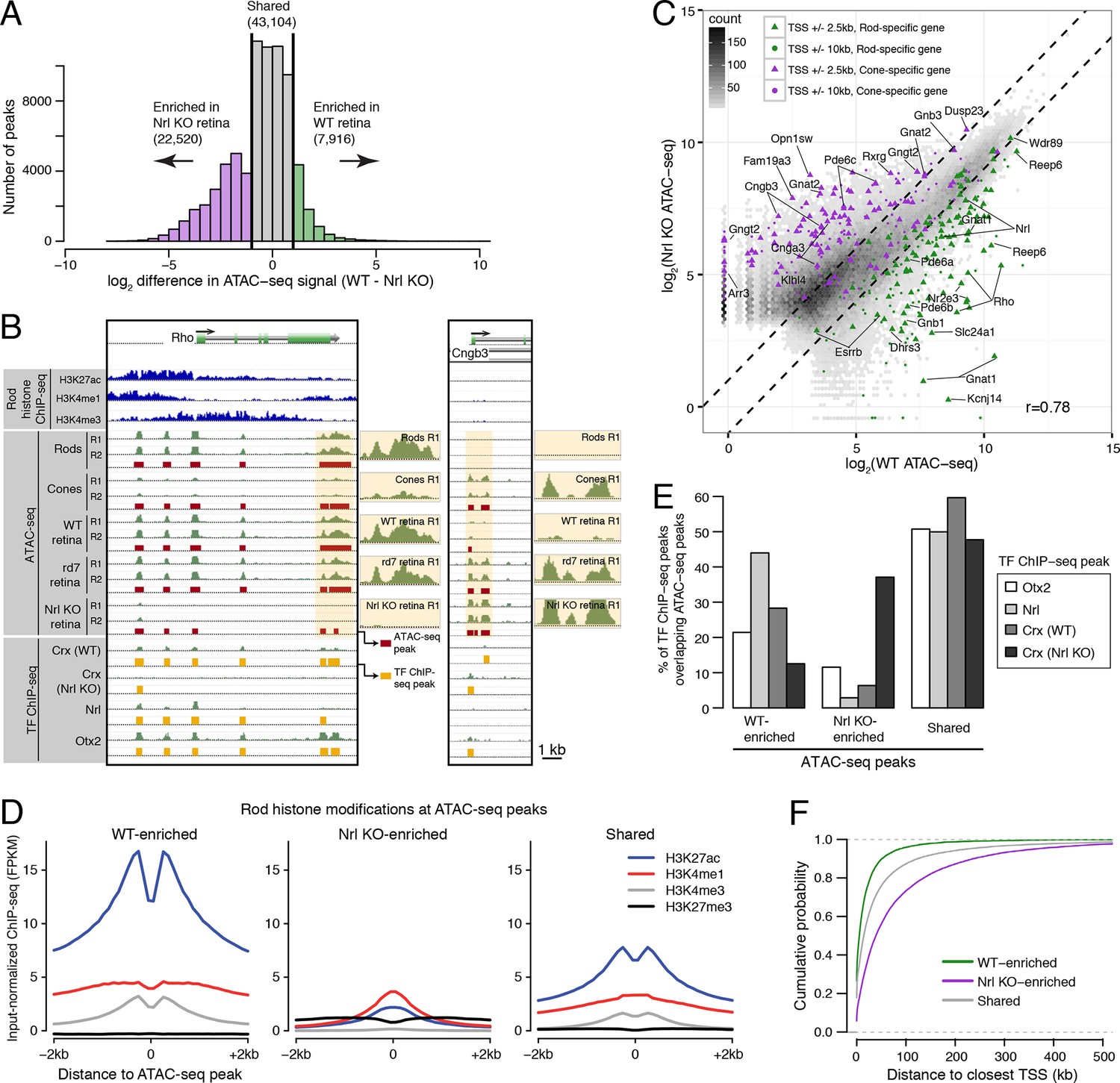
Distinctive features of WT-enriched versus Nrl KO-enriched accessible chromatin.
(A) Histogram showing that the WT retina has nearly three-fold fewer number of enriched ATAC-seq peaks compared to the Nrl KO retina. (B) Browser images showing histone modification ChIP-seq signals (rods, top), ATAC-seq signals (middle), and TF ChIP-seq signals (bottom) near Rho, a rod-specific gene (left) and near Cngb3, a cone-specific gene (right). Enlarged images of the ATAC-seq signals in the highlighted area are shown for one replicate of each cell or tissue type. Bars below the raw data indicate locations identified as ATAC-seq peaks or TF ChIP-seq peaks. (C) Peaks near rod genes (green; e.g., Nrl, Gnat1) generally show higher ATAC-seq signals in WT than in Nrl KO retina. Peaks near cone genes (purple; e.g., Pde6h, Pde6c) generally show higher ATAC-seq signals in Nrl KO than in WT retina. Colored points show all ATAC-seq peaks that fall within 2.5 kb (triangle) or 10 kb (circle) of the TSS. Selected peaks are labeled by their associated gene. r, Pearson correlation. (D) Line plots showing that WT-enriched ATAC-seq peaks have higher mean levels of active rod histone modifications (H3K27ac, H3K4me1, and H3K4me3) compared to Nrl KO-enriched peaks. (E) Barplot showing the percentage of TF ChIP-seq peaks that overlap each category of ATAC-seq peak. (F) WT-enriched ATAC-seq peaks are distributed closer to the TSS than Nrl KO-enriched and shared ATAC-seq peaks.
Figure 3—figure supplement 1
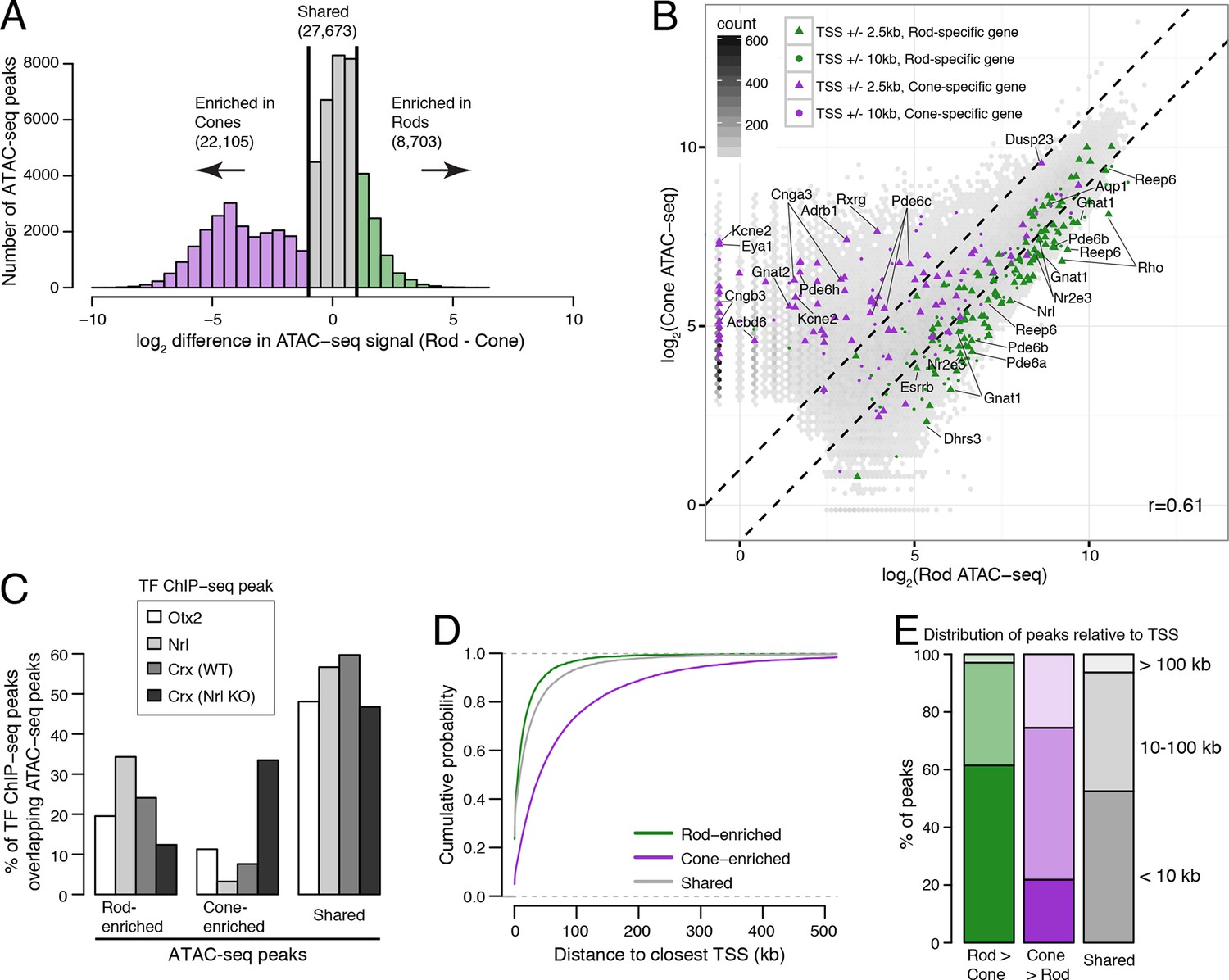
Comparisons of ATAC-seq signals between purified rod and cone nuclei.
