Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling
- Stanford University, United States
- University of Chicago, United States
- Howard Hughes Medical Institute, Stanford University, United States
Figures
Figure 1 with 7 supplements
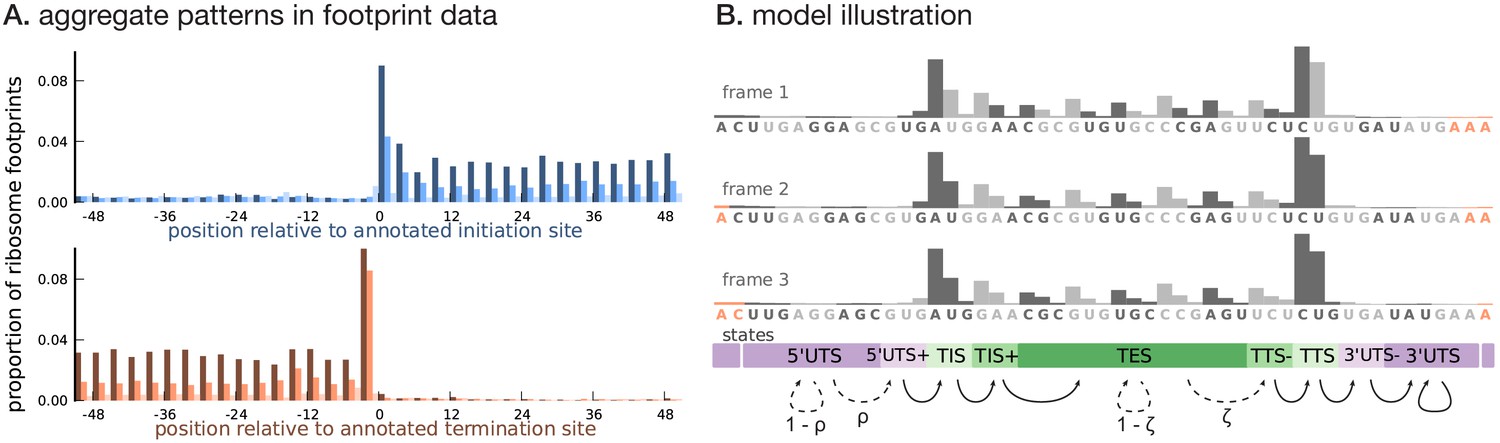
Illustrating the model.
(A) Proportion of footprint counts aggregated across 1000 highly expressed annotated coding transcripts, centered at their translation initiation (blue) and termination (orange) sites. In aggregate, RPF count data have higher abundance within the CDS than the UTRs and exhibit a 3-base periodicity within the CDS. (B) Each transcript belongs to one of three unobserved reading frames, and is represented as a sequence of base-triplets (highlighted by differing shades of gray) that depends on the reading frame. Each triplet belongs to one of nine unobserved states. The state sequence shown corresponds to frame 3 and varying shades from purple to green highlight the different states. Base positions marked in orange are modeled independently and always belong to the relevant UTS state. Transitions with nonzero probabilities are indicated by arrows, with solid arrows denoting a probability of 1 and dotted arrows denoting probabilities that are a function of the underlying sequence.
Figure 1—figure supplement 1
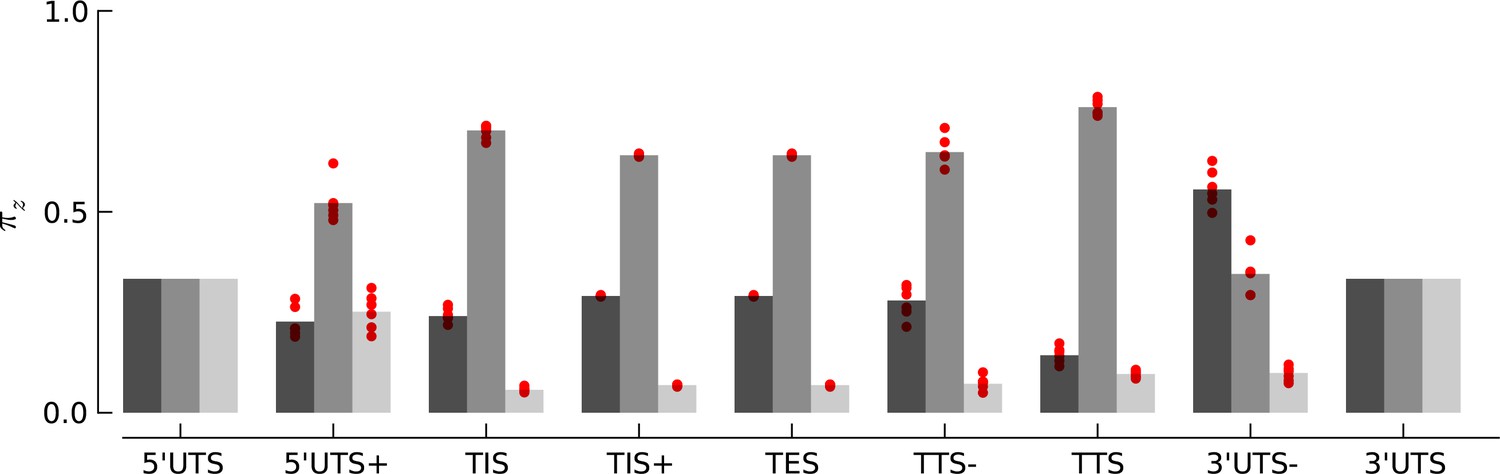
Robustness of periodicity parameter estimates.
Bar plots of the periodicity parameters , for 29 bp long footprints, estimated using the top 5000 expressed genes. Each red circle indicates the parameter values estimated by using a random set of 1000 genes.
