Tracking interspecies transmission and long-term evolution of an ancient retrovirus using the genomes of modern mammals
- Boston College, United States
Figures
Figure 1

Schematic representation of the major features of ERV-Fc proviruses.
The region colored in blue indicates gag, brown indicates pol, and yellow represents env coding regions. The gray-colored regions indicate the two long terminal repeat (LTR) regions. Vertical lines within these regions indicate where proteolytic cleavage would occur between protein subunits. The identity of these subunits is indicated below the schematic: MA = matrix, CA = capsid, NC = nucleocapsid, PR = protease, RT = reverse transcriptase, IN = integrase, SU = surface, TM = transmembrane, PPT = polypurine tract. The probable location of the viral RNA packaging motif is indicated by ψ. At the termini of the retroviral LTR sequences is shown the canonical TG/CA dinucleotides as well as the 5 nucleotide target site duplications (TSDs) flanking the provirus. ERV, endogenous retrovirus.
Figure 2
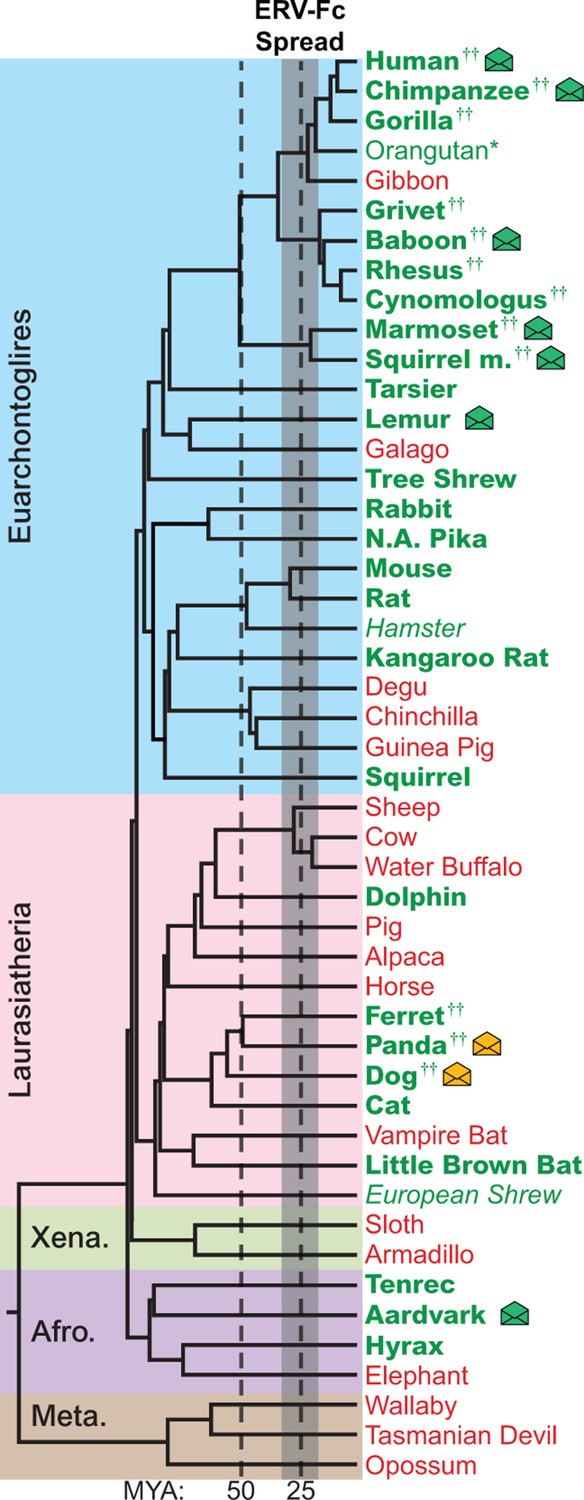
The genomes of most Eutherian mammals harbor ERV-Fc.
A mammalian phylogeny (adapted from [(Bininda-Emonds et al., 2007]) including species whose genomes were examined for the presence of ERV-Fc. Species lacking ERV-Fc are depicted in red, while those found to harbor ERV-Fc are depicted in green. Bold font indicates that coding potential in one or more gene regions could be reconstructed, italics indicates that ERV-Fc fragments were identified but coding potential could not be reconstructed; * indicates that only a solo LTR was identified; and †† indicates that a species harbors two genetically distinct ERV-Fc lineages. Background shading indicates higher-order taxonomic relationships: blue = Euarchontoglires, pink = Laurasiatheria, green = Xenarthra, purple = Afrotheria, brown = Metatheria. Envelope icons indicate species in which ERV-Fc env open reading frame(s) were identified, and the icons colored green indicate env with homology to HERV-Fc; yellow icons indicate the env had greater similarity to HERV-W. ERV, endogenous retrovirus. HERV, human ERV.
-
Figure 2—source data 1
Genome sequence database summary.
List of the genome database builds, fold coverage, and method of sequence acquisition for all species included in this study.
- https://doi.org/10.7554/eLife.12704.005
-
Figure 2—source data 2
Overview of recovered ERV-Fc sequences.
Summary of ERV-Fc lineages identified, our designations for these lineages, which viral sequences were recovered, and correlation to their RepBase designation (if applicable). Also included is basic taxonomic information about the hosts from which these viral sequences were retrieved. ERV, endogenous retrovirus.