(A) Histogram showing that rods have ~2.5-fold fewer number of enriched peaks compared to cones. (B) Similar to Figure 3C, peaks near rod genes (green; e.g., Nrl, Gnat1) generally show higher ATAC-seq signals in rods than in cones. In contrast, peaks near cone genes (purple; e.g., Pde6h, Pde6c) generally show higher ATAC-seq signals in cones than in rods. Colored points show ATAC-seq peaks which fall within 2.5 kb (triangle) or 10 kb (circle) of the TSS. Selected peaks are labeled by their associated gene. r, Pearson correlation. (C) Barplot showing the percentage of TF ChIP-seq peaks that overlap rod-enriched, cone-enriched, and shared ATAC-seq peaks. (D–E) Rod-enriched ATAC-seq peaks are distributed closer to the TSS than cone-enriched and shared ATAC-seq peaks. The majority of cone-enriched ATAC-seq peaks fall >10 kb from a TSS, whereas the majority of rod-enriched ATAC-seq peaks are TSS-proximal.
Figure 3—figure supplement 2
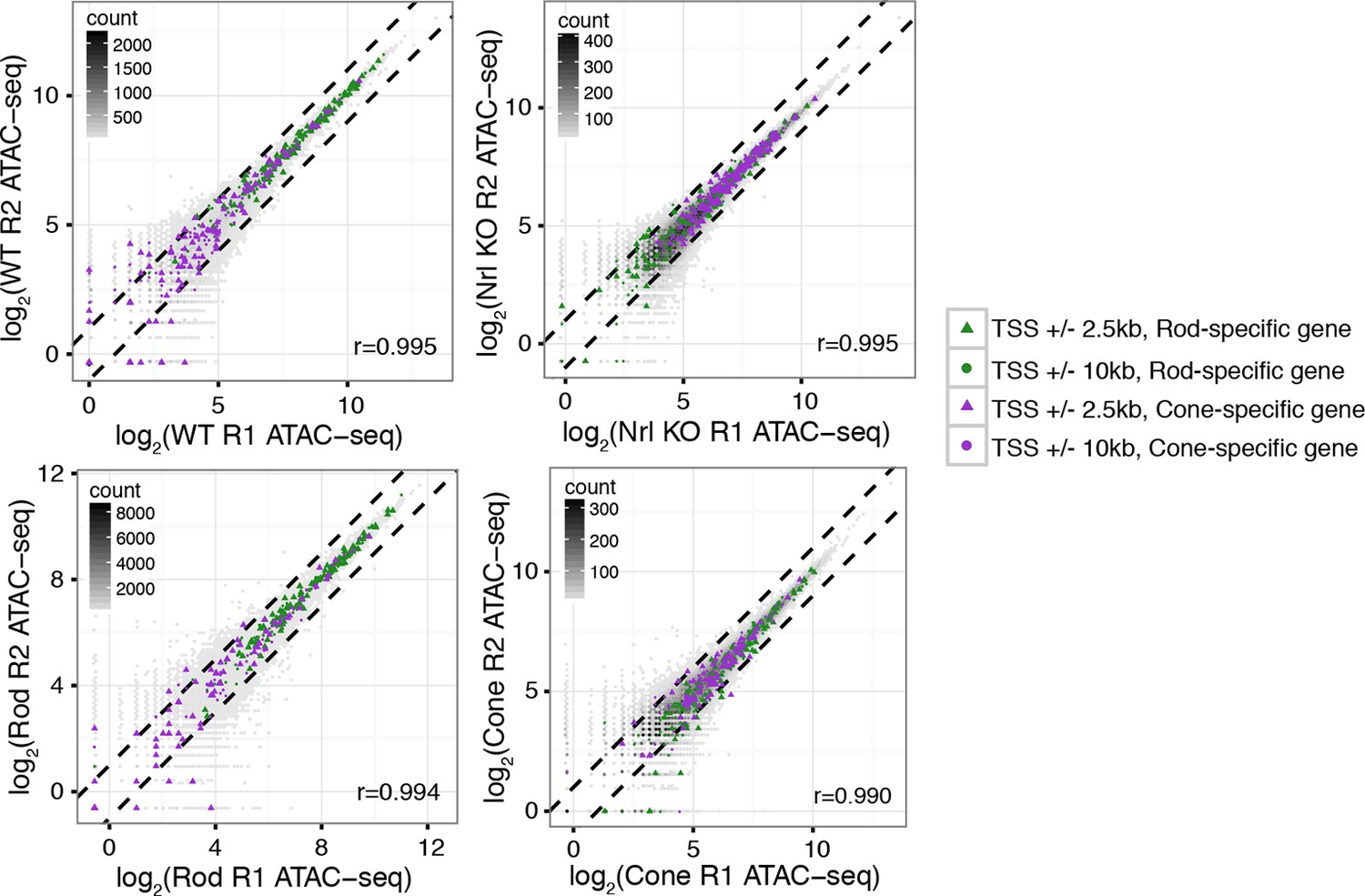
ATAC-seq signals between biological replicates.
ATAC-seq signals are well-correlated between biological replicates at ATAC-seq peaks. Biological replicates (R1, R2).
Figure 3—figure supplement 3
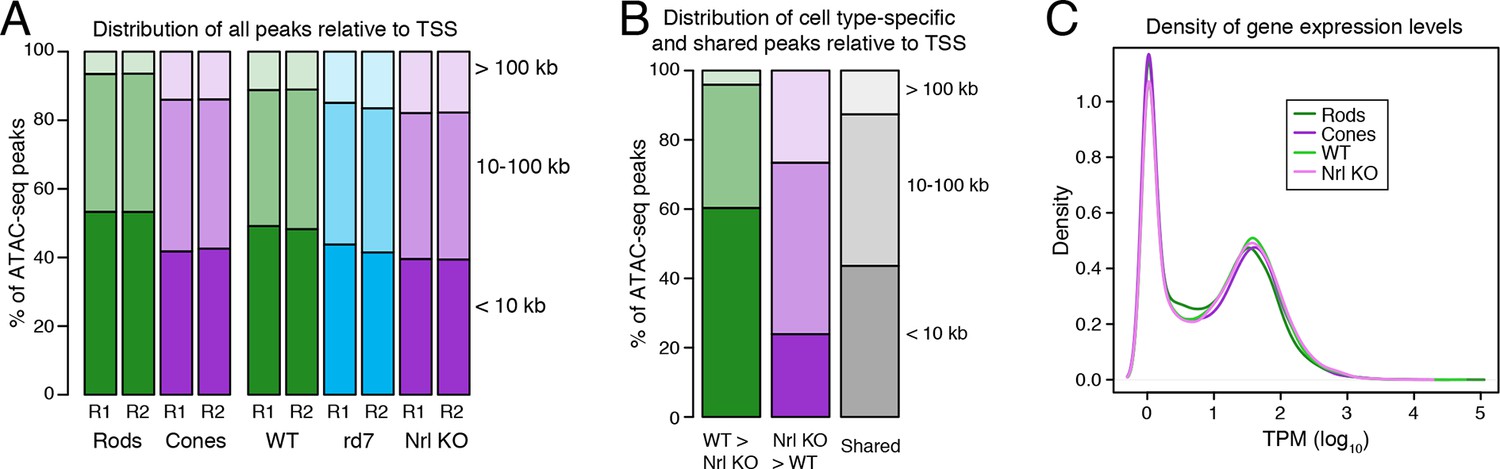
Cell type-specific differences in ATAC-seq peak distribution are not reflected by gene expression.
(A–B) Barplot showing the percentage of all ATAC-seq peaks in each sample that fall proximal (<10 kb) or distal (10–100 kb and >100 kb) to a TSS (A). For cell type-specific peaks (B), WT-enriched ATAC-seq peaks are distributed closer to the TSS than Nrl KO-enriched peaks. (C) Density plot showing that all samples have similar distributions of gene expression levels.
Figure 4 with 1 supplement
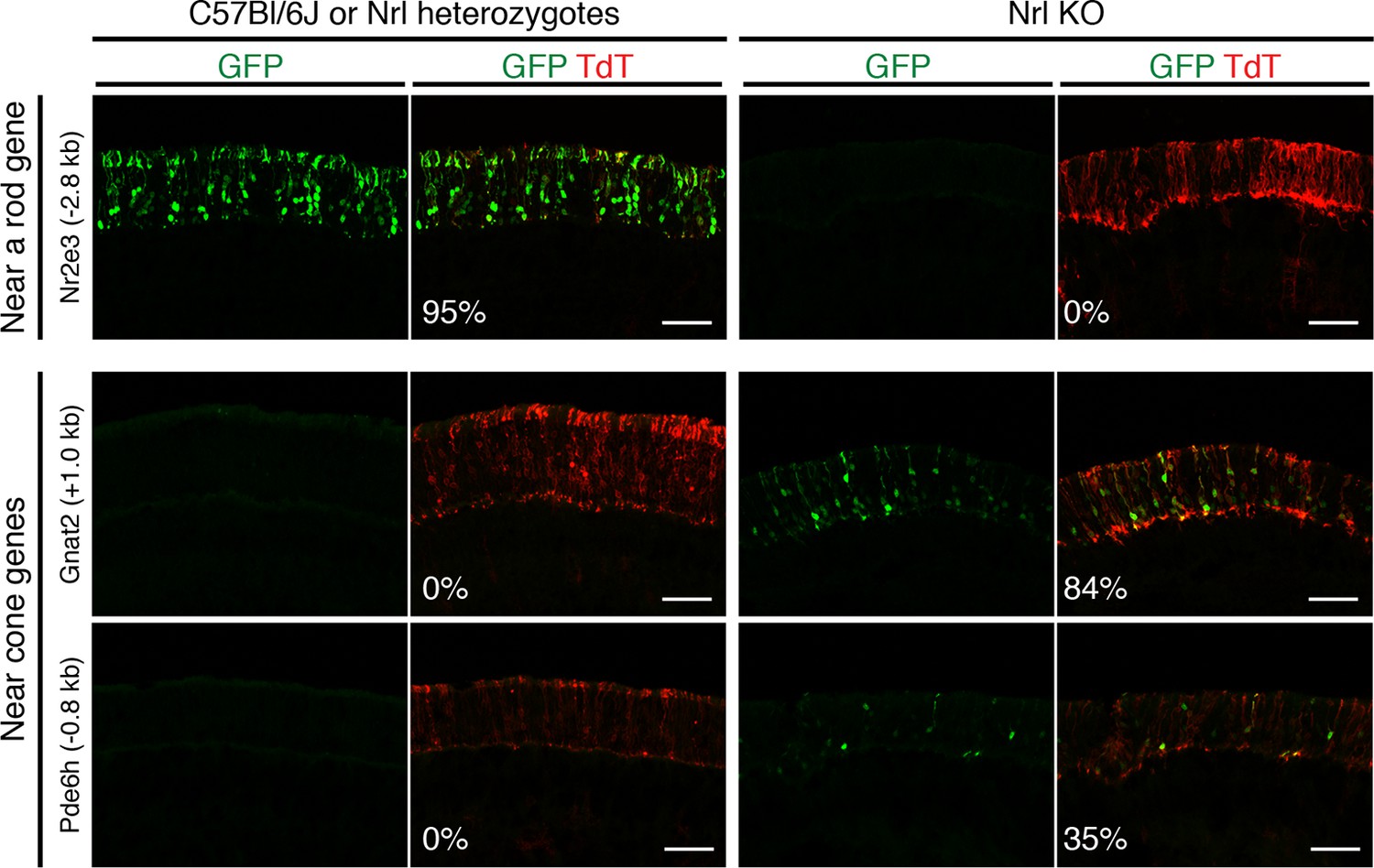
In vivo retinal electroporation of putative regulatory elements.