Figure 1—figure supplement 2
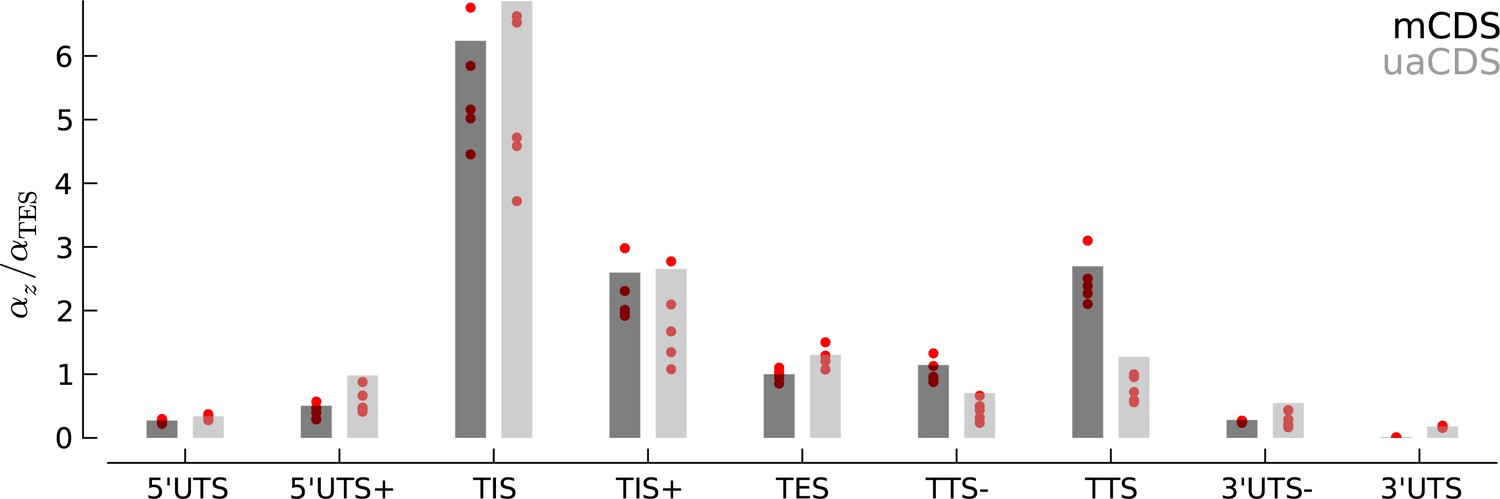
Robustness of occupancy parameter estimates.
Bar plots of the maximum likelihood estimates of the occupancy parameters in each state, (scaled by for mCDS). For mCDS (dark gray), the parameters were estimated using the top 5000 expressed genes, and the red circles indicate parameter values estimated using a random set of 1000 genes. For uaCDS (light gray), the parameters were estimated using a random set of 5000 upstream-restricted transcripts, and each red circle indicates values estimated using random sets of 1000 upstream-restricted transcripts.
Figure 1—figure supplement 3

Decision rules to identify matches and mismatches of mCDS to annotation.
Illustrating the decisions by which mCDS inferred on each transcript are identified as an exact match, a frame match) or a mismatch to annotated coding sequences. Matches and mismatches are only defined when the transcript is annotated by GENCODE as coding and the classification depends on agreement with the annotated CDS. A gene is considered to have an exact (or frame) match if at least one of its isoforms is labeled an exact (or frame) match. In all other cases, the inference for the coding gene is considered a mismatch.
Figure 1—figure supplement 4

Model accuracy.
Quantifying model accuracy in terms of the fraction of previously annotated CDSs accurately recovered as a function of total footprint sequencing depth. We performed the entire analysis on the same set of assembled transcripts (parameter estimation and inference) after subsampling the data. Starting with inferences using the complete footprint data set that exactly match annotated CDSs (top subpanel), we show the fraction of these CDSs that were accurately (blue) and inaccurately (brown) recovered with a high posterior for varying sequencing depths. Starting with inferences that only match the frame of annotated CDSs (bottom subpanel), we show the fraction of accurately and inaccurately recovered CDSs for varying sequencing depths.
Figure 1—figure supplement 5
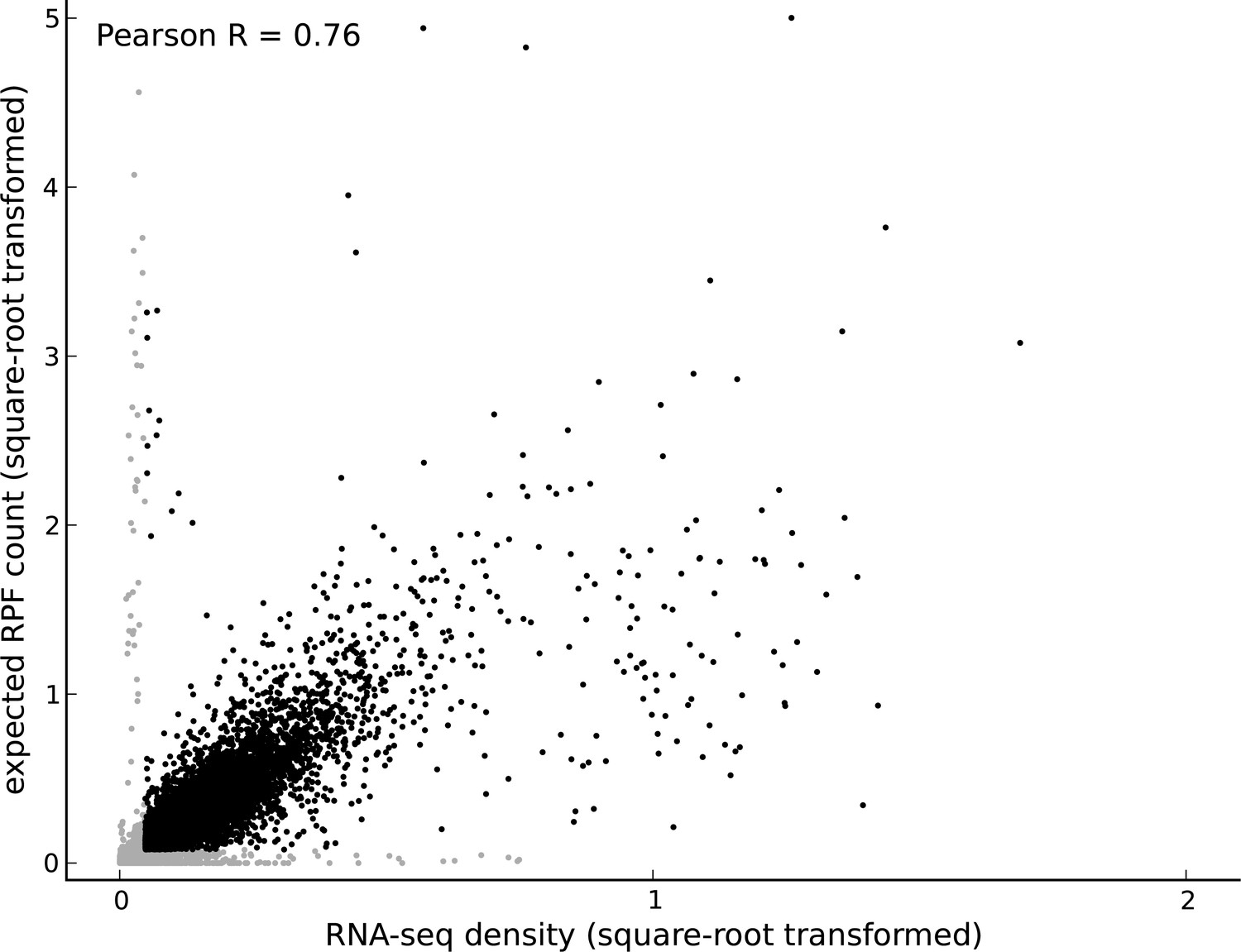
Comparing footprint abundance and gene expression.