- https://doi.org/10.7554/eLife.12704.006
-
Figure 2—source data 3
Sequences of ERV-Fc primer binding sites.
ERV, endogenous retrovirus
- https://doi.org/10.7554/eLife.12704.007
Figure 3 with 2 supplements
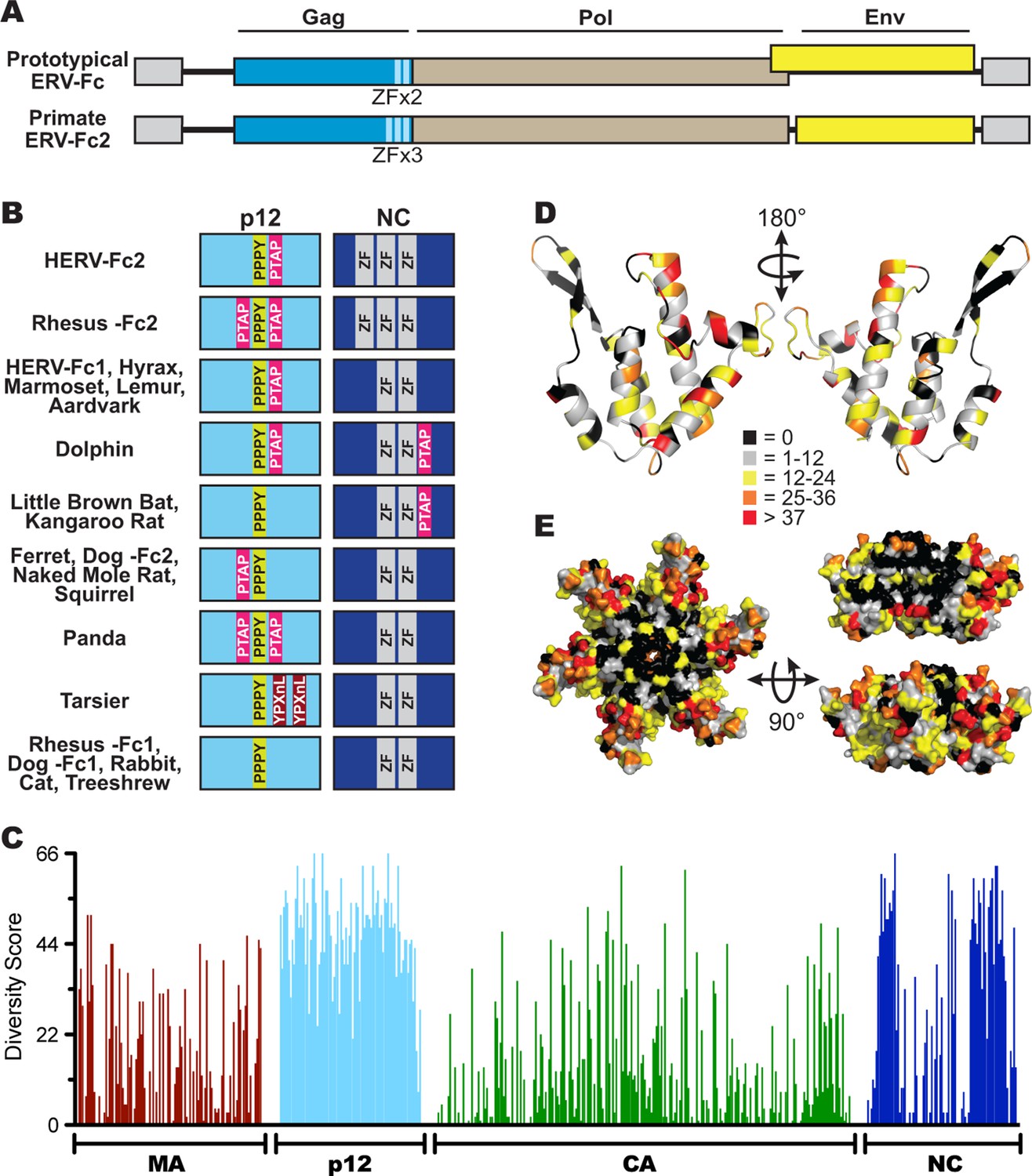
Sequence diversity in ERV-Fc is consistent with an extended period of exogenous replication.
(A) Structures of ERV-Fc genomes with blue, brown, and yellow boxes indicating gag, pol, and env coding sequences, respectively. Light blue regions indicate the multiple zinc finger motifs in the NC subunit. (B) Organization of late domains (PPPY, PTAP, YPXnL) and zinc finger domains within the p12 and NC subunits of ERV-Fc Gag, respectively. (C) Plot of amino acid diversity across ERV-Fc Gag with the diversity score calculated by summation of 'match scores' (Smith and Smith, 1990) in pairwise comparisons of ERV-Fc sequences to a global consensus sequence. (D) Ribbon diagram of ERV-Fc consensus monomeric CA model, with the residues highlighted according to their diversity score. (E) Surface view of hexameric ERV-Fc consensus N-terminal CA domain model: left = view of cytoplasmic exposed surface; top right = cross-sectional view through hexamer, with three monomers removed; bottom right = surface view of monomers available for interhexamer interactions. In both right panels, the figure is oriented such that the cytoplasmic exposed surface of CA is at the top.