Cryosections of C57Bl/6J or Nrl heterozygote retinas (left) and Nrl KO retinas (right) from 3–4 week old mice after in vivo retinal electroporation at P0 of a putative rod regulatory element near Nr2e3 (top row) or putative cone regulatory elements near cone-specific genes (bottom rows). The element near Nr2e3 induces GFP reporter expression only in WT retina but not in Nrl KO retina. Elements near Gnat2 and Pde6h induce GFP reporter expression in Nrl KO retina but not in WT retina. The TdT signal is a control for electroporation efficiency. The average % of electroporated (TdT+) cells that are GFP+ is shown. Coordinates of electroporated elements are listed in Supplementary file 6.
Figure 4—figure supplement 1
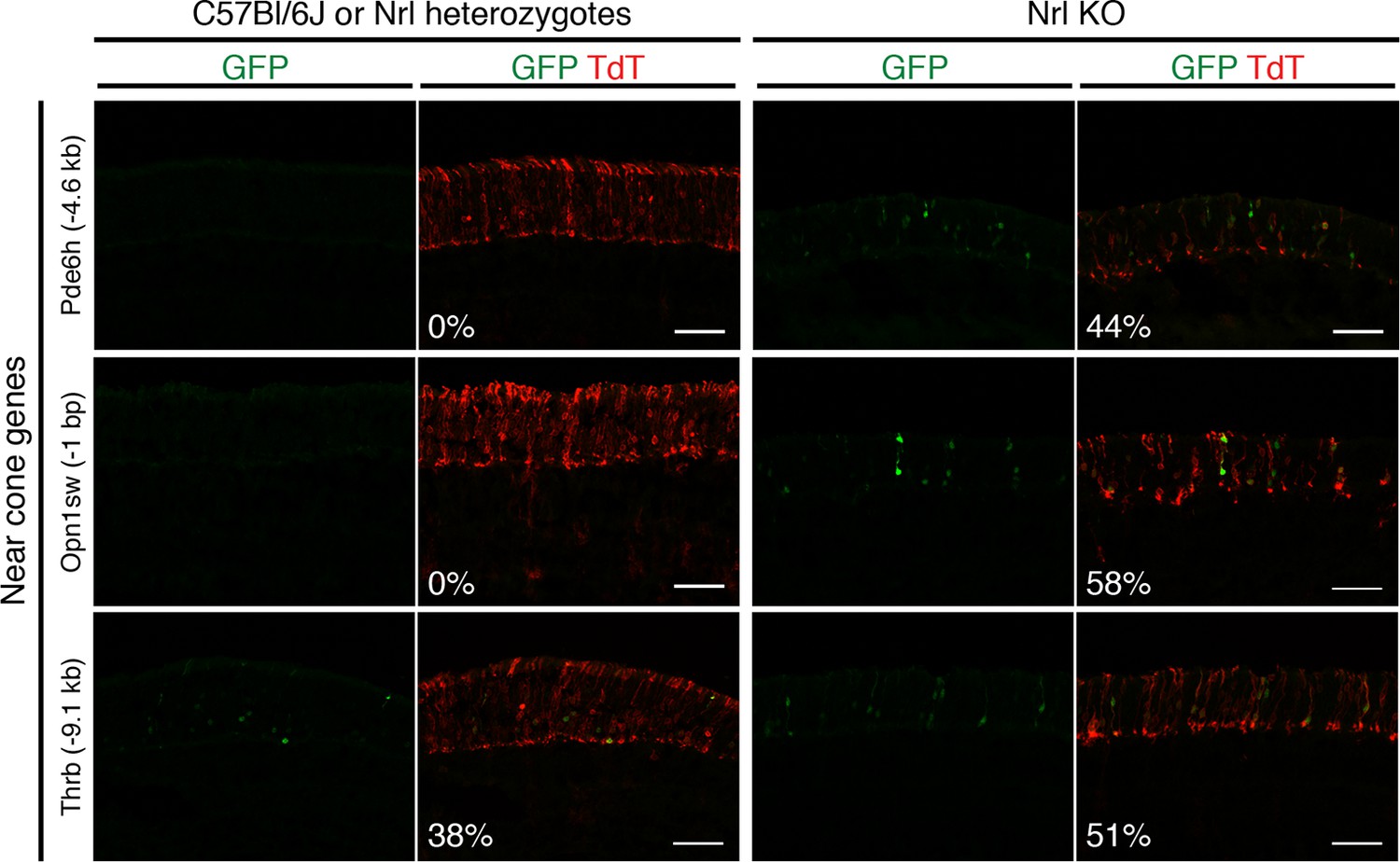
Additional examples of in vivo retinal electroporation of putative cone regulatory elements.
Cryosections of C57Bl/6J or Nrl heterozygote retinas (left) and Nrl KO retinas (right) from 3–4 week old mice after in vivo retinal electroporation at P0 of putative cone regulatory elements near cone-specific genes. Elements near Pde6h and Opn1sw induce GFP reporter expression in Nrl KO retina but not in WT retina. An element near Thrb induces GFP reporter expression in both WT and Nrl KO retinas. The TdT signal is a control for electroporation efficiency. The average % of electroporated (TdT+) cells that are GFP+ is shown. Coordinates of electroporated elements are listed in Supplementary file 6.
Figure 5 with 2 supplements
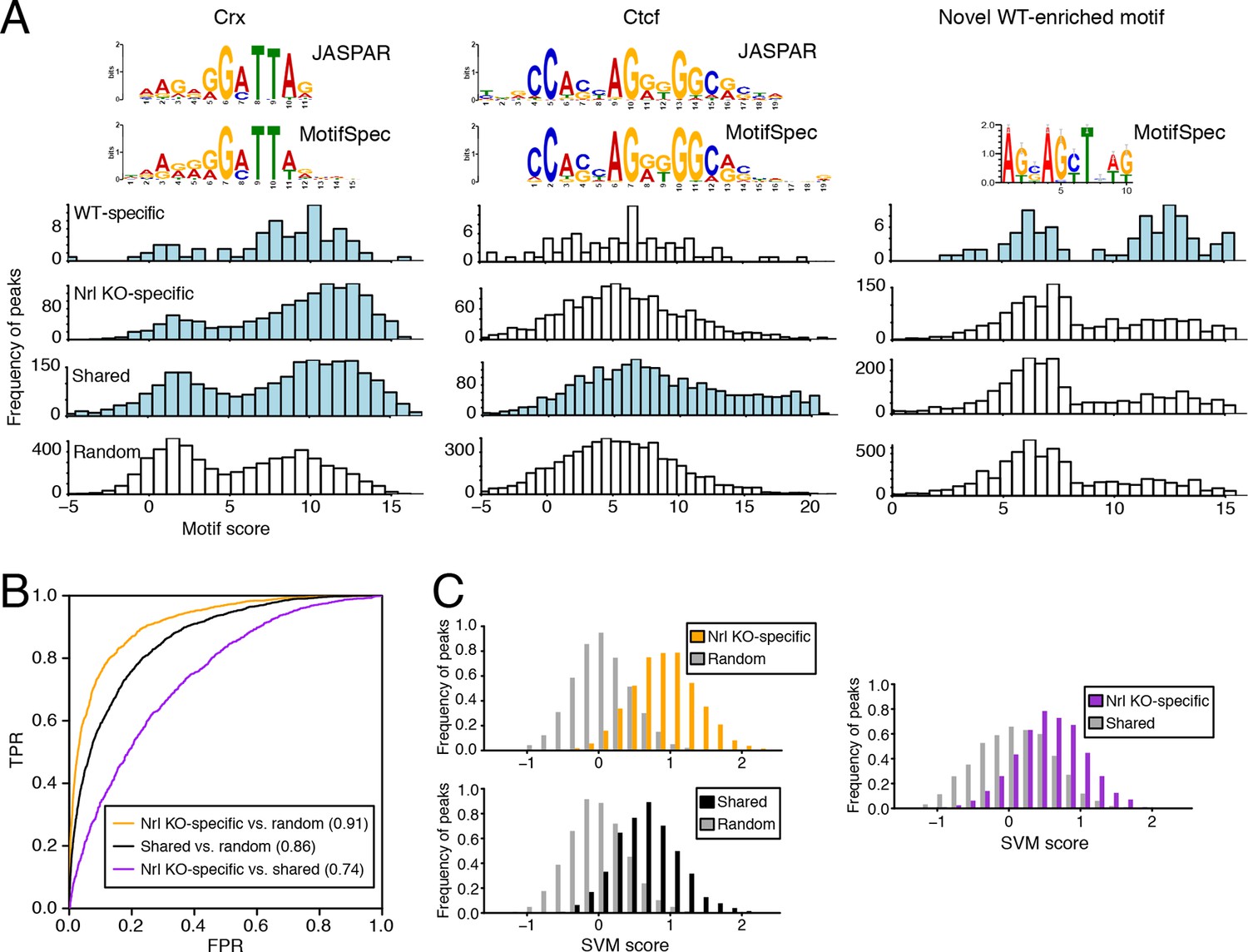
Machine learning identifies DNA sequence features of photoreceptor accessible chromatin.