Scatter plot of the RNA-seq density (reads per base per million sequenced reads) within annotated coding transcripts vs. the expected ribosome footprint count (normalized by sequencing depth) in triplets within the CDS of the transcripts. The top 50th percentile of expressed genes is shown in black and the bottom 50th percentile is shown in gray. For highly expressed genes, it is reasonable to assume that the expected footprint count in a triplet scales linearly with RNA-seq density. However, for lowly expressed genes and outlier genes (i.e., genes with high expected footprint count and low RNA-seq density, and genes with high RNA-seq density and low expected footprint count), this assumption may not be valid.
Figure 1—figure supplement 6
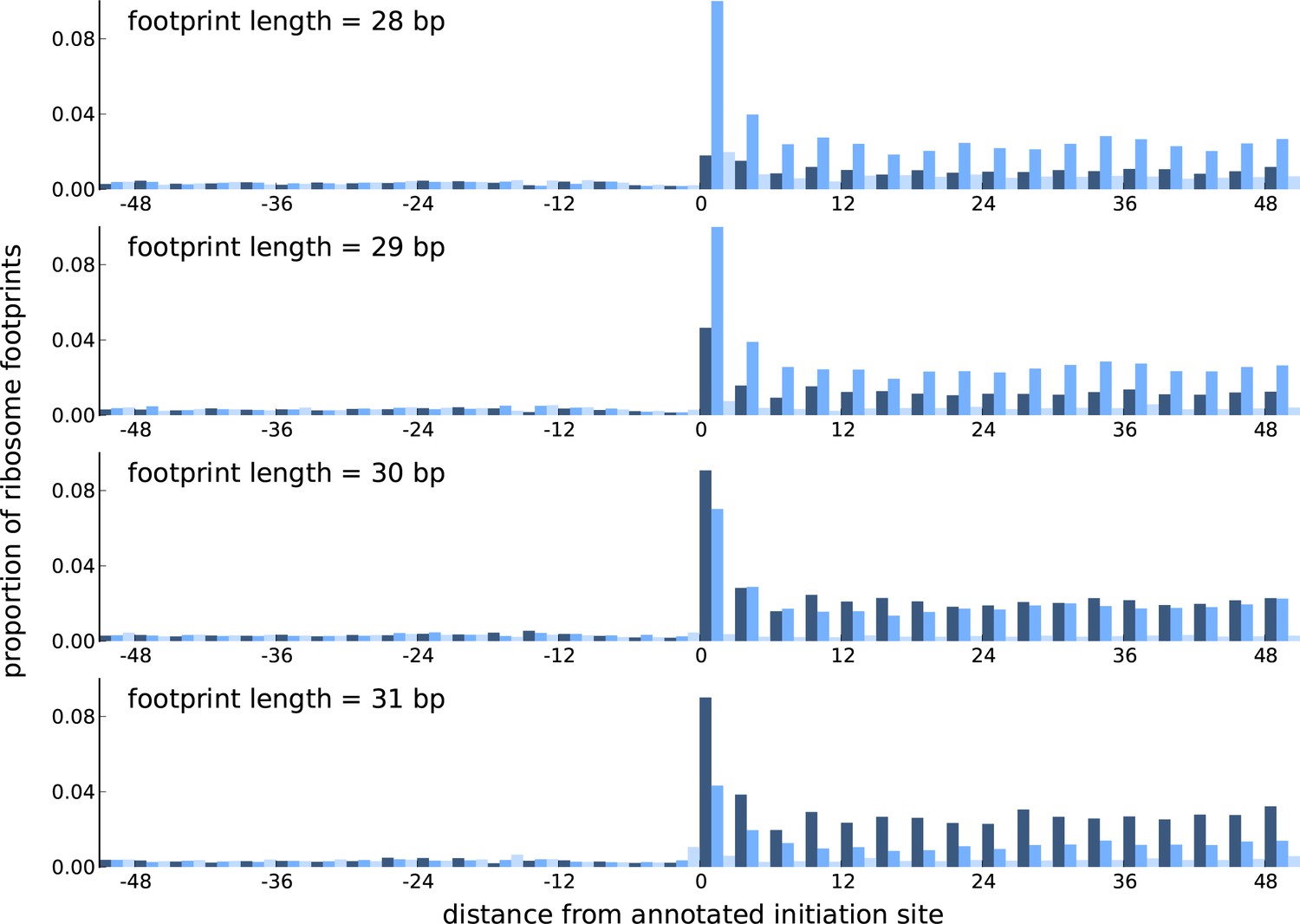
Comparing the periodicity in ribosome footprint counts for footprints of different lengths.
Proportion of footprint counts aggregated across 1000 highly expressed annotated coding transcripts, centered at their translation initiation sites. Each subplot illustrates the proportions for footprints of a given length, with the periodicity in the proportions showing distinct features between footprints of different lengths.