Figure 3—figure supplement 1
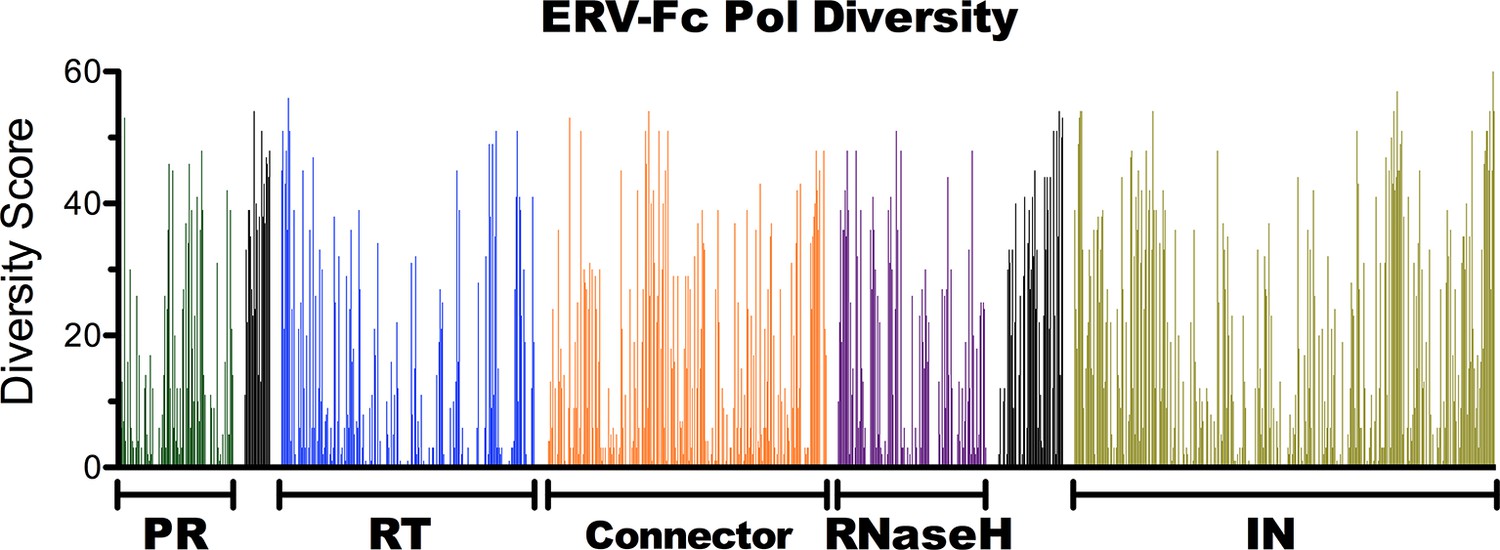
Amino acid diversity in ERV-Fc Pol.
Plot of amino acid diversity across ERV-Fc Pol with the diversity score calculated by summation of 'match scores' [Smith and Smith, 1990] in pairwise comparisons of ERV-Fc sequences to a global consensus sequence.
Figure 3—figure supplement 2

Amino acid diversity in ERV-Fc Env.
Plot of amino acid diversity across ERV-Fc Env with the diversity score calculated by summation of 'match scores' [Smith and Smith, 1990] in pairwise comparisons of ERV-Fc sequences to a global consensus sequence.
Figure 4 with 1 supplement
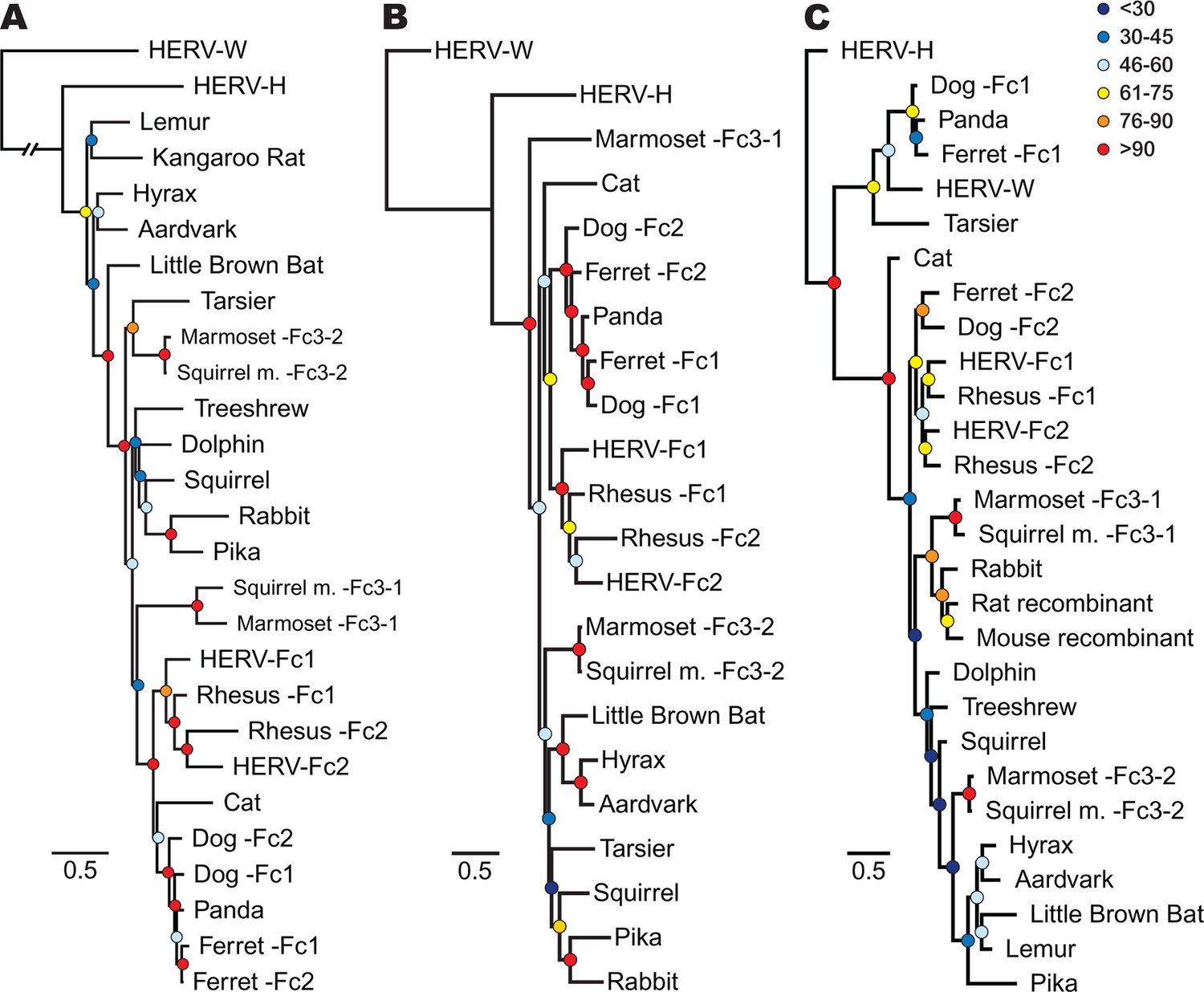
Phylogenetic relationship between ERV-Fc sequences.
Maximum likelihood amino acid trees of (A) Gag (B) Pol and (C) TM generated using the LG substitution matrix. In each panel, HERV-H and HERV-W sequences were included as outgroups. Boostrap confidence values of nodes are depicted by colored