(A) Barplots showing score distributions of the strongest single motifs detected by a discriminative algorithm (MotifSpec) used to identify differentially enriched motifs. CRX binding sites are enriched in all sets of peaks relative to GC-matched random genomic sequences (left). CTCF is enriched at shared peaks (middle), and a novel motif is enriched at WT-specific peaks (right). In each peak set, the distribution of motif scores which are predictive above AUC = 0.6 versus random sequence is shown in blue. (B–C) ROC curves showing that gapped k-mer SVM can classify ATAC-seq peaks using regulatory sequence features (B). When trained versus GC-matched random genomic sequences, gkm-SVM auROC is high (in parentheses). Distinguishing Nrl KO-specific ATAC-seq peaks from shared peaks is more challenging (C, right) than distinguishing between Nrl KO-specific (C, top left) and shared (C, bottom left) peaks from random regions. Nevertheless, the sequence-based SVM score, a weighted sum of k-mer counts, is still able to distinguish Nrl KO-specific peaks from shared peaks based on sequence features.
Figure 5—figure supplement 1

Retinal k-mers.
(A) For regions used in the k-mer analysis, WT-specific ATAC-seq peaks (left) are distributed near the TSS of rod-enriched (>2 fold) genes, whereas Nrl KO-specific ATAC-seq peaks (right) are closer to the TSS of cone-enriched genes. (B) Boxplots showing the log-transformed, normalized ATAC-seq signal (left two columns) and TF ChIP-seq signal (right four columns) in different classes of ATAC-seq peaks. (C) Lists of the most predictive sequence features for distinguishing ATAC-seq peaks. The largest gkm-SVM weights for Nrl KO-specific peaks (+) versus random regions (-) (left), shared peaks (+) versus random regions (-) (middle), and Nrl KO-specific (+) versus shared peaks (-) (right) are listed.
Figure 5—figure supplement 2
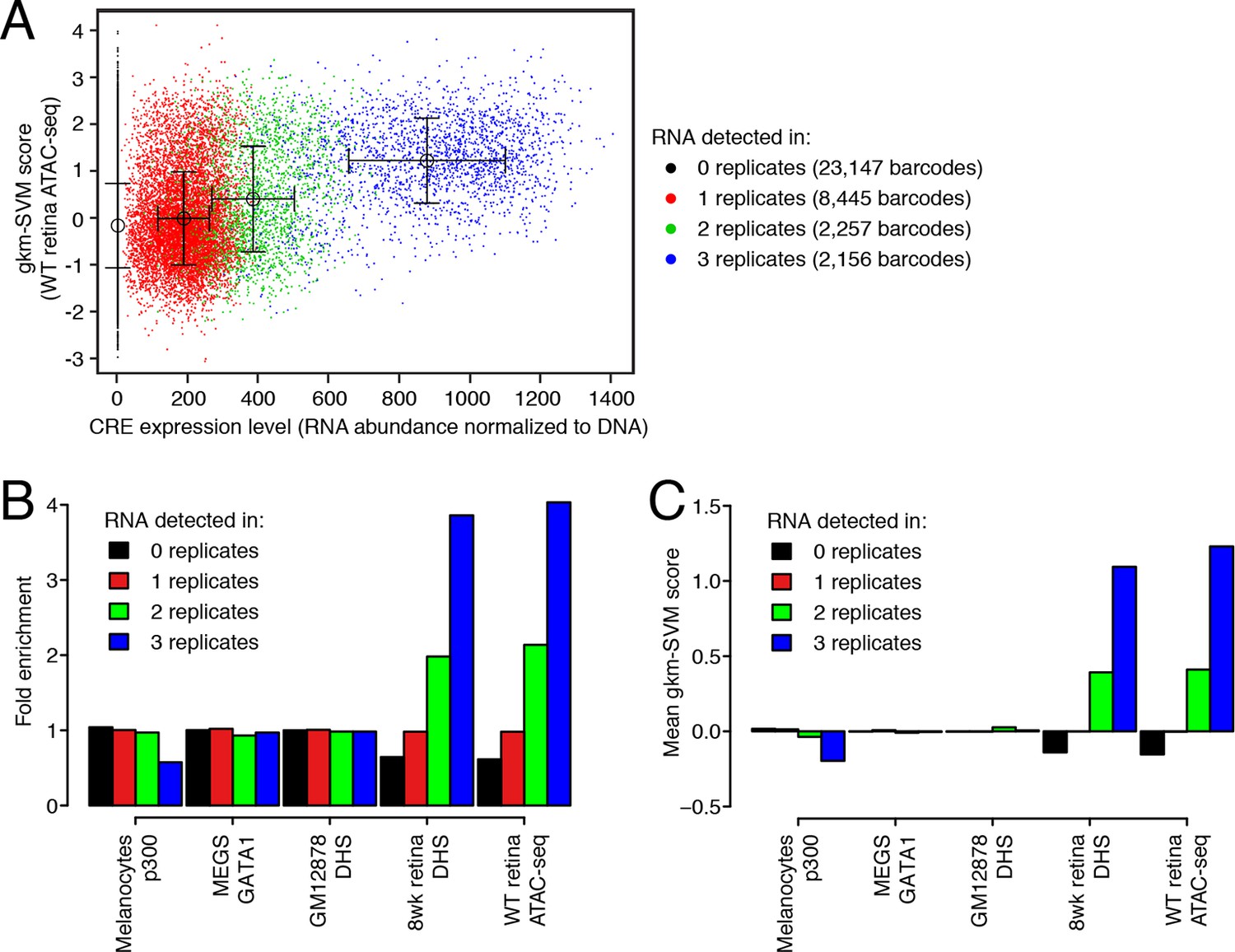
DNA regulatory sequences inferred from retinal chromatin accessibility yield gkm-SVM scores which predict enhancer activity in a massively parallel reporter assay.
(A) A scatterplot showing that retinal expression levels of >3000 candidate retina, brain, heart, and liver CREs (Shen et al., 2016) are strongly correlated with the number of replicates in which each candidate CRE barcode was detected in the RNA sample. In addition, candidate CREs with high expression have higher scores using the regulatory vocabulary trained on the WT retina ATAC-seq dataset. Error bars show mean +/- 1 S.D. for each of the four sets of data points. (B) Relative to all candidate CREs, the top-scoring 10% of 36,005 candidate CRE constructs are strongly enriched in highly expressed sequences (i.e., those detected in all three replicates) when gkm-SVM is trained on retinal ATAC-seq peaks or DHS. There is no enrichment when training is performed with chromatin features from non-retinal cell types: p300-bound enhancers in melanocytes (Gorkin et al., 2012), GATA1-bound enhancers in megakaryocytes (Pimkin et al., 2014), and DHSs in lymphoblasts (Lee et al., 2015). (C) For retinal ATAC-seq or DHS, the mean gkm-SVM score is higher for sequences detected in all three replicates, than for sequences detected in zero, one, or two replicates, or when gkm-SVM is trained on chromatin features from non-retinal cell types.