Figure 1—figure supplement 7
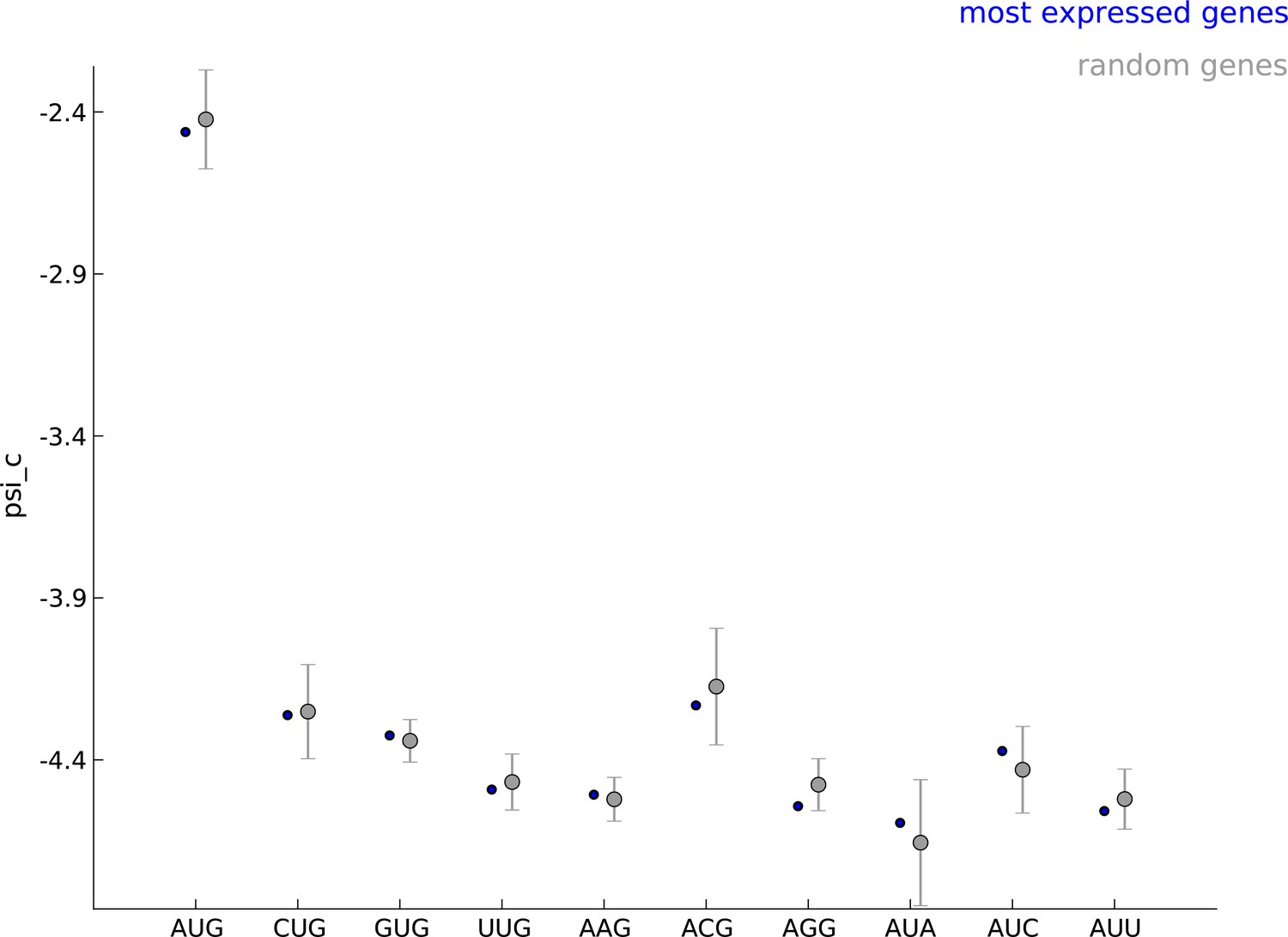
Robustness of parameters for start codon usage to choice of learning set.
The estimated values of the parameter when using the 5000 most expressed genes (blue) and random sets of 5000 genes (gray) show that start codon usage is robust to the choice of the learning set.
Figure 2 with 1 supplement
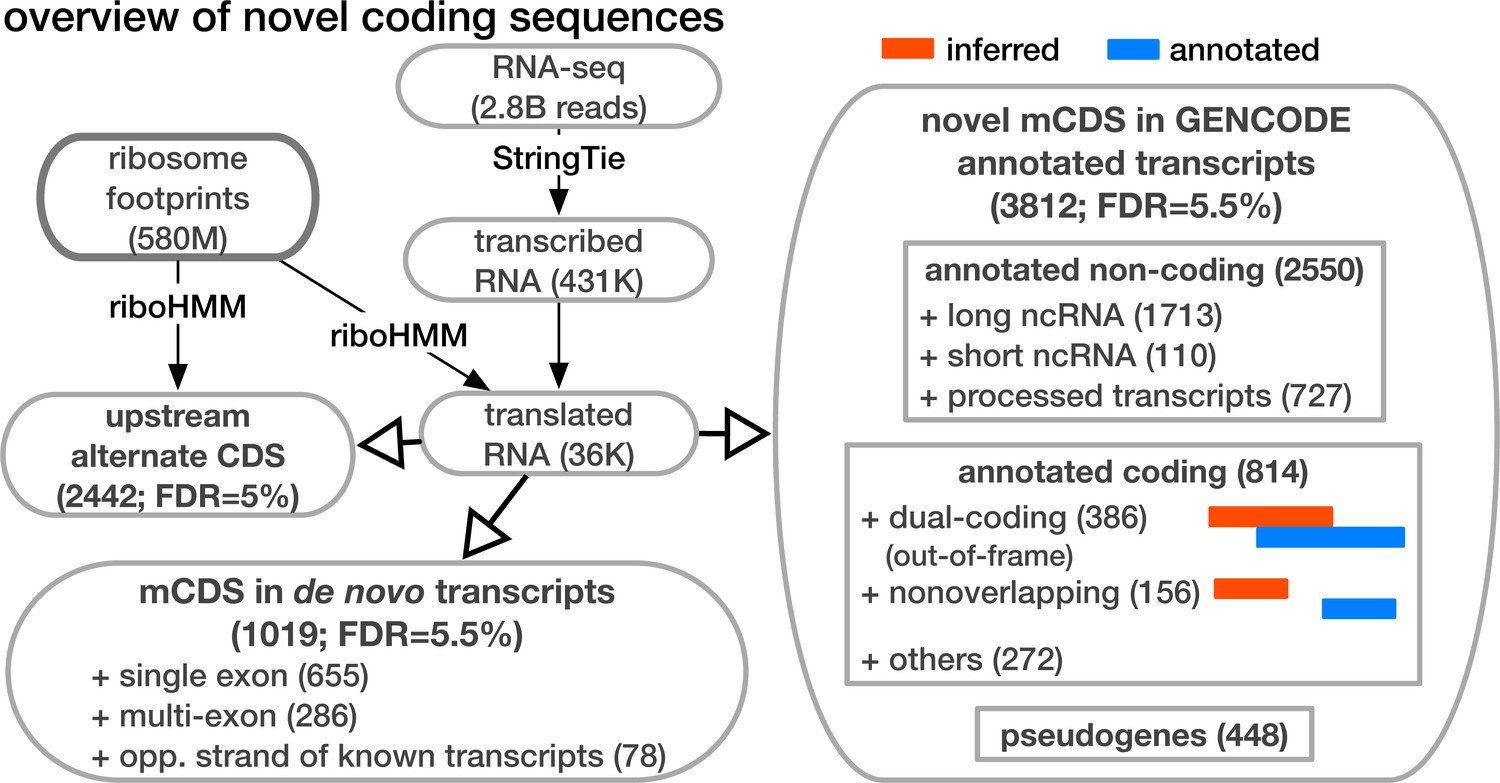
Overview of novel coding sequences.
The analysis workflow indicates the size of the data (in read/footprint depth, or number of transcripts) at each step and the numbers and classes of transcript within which novel translated sequences were identified. Transcripts assembled by StringTie that do not overlap any annotated gene are called 'novel transcripts'. Long non-coding RNA includes lincRNAs, antisense transcripts and transcripts with retained introns, short non-coding RNA includes snRNA, snoRNA and miRNA, processed transcripts are transcripts without a long, canonical ORF, and pseudogenes include all subclasses of such genes annotated by GENCODE.