spheres. In order to save space, a distance of approximately 0.6 was removed from the HERV-W outgroup branch in the Gag phylogeny (A), as indicated by the broken line.
-
Figure 4—source data 1
Full-length ERV-Fc Gag alignment.
The phylogeny shown in Figure 4A is based on this alignment excluding the p12 region (as described in the Materials and methods).
- https://doi.org/10.7554/eLife.12704.012
-
Figure 4—source data 2
ERV-Fc CA alignment.
- https://doi.org/10.7554/eLife.12704.013
-
Figure 4—source data 3
Full-length ERV-Fc Pol alignment.
The phylogeny shown in Figure 4B is based on this alignment.
- https://doi.org/10.7554/eLife.12704.014
-
Figure 4—source data 4
ERV-Fc RT alignment.
- https://doi.org/10.7554/eLife.12704.015
-
Figure 4—source data 5
Full-length ERV-Fc Env sequences, including all recovered open reading frames.
- https://doi.org/10.7554/eLife.12704.016
-
Figure 4—source data 6
ERV-Fc TM alignment.
The phylogeny shown in Figure 4C is based on this alignment.
- https://doi.org/10.7554/eLife.12704.017
-
Figure 4—source data 7
Alignment of ERV-Fc Pol including both inferred and strict consensus sequences.
The phylogeny shown in Figure 4—figure supplement 1 is derived from this alignment.
- https://doi.org/10.7554/eLife.12704.018
Figure 4—figure supplement 1
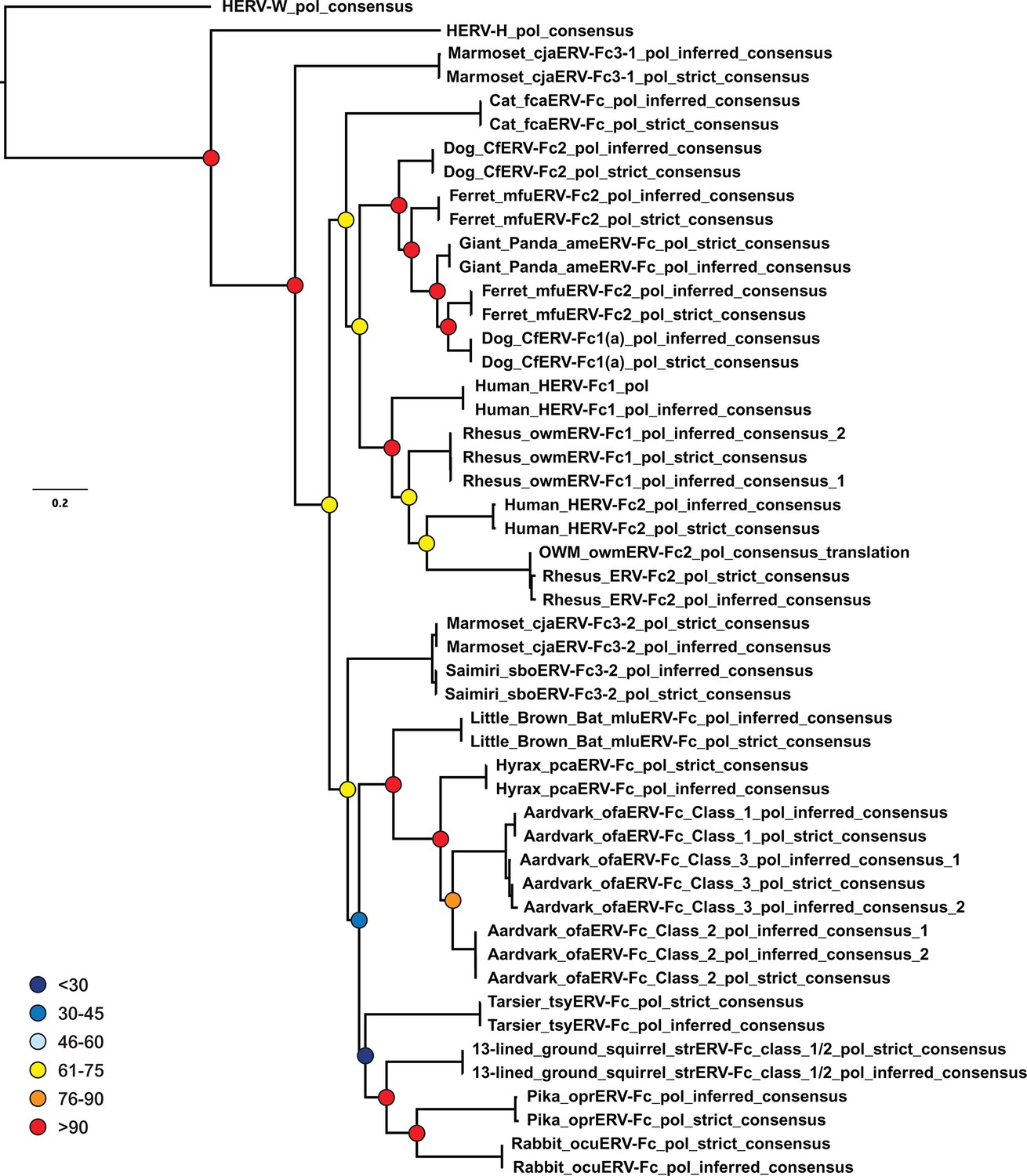
Inferences made in deriving ERV-Fc consensus sequences do not significantly affect phylogenetic relationships.
A ML phylogeny, generated via RAxML, comparing the relationship between our inferred Pol sequences and strict consensus sequences. HERV-H and HERV-W sequences were included as outgroups. Boostrap confidence values of nodes are depicted by colored spheres. HERV, human endogenous retrovirus; ML, maximum likelihood.
Figure 5 with 1 supplement
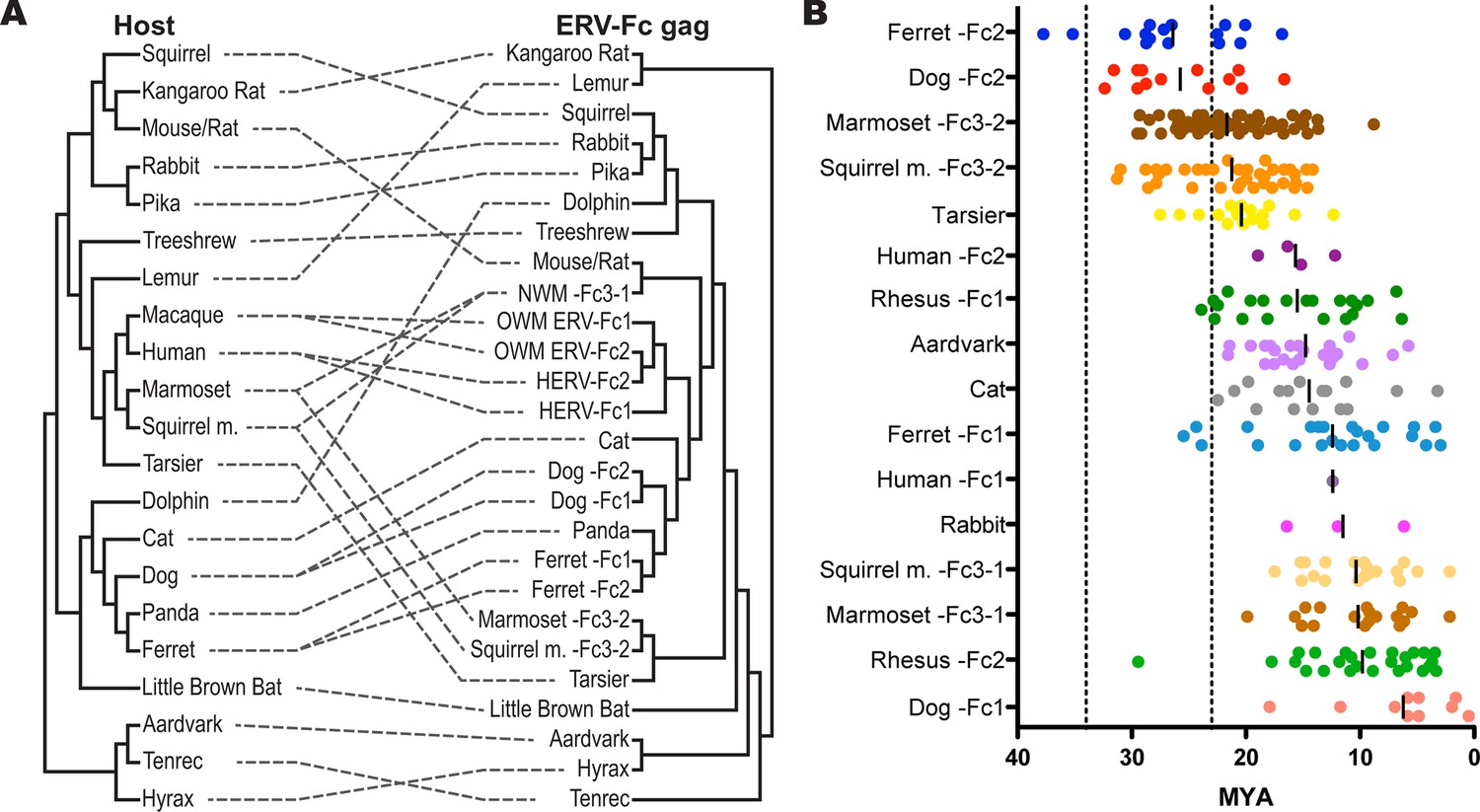
ERV-Fc has a multimillion-year history of replication with multiple cross-species transmissions.