Figure 6 with 3 supplements
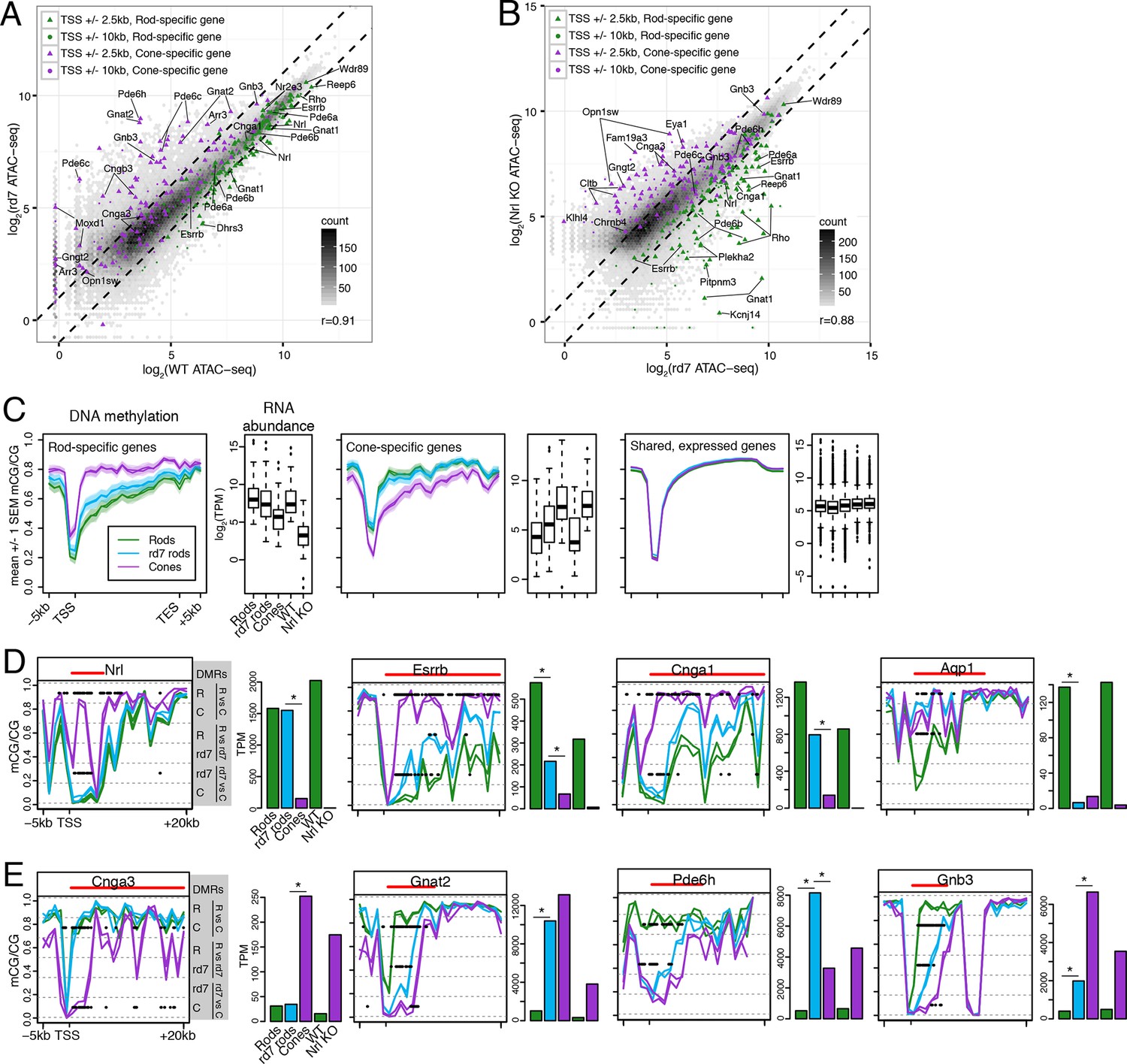
rd7 rods show intermediate epigenomic profiles compared to WT rods and cones.
(A–B) (A) Peaks near rod-specific genes (green) generally show equivalent ATAC-seq signals in WT and rd7 retinas. A subset of peaks near cone-specific genes (purple) have higher signals in rd7 retina than in WT retina. (B) Peaks near rod-specific genes (green) generally show higher ATAC-seq signal in rd7 retinas than in Nrl KO retinas. Peaks near cone-specific genes (purple) show either similar ATAC-seq signals in rd7 retina and Nrl KO retina or higher ATAC-seq signal in Nrl KO retina. For both (A) and (B), colored points show ATAC-seq peaks that fall within 2.5 kb (triangle) or 10 kb (circle) of a rod-specific gene (green) or a cone-specific gene (purple). Selected peaks are labeled by their associated gene. r, Pearson correlation. (C) Genes that are up-regulated in rods (left) and cones (middle) show lower levels of CG DNA methylation in rods and cones, respectively. SEM, standard error of the mean. (D–E) At individual rod-specific (D) and cone-specific (E) genes, line plots showing CG DNA methylation levels in a region between -5 kb and +20 kb around the TSS. Biological replicates are shown as separate lines (WT rods, green; rd7 rods, blue; cones, purple). Pairwise DMRs are indicated with black lines. R, WT rods; C, cones; rd7, rd7 rods. The gene body is indicated with a red line. Barplots showing RNA abundances. All genes are differentially expressed between WT rods and cones and between WT retina and Nrl KO retina. Asterisks indicate differentially expressed genes between rd7 rods and WT rods or between rd7 rods and cones.
Figure 6—figure supplement 1

Epigenomic patterns of WT rods, rd7 rods, and cones near photoreceptor genes.
Browser images showing ATAC-seq signals (top), CG DNA methylation (middle), and TF ChIP-seq signals (bottom) at examples of rod-specific (Nrl, Nr2e3, Pde6b) and cone-specific (Pde6c, Cnga3) genes.
Figure 6—figure supplement 2

CG DNA methylation around rod-specific genes.
Line plots showing CG DNA methylation levels in a region between -5 kb and +20 kb around the TSS of rod-specific genes. Biological replicates are shown as separate lines. Hypo-DMRs are indicated with black lines. R, WT rods; C, cones; rd7, rd7 rods. The gene body is indicated with a red line. Barplots showing RNA abundances. All genes are differentially expressed between WT rods and cones and between WT retina and Nrl KO retina. Asterisks indicate differentially expressed genes between rd7 rods and WT rods or between rd7 rods and cones.
Figure 6—figure supplement 3
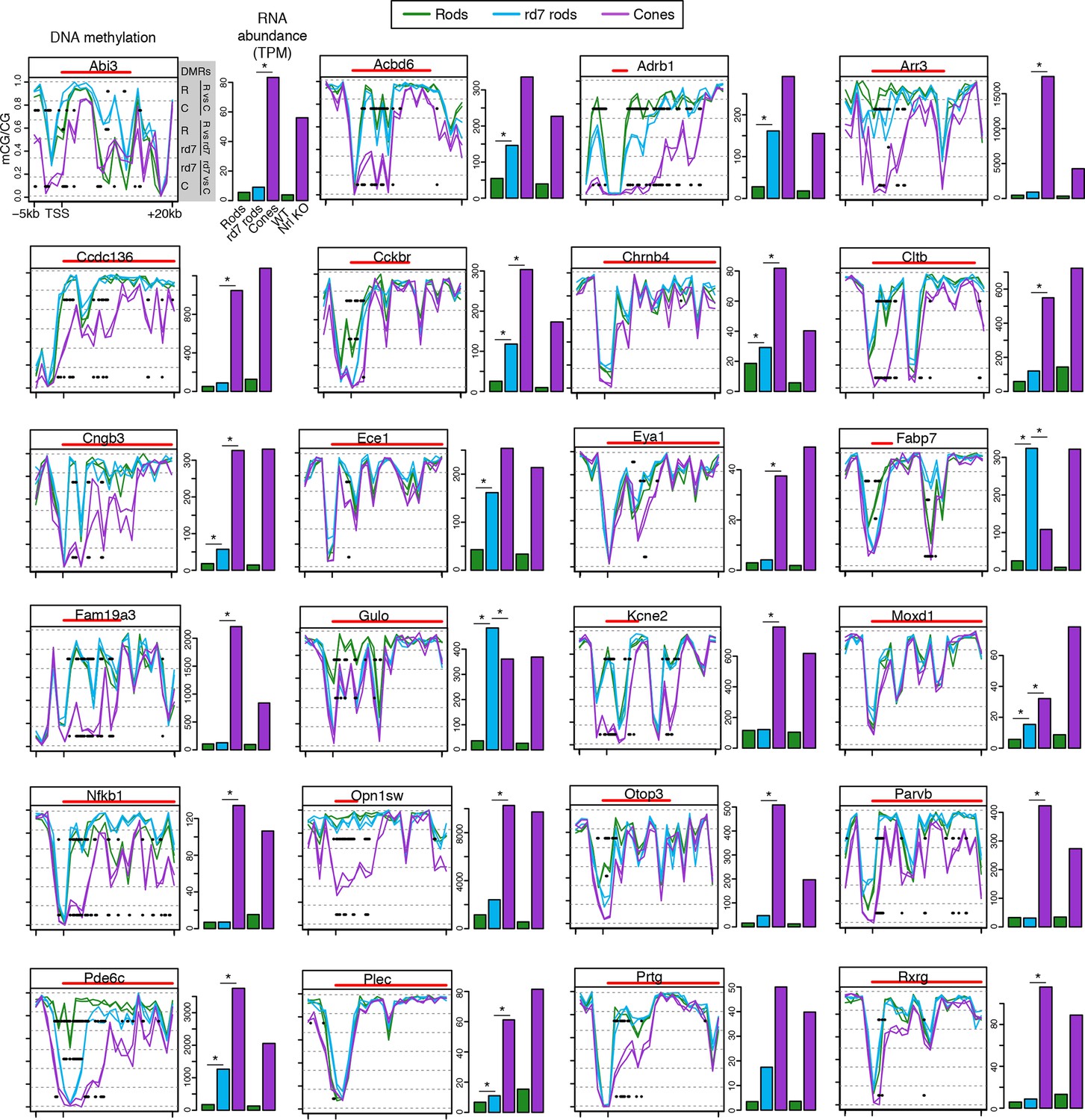
CG DNA methylation around cone-specific genes.
Line plots showing CG DNA methylation levels in a region between -5 kb and +20 kb around the TSS of cone-specific genes. Biological replicates are shown as separate lines. Hypo-DMRs are indicated with black lines. R, WT rods; C, cones; rd7, rd7 rods. The gene body is indicated with a red line. Barplots showing RNA abundances. All genes are differentially expressed between WT rods and cones and between WT retina and Nrl KO retina. Asterisks indicate differentially expressed genes between rd7 rods and WT rods or between rd7 rods and cones.