Figure 2—figure supplement 1
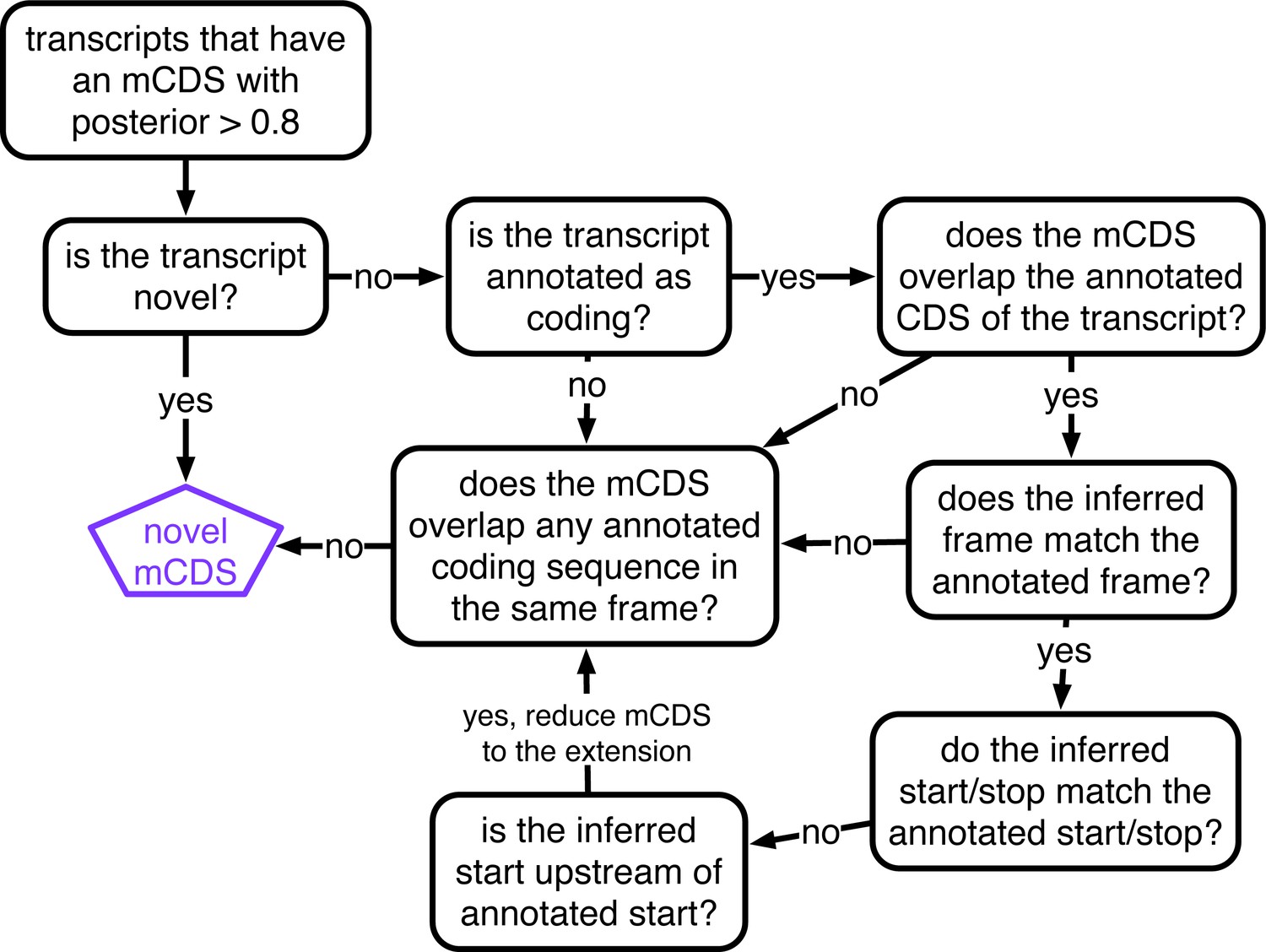
Decision rules to identify novel mCDS.
Illustrating the decisions that identify novel mCDS. The mCDS that do not overlap any known CDS (pooling GENCODE, UCSC and CCDS annotations) are labeled as novel mCDS -- these include mCDS from both novel and annotated transcripts.
Figure 3 with 4 supplements
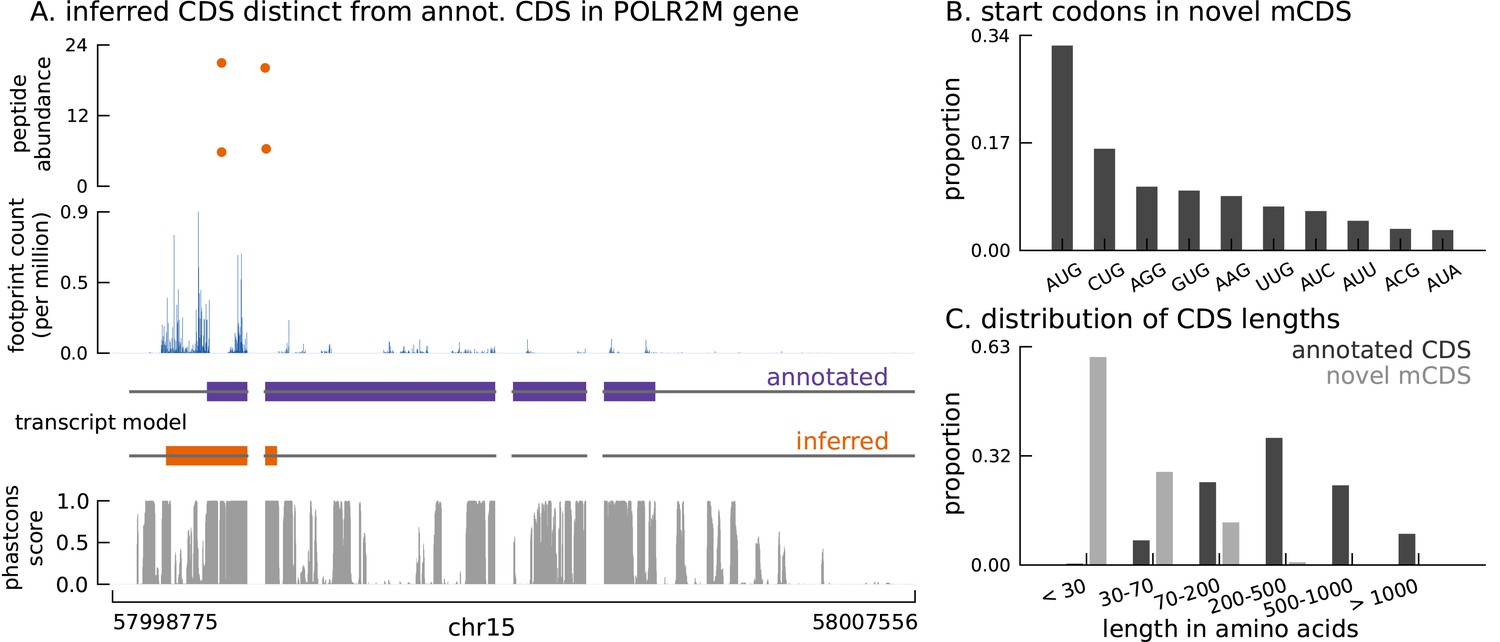
Thousands of novel translated sequences identified in annotated and novel transcript isoforms.