(A) Tanglegram comparison of host (left) and ERV-Fc phylogenies (right); dashed lines match species and the ERV-Fc found within their genome. The host phylogeny was adapted from (Bininda-Emonds et al., 2007), while the ERV-Fc phylogeny is a supertree generated using Matrix Representation Parsimony (MRP) based on CA and Gag amino acid phylogenies. (B) LTR-derived age estimates of ERV-Fc loci derived by applying a neutral evolution rate of 4.5×10-9 substitutions per site per year to the nucleotide divergence between the 5’ and 3’ LTRs. Each plotted point represents the age estimate of a single genomic locus. Loci that show clear signatures of gene conversion or recombination have been omitted from this analysis. The average age is indicated by black vertical lines. Dotted lines indicate the approximate boundaries of the Oligocene epoch (~33.9 to ~23 MYA). ERV, endogenous retrovirus.
Figure 5—figure supplement 1
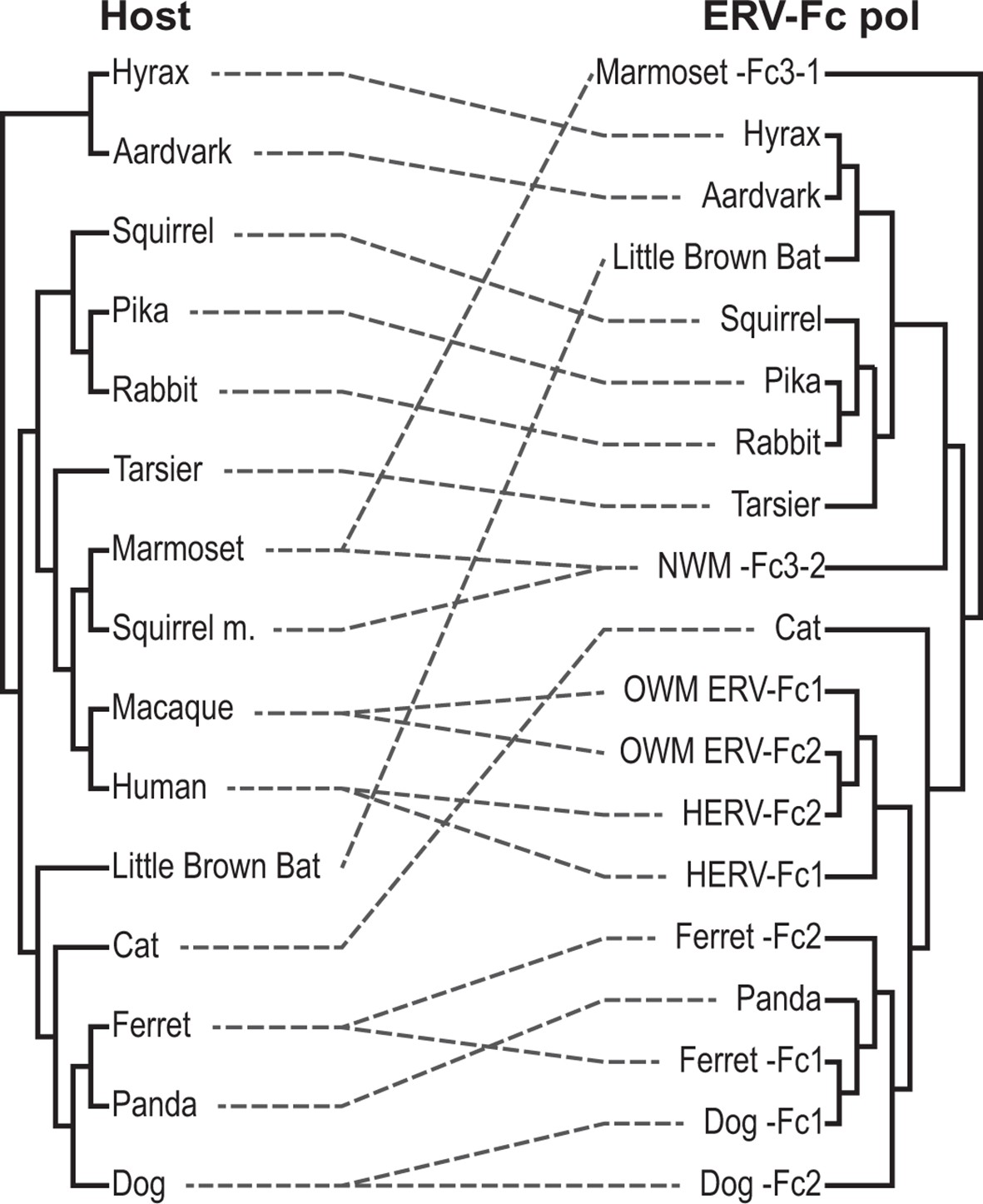
Tanglegram comparison of host (left) and ERV-Fc phylogenies (right).