Figure 7 with 1 supplement
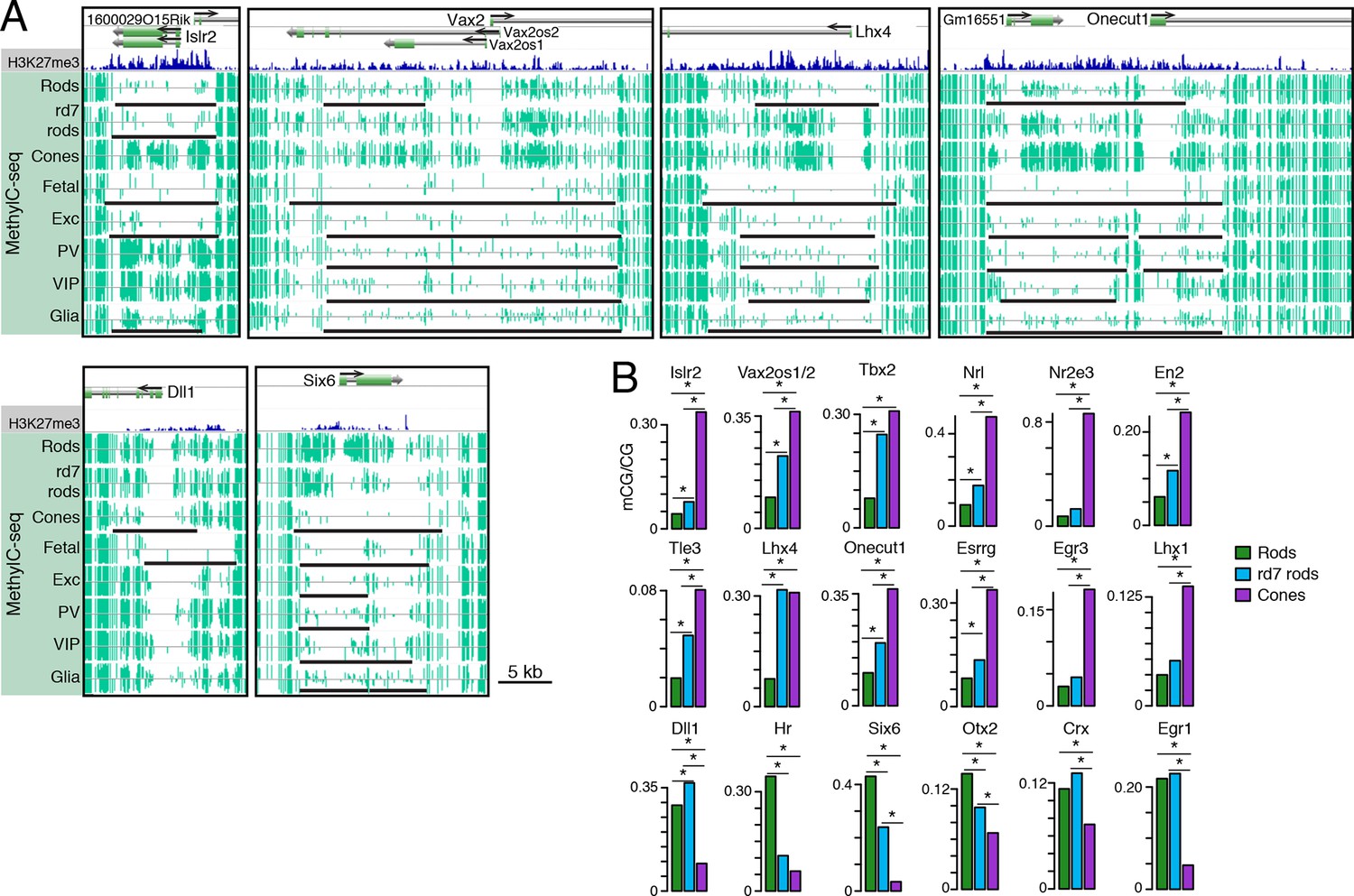
Retinal photoreceptors show distinct methylation patterns at DNA methylation valleys.
(A) Browser images showing rod H3K27me3 (top track, blue) and CG methylation levels in retinal and cortical methylomes (bottom tracks, green). A variety of cell type-specific mCG patterns are shown at these regions, including hyper-methylation in all retinal samples compared to all cortical samples (e.g., Vax2/Vax2os1/2) and hyper-methylation in a subset of retinal and cortical samples (e.g., Islr2). Rod H3K27me3+ DMVs overlapping Islr2, Vax2os1/2, Lhx4, and Onecut1 show higher levels of methylation in cones compared to rods. In contrast, rod H3K27me3+ DMVs overlapping Dll1 and Six6 show higher levels of methylation in rods compared to cones. Black lines indicate DNA methylation valleys identified in each cell type. (B) Barplots showing the levels of CG methylation in rods, rd7 rods, and cones at DMVs overlapping individual TF genes. Asterisks indicate significance at FDR <1 x 10–10 (Fisher’s Exact Test).
Figure 7—figure supplement 1
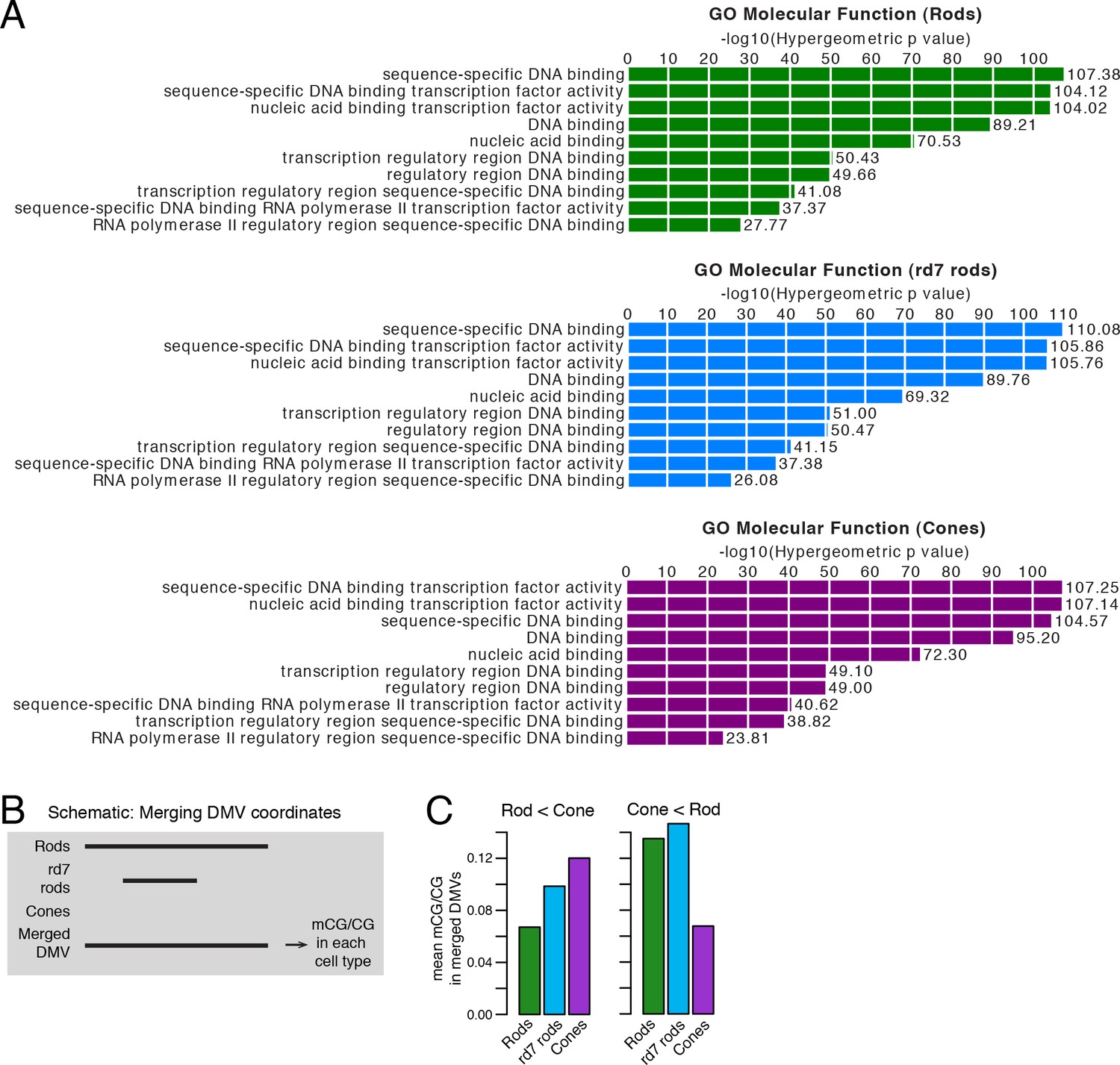
CG DNA methylation at DNA methylation valleys.
(A) GO analysis from GREAT (McLean et al., 2010) showing that genes associated with DMVs are highly enriched for DNA-binding factors. The top ten terms from GO Molecular Function are displayed. (B) A schematic showing how DMV coordinates were merged across WT rod, rd7 rod, and cone methylomes. (C) Barplots showing the mean levels of WT rod, rd7 rod, and cone CG methylation at DMVs with higher methylation in cones than in WT rods (left) or at DMVs with higher methylation in WT rods than in cones (right).