(A) The inferred CDS for an isoform of the POLR2M gene overlaps its annotated CDS and is in a different frame. All four distinct peptides uniquely mapping to this gene match the inferred CDS (protein-level posterior error probability = 3 × 10−35). (The introns and the last exon have been shortened for better visualization.) (B) Distribution of start codon usage across all novel mCDS. (C) Distribution of the lengths of the novel mCDS (gray) compared with the lengths of GENCODE annotated CDSs (black).
Figure 3—figure supplement 1
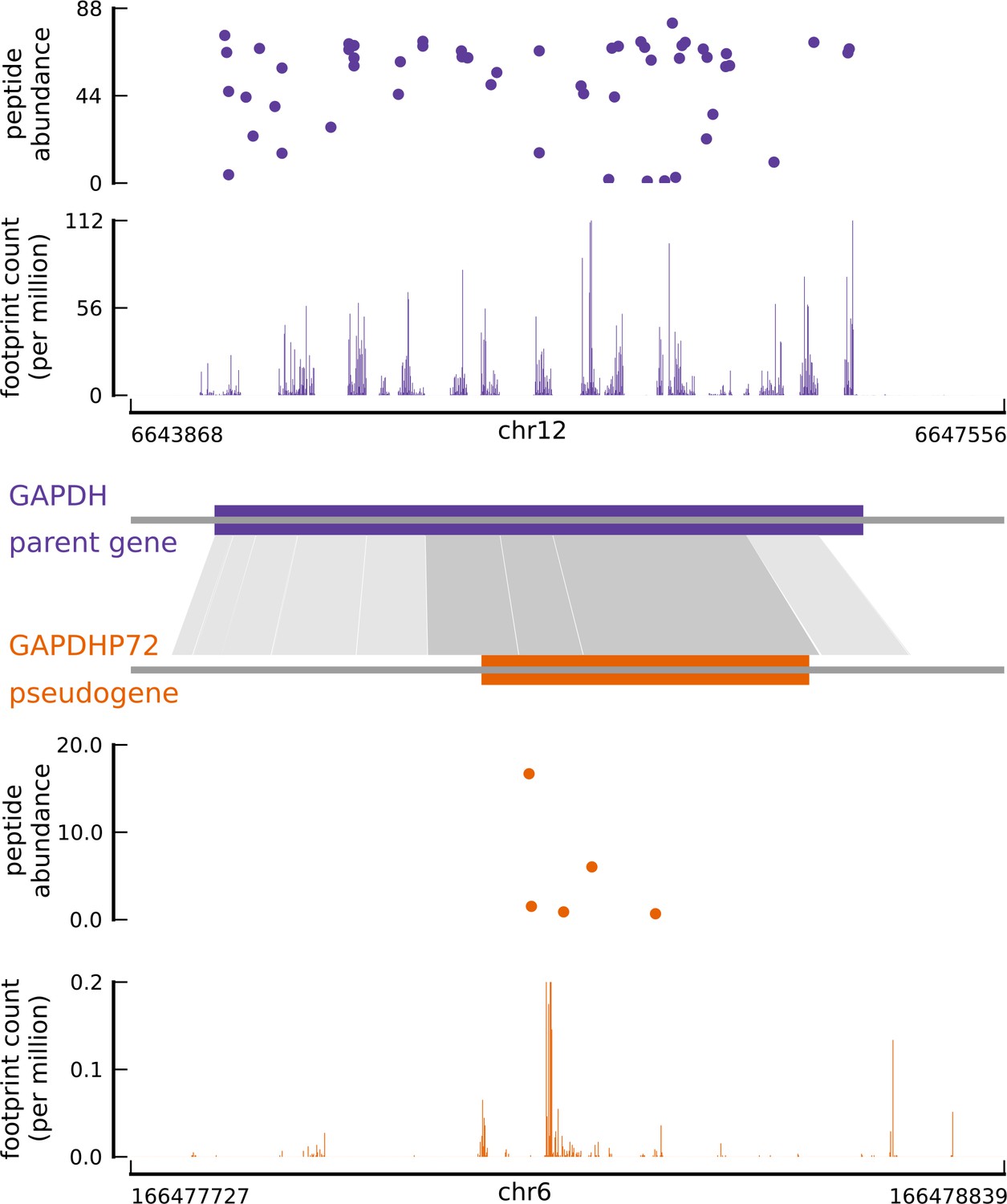
Translated coding sequences identified in hundreds of pseudogenes.
Comparing the inferred CDS in the pseudogene GAPDHP72 (orange) with the annotated CDS (purple) of its parent gene GAPDH (introns have been removed for better visualization). Gray shaded boxes show the alignment between the pseudogene and parent gene transcripts, and dark shaded boxes indicate that the inferred coding frame of the pseudogene matches that of the parent gene. Although the pseudogene and parent gene share coding frames, the underlying sequences are sufficiently different.