The host phylogeny was adapted from (Bininda-Emonds et al., 2007). The ERV-Fc phylogeny is the ERV-Fc Pol tree shown in Figure 4B. Dashed lines match ERV-Fc lineages and the species from which they were identified. ERV, endogenous retrovirus.
Figure 6
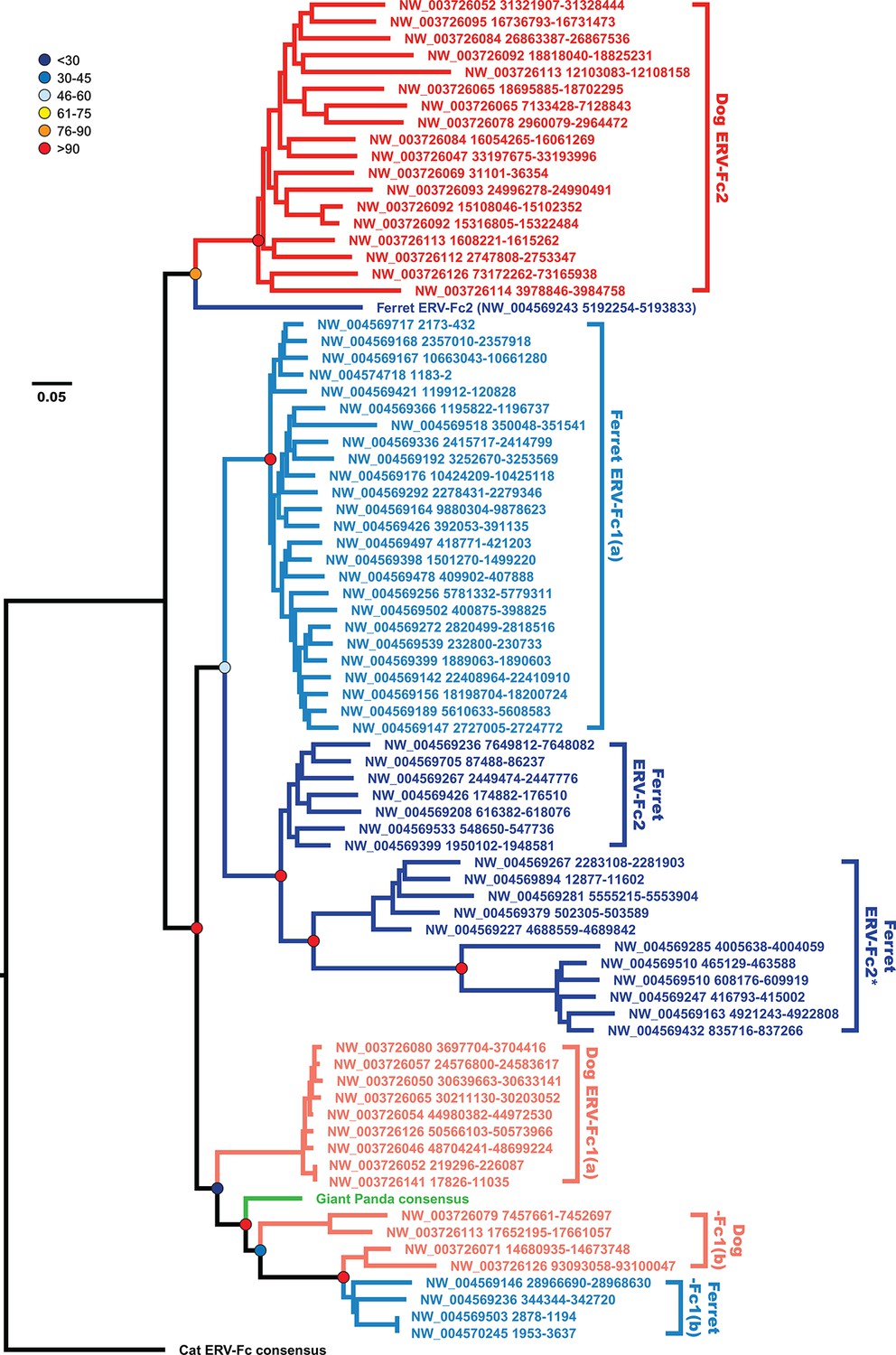
The evolutionary history of carnivore ERV-Fc1 includes numerous cross-species transmission events and at least one recombination event.