Figure 8 with 2 supplements
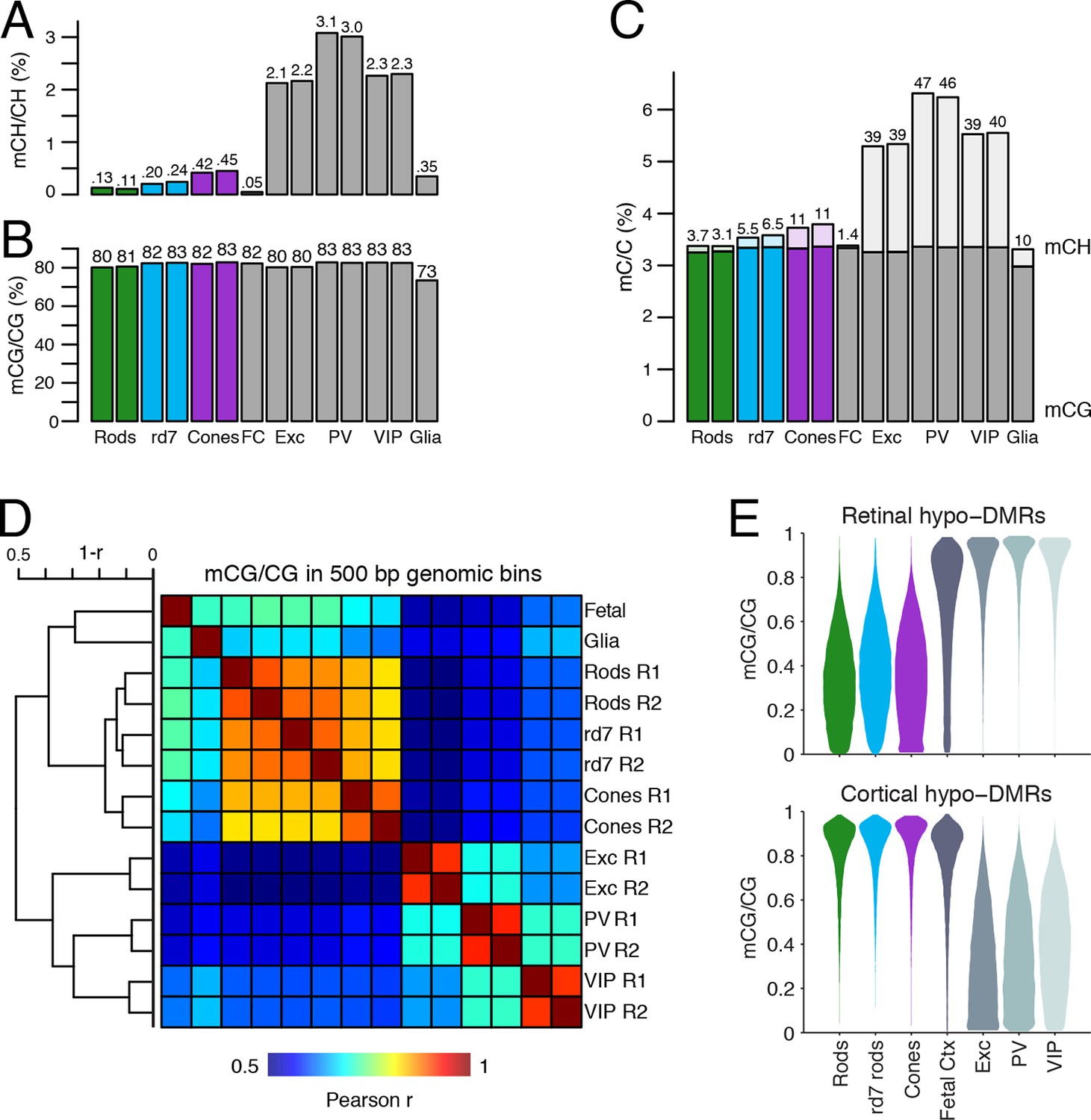
DNA methylation at retinal photoreceptors versus cortical neurons.
(A–B) The levels of CH (A) and CG (B) DNA methylation for retinal and cortical cell types. FC, E13 fetal cerebral cortex. (C) The total level (CG and CH) of DNA methylation. The percentage of all methylcytosines that are in the CH context (top) is shown. (D) Heatmap showing the pairwise Pearson correlation (r) of CG methylation levels in 500 bp genomic bins among retinal and cortical samples. The dendrogram shows hierarchical clustering using 1-r as the distance measure. Biological replicates (R1, R2). (E) The fetal cortex shows a lower distribution of mCG/CG at pan-retinal hypo-DMRs (top) compared to pan-cortical hypo-DMRs (bottom).
Figure 8—figure supplement 1
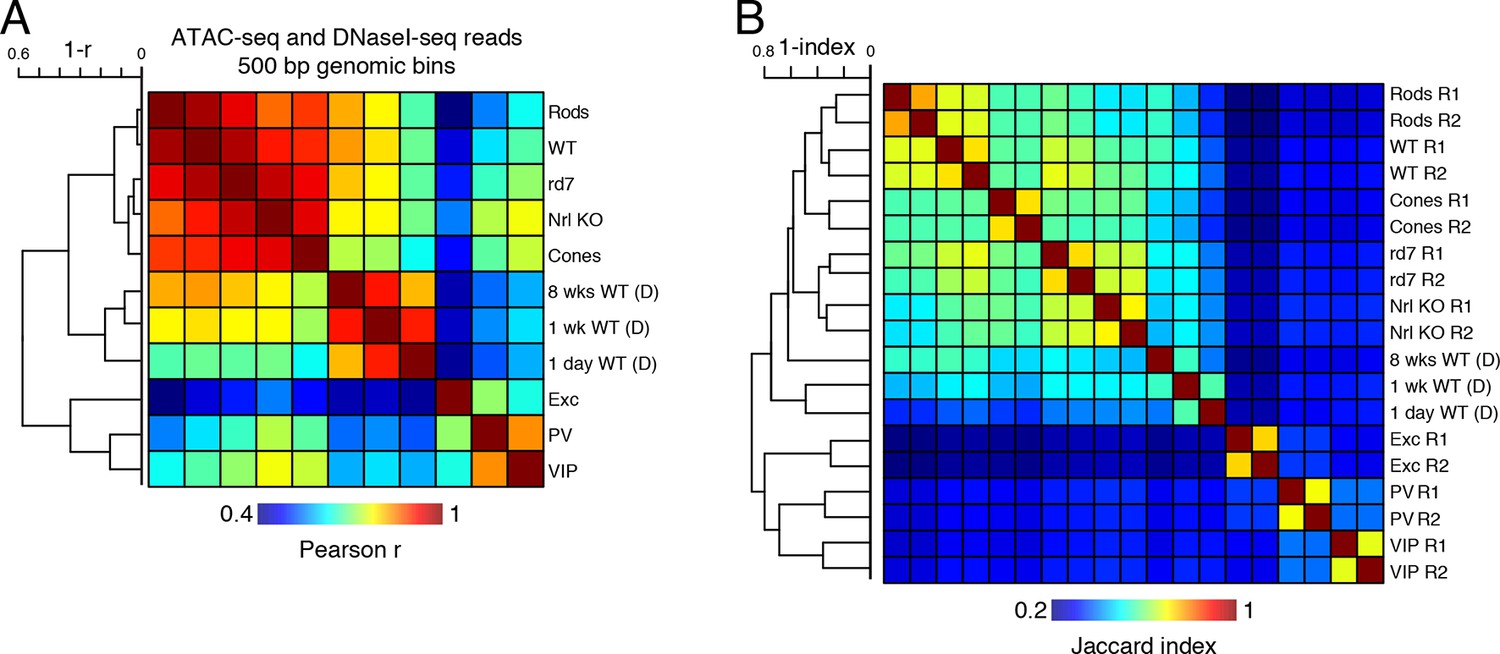
Correlations of accessible chromatin among retinal and cortical samples.
(A) Heatmap showing pairwise Pearson correlation (r) for ATAC-seq and DNaseI-seq (D) read densities in 500 bp genomic bins among retinal and cortical samples. The dendrogram shows hierarchical clustering using 1 - r as the distance measure. (B) Heatmap showing the Jaccard index for ATAC-seq and DNaseI-seq (D) peaks among retinal and cortical samples. The dendrogram shows hierarchical clustering using 1 - Jaccard index as the distance measure. Biological replicates (R1, R2).