Figure 3—figure supplement 2
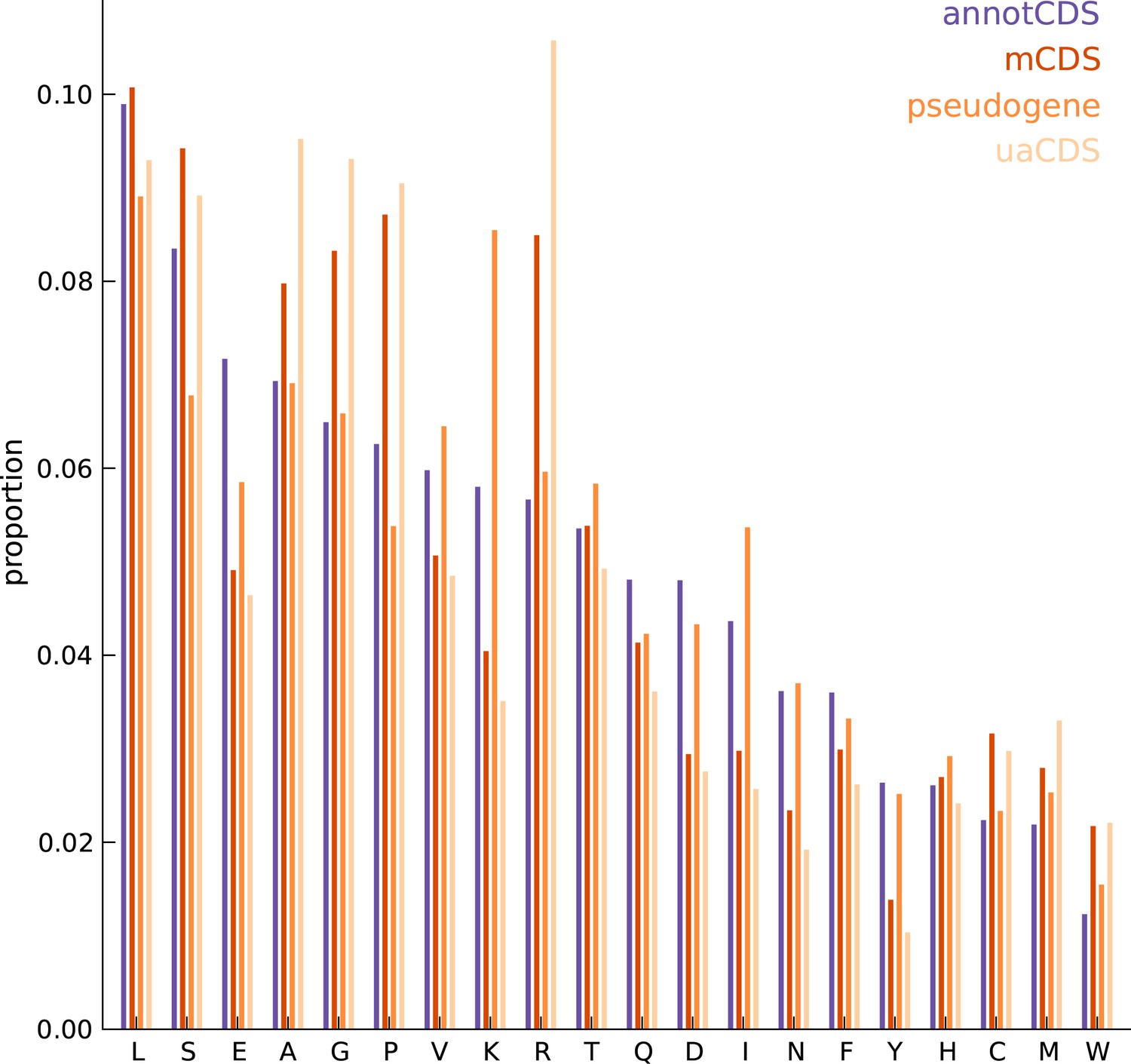
Comparing the amino acid content between annotated and novel CDS.
The overall amino acid content is largely similar between annotated CDSs and novel CDSs (mCDS, pseudogenes and uaCDS), with a substantially higher proportion of lysine and isoleucine residues in CDSs within pseudogenes, and a higher proportion of alanine, arginine, glycine, and proline residues within mCDS and uaCDS.
Figure 3—figure supplement 3
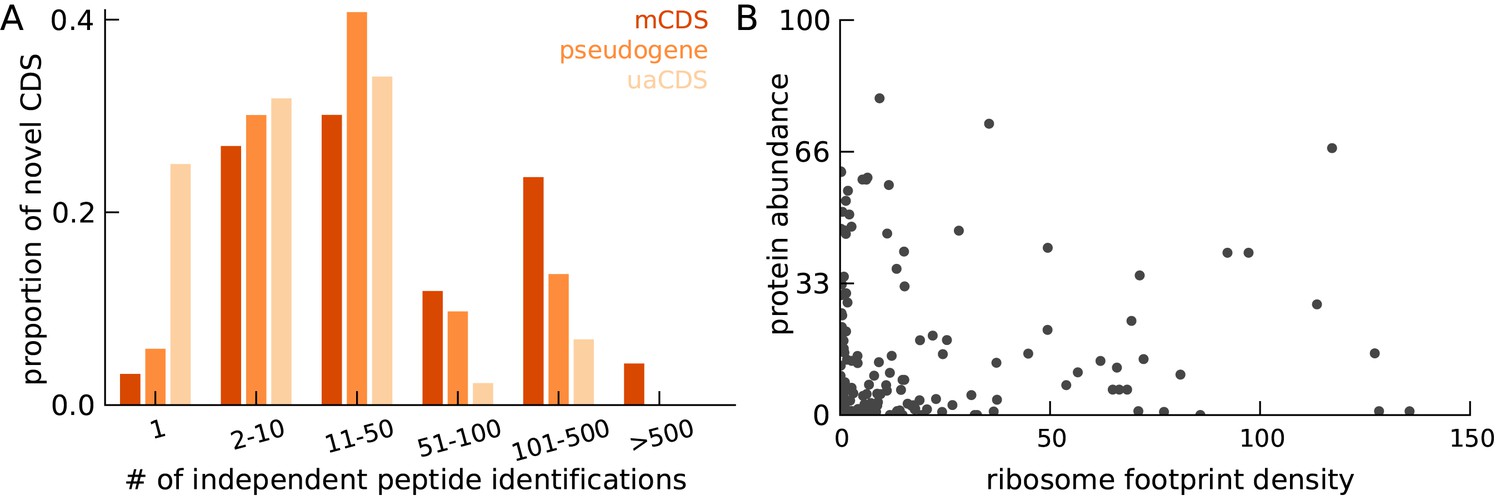
Characteristics of peptides matched to novel CDS.
(A) Histogram of number of independent identification of unique peptides across novel mCDS that have at least one peptide match. (B) Scatter plot of the untreated ribosome footprint density and the protein abundance (median abundance across all unique peptides) across all novel mCDS that have at least one peptide match (Spearman R = 0.24; ).