Maximum likelihood phylogenetic analysis of carnivore ERV-Fc gag nucleotide sequences: sequences from the dog genome are colored in a shade of red, those from the ferret genome are colored in a shade of blue, and the panda consensus gag is colored in green. The feline ERV-Fc consensus gag sequence has been included as an outgroup and is colored black. For sequences from the dog and ferret genomes, the darker colored taxa are ERV-Fc2 sequences (defined based on their association with an ERV-Fc envelope sequence), while the lighter colored taxa are ERV-Fc1 sequences (defined by an association with ERV-W envelope sequence). Lineages where a large portion of the gag sequence has been replaced with heterologous non-coding sequence is denoted by * in the name. Boostrap confidence values of ancestral nodes are depicted by colored spheres. ERV, endogenous retrovirus.
-
Figure 6—source data 1
Nucleotide alignment of carnivore ERV-Fc gag sequences.
The phylogeny shown in Figure 6 is derived from this alignment.
- https://doi.org/10.7554/eLife.12704.023
Figure 7
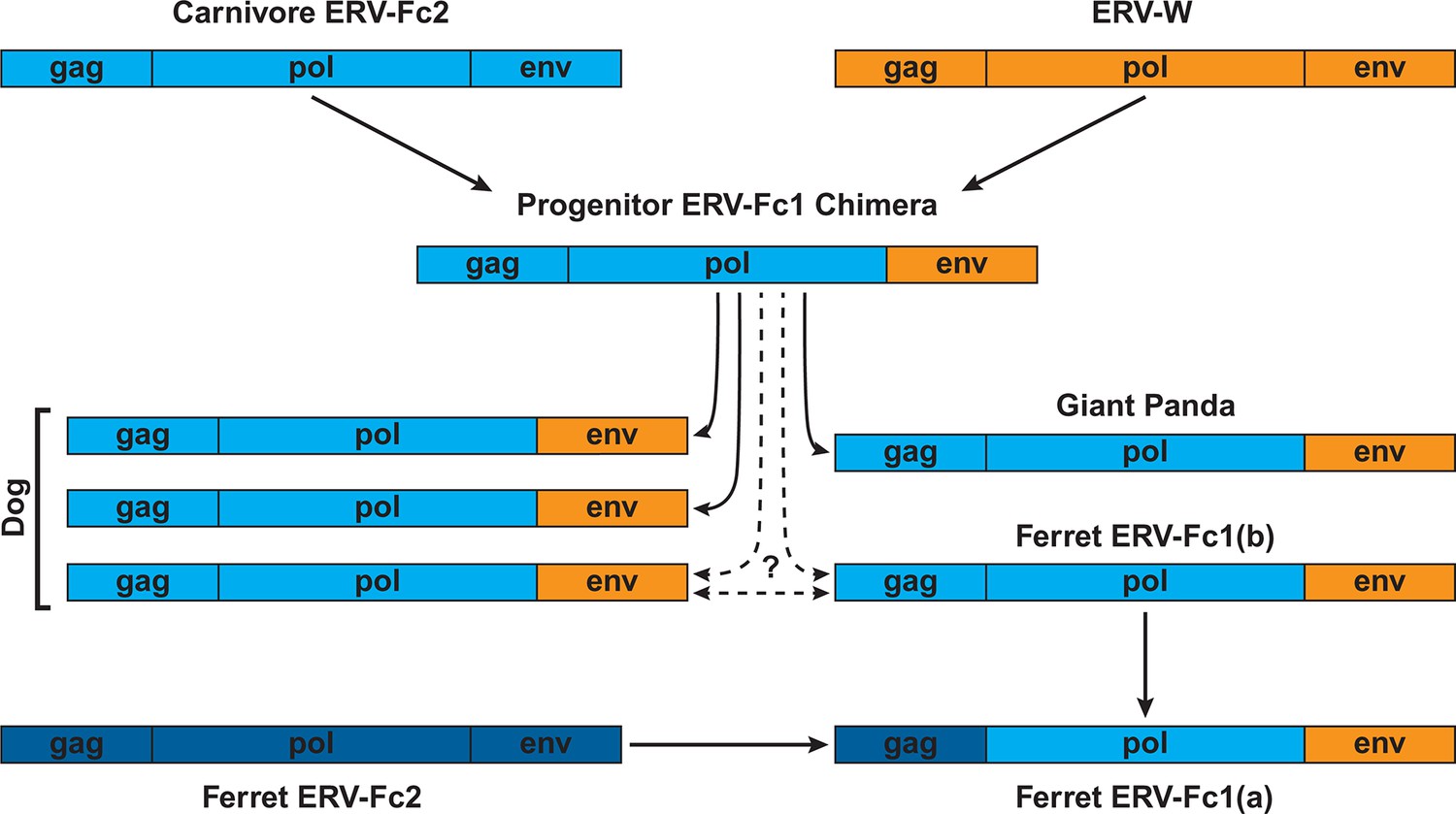
Proposed recombination and transmission sequence involving carnivore ERV-Fc1.
ERV-Fc sequences are depicted in blue, while ERV-W sequences are depicted in orange. See text for a detailed explanation of the arrows.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Tracking interspecies transmission and long-term evolution of an ancient retrovirus using the genomes of modern mammals
eLife 5:e12704.
https://doi.org/10.7554/eLife.12704




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}