Figure 8—figure supplement 2
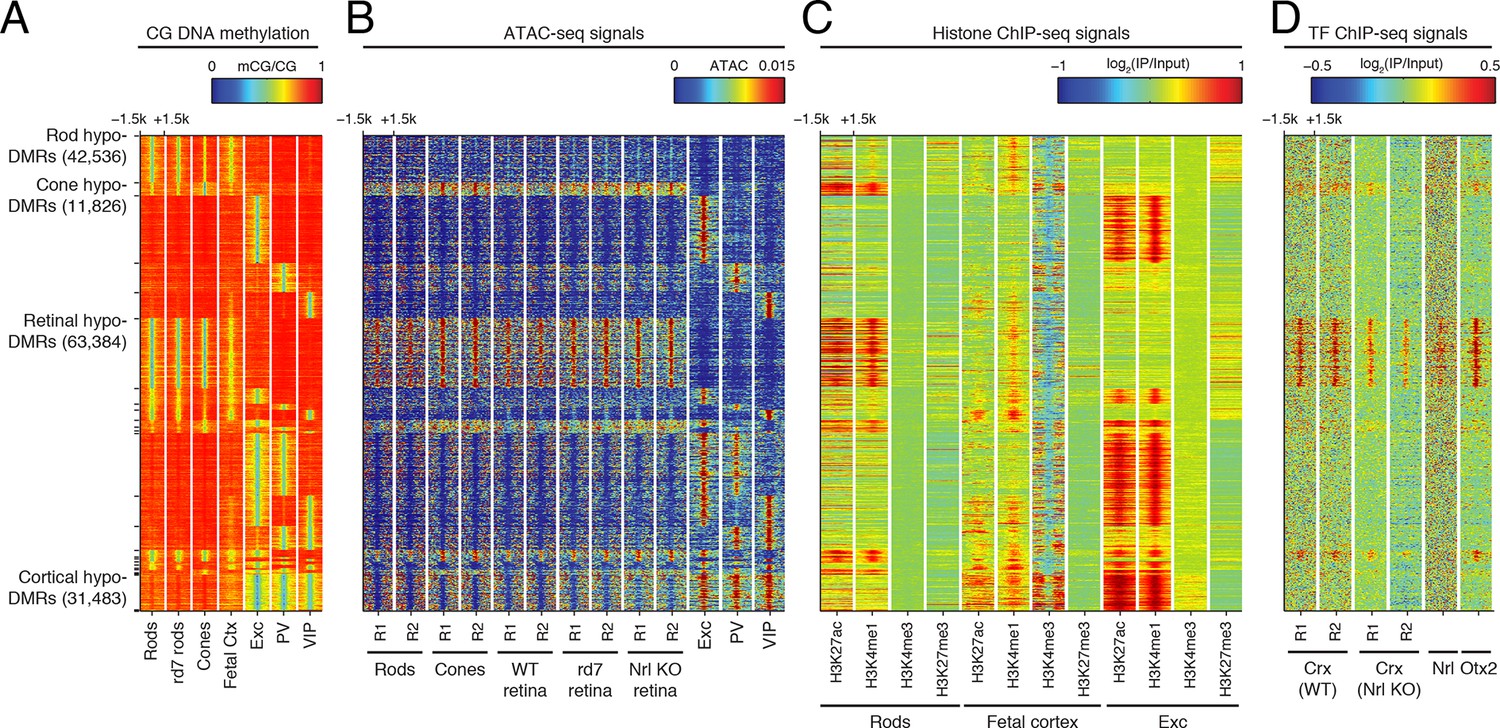
Retinal versus cortical hypo-DMRs.
(A–D) Heatmap showing CG DNA methylation levels at retinal versus cortical hypo-DMRs (A). At the same genomic regions, the following were plotted: ATAC-seq signals (B), histone ChIP-seq signals (C), and retinal TF ChIP-seq signals (D).
Figure 9
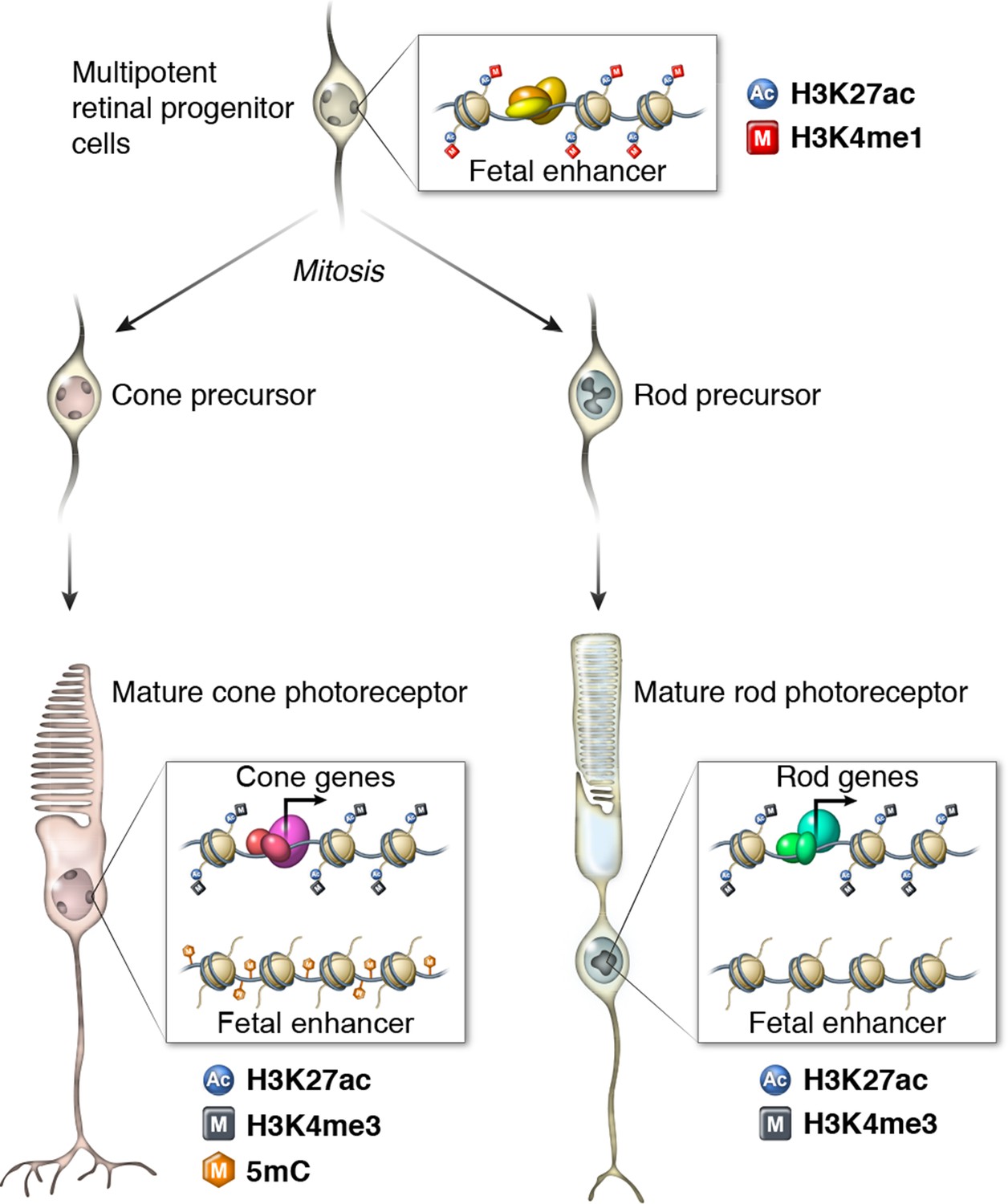
Epigenomic model of rod and cone photoreceptor development.
Enhancers that are active only in progenitor cells (termed 'fetal-only', as the fetal brain was used as a rich source of generic neural progenitors) have low levels of DNA methylation and are enriched for H3K27ac and H3K4me1 histone modifications. In mature cones, histones near fetal-only enhancers lose H3K27ac and H3K4me1 and there is a gain of DNA methylcytosines. In contrast, in mature rods, fetal-only enhancers lose H3K27ac and H3K4me1 but the DNA remains unmethylated, potentially due to the barrier to cytosine methyltransferases posed by their high level of chromatin condensation. In both rods and cones, expressed genes, including rod- and cone-specific photoreceptor genes, have promoters marked by low DNA methylation, high chromatin accessibility, and enrichment for H3K27ac and H3K4me3. Active enhancers are marked by low DNA methylation, high chromatin accessibility, and enrichment for H3K27ac and H3K4me1 (not shown).
Author response image 1

Additional files
-
Supplementary file 1
Characteristics of each sequencing sample
- https://doi.org/10.7554/eLife.11613.028
-
Supplementary file 2
Hypo-methylated features in each sample.
(A-C) UMRs for each methylome sample; (D-F) LMRs for each methylome sample
- https://doi.org/10.7554/eLife.11613.029
-
Supplementary file 3
Accessible chromatin peaks in each sample
(A-J) ATAC-seq peaks for each sample (narrowPeak files from MACS2).
- https://doi.org/10.7554/eLife.11613.030
-
Supplementary file 4
Differentially methylated regions.
(A) Pairwise DSS DMRs between rods and cones, hypo-DMR in rods; (B) Pairwise DSS DMRs between rods and cones, hypo-DMR in cones; (C) Pairwise DSS DMRs between WT rods and rd7 rods, hypo-DMR in WT rods; (D) Pairwise DSS DMRs between WT rods and rd7 rods, hypo-DMR in rd7 rods; (E) Pairwise DSS DMRs between rd7 rods and cones, hypo-DMR in rd7 rods; (F) Pairwise DSS DMRs between rd7 rods and cones, hypo-DMR in cones; (G) DMRs between retina and cortex samples identified using Methylpy. In column D, a 1 denotes the hypo-methylated sample(s), in the following order: neocortical excitatory neurons, PV neurons, and VIP neurons (from Mo et al., 2015), Cones, WT rods, and rd7 rods (this study).
- https://doi.org/10.7554/eLife.11613.031
-
Supplementary file 5
Gene expression data.
(A) RNA abundances (in TPM) for each RNA-seq sample (rods, rd7 rods, and cones from this study; re-analysis of WT retina and Nrl KO retina from Brooks et al., 2011). (B-E) Pairwise lists of differentially expressed genes.
- https://doi.org/10.7554/eLife.11613.032
-
Supplementary file 6
Summary of regions selected for in vivo electroporation
- https://doi.org/10.7554/eLife.11613.033
-
Supplementary file 7
Characteristics of DNA methylation valleys
- https://doi.org/10.7554/eLife.11613.034
-
Supplementary file 8
Rod histone modifications.
(A-D) SICER peaks for H3K27ac (A), H3K4me1 (B), H3K4me3 (C), and H3K27me3 (D) enrichment in WT rods.
- https://doi.org/10.7554/eLife.11613.035
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Epigenomic landscapes of retinal rods and cones
eLife 5:e11613.
https://doi.org/10.7554/eLife.11613




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}