Figure 3—figure supplement 4
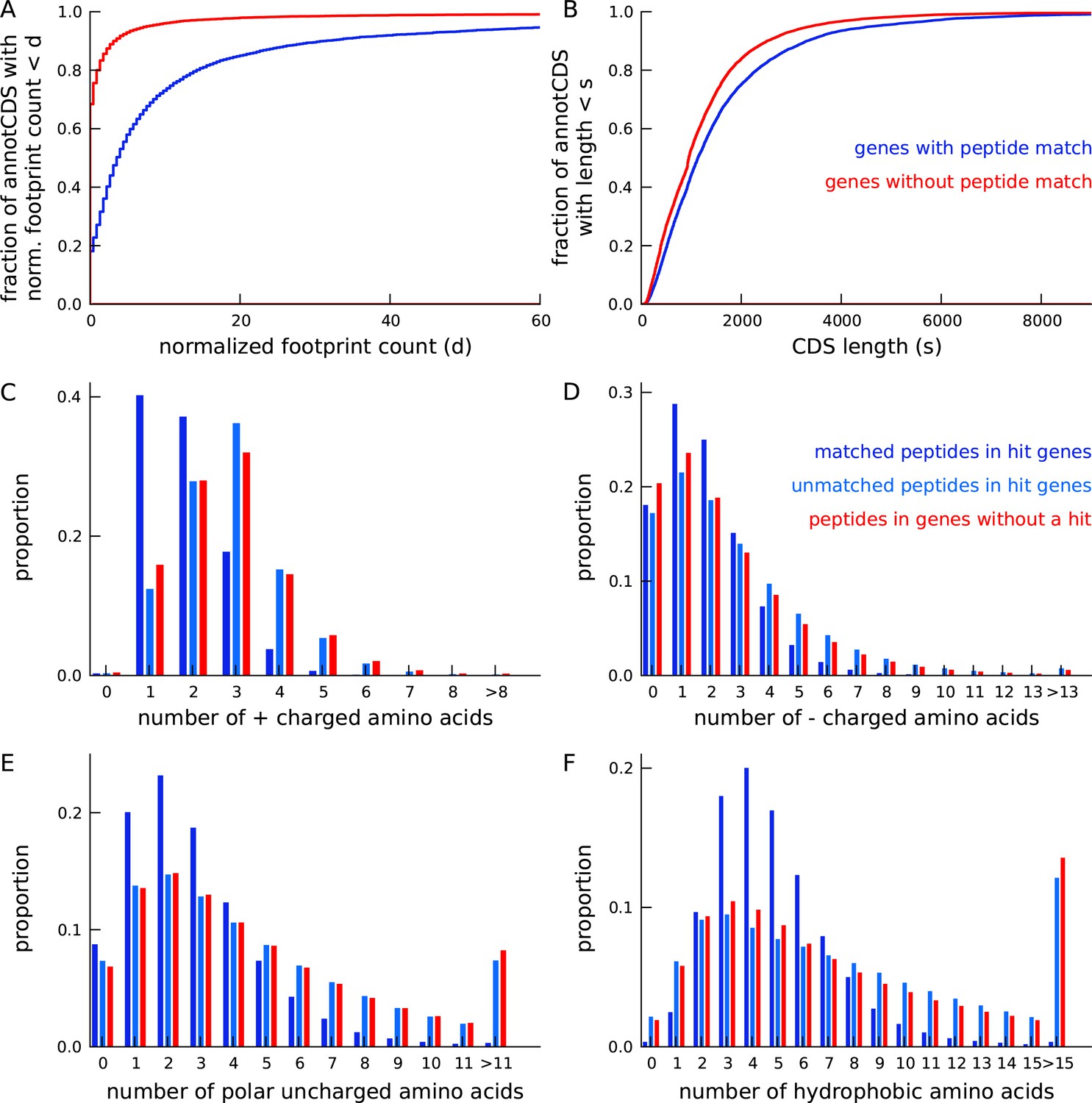
Annotated genes with peptide hits tend to be longer, have higher expression and a distinct amino acid composition.
(A) Cumulative distribution of footprint density for genes with at least one unique peptide hit (blue) and genes with no unique peptide hit (red). The median footprint density of genes with a peptide hit is about 125 fold higher than the median footprint density of genes without a peptide hit. (B) Cumulative distribution of protein length for genes with at least one unique peptide hit (blue) and genes with no unique peptide hit (red). Genes with a peptide hit tend to code for proteins that are 20% longer than proteins encoded by genes without a peptide hit. (C-F) Comparing amino acid composition within tryptic peptides with a mass-spectrum match and tryptic peptides without a mass-spectrum match. Amino acids, grouped by their electrostatic properties, have distinct compositions between matched and unmatched peptides. Matched peptides tend to be significantly shorter than unmatched peptides, and have a distinct composition of charged amino acids.
Figure 4 with 1 supplement
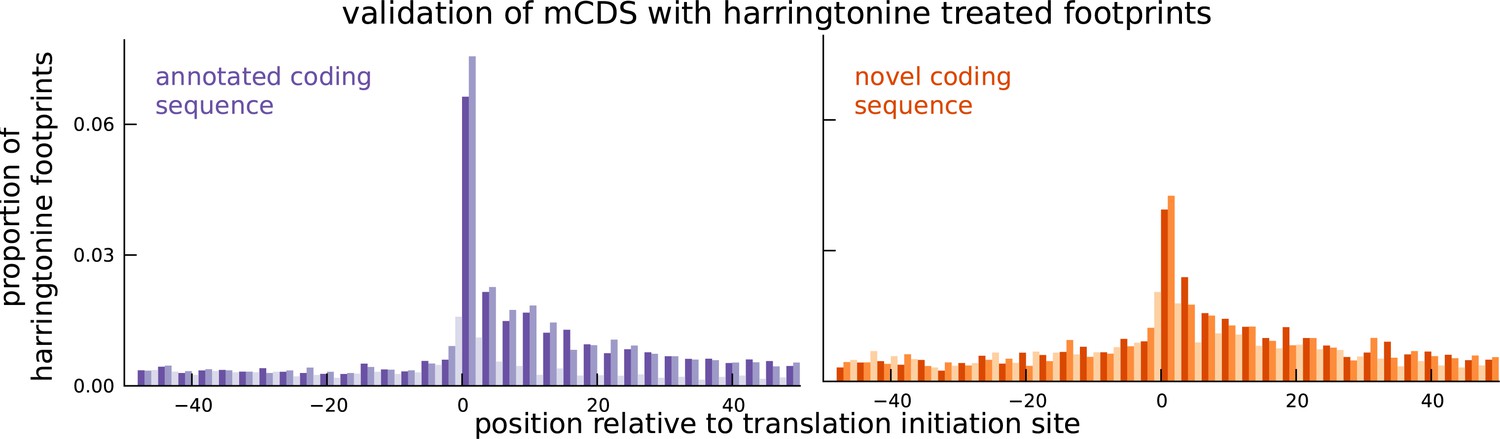
Validation of novel mCDS using harringtonine-treated ribosome profiling data.
Harringtonine-treated ribosome footprints show enrichment at the inferred translation initiation sites, when aggregated across novel mCDS (orange), similar to the enrichment at the initiation sites of a matched number of mCDS that agreed exactly with the annotated CDS (purple), suggesting that ribosomes do initiate translation of the novel mCDS.
Figure 4—figure supplement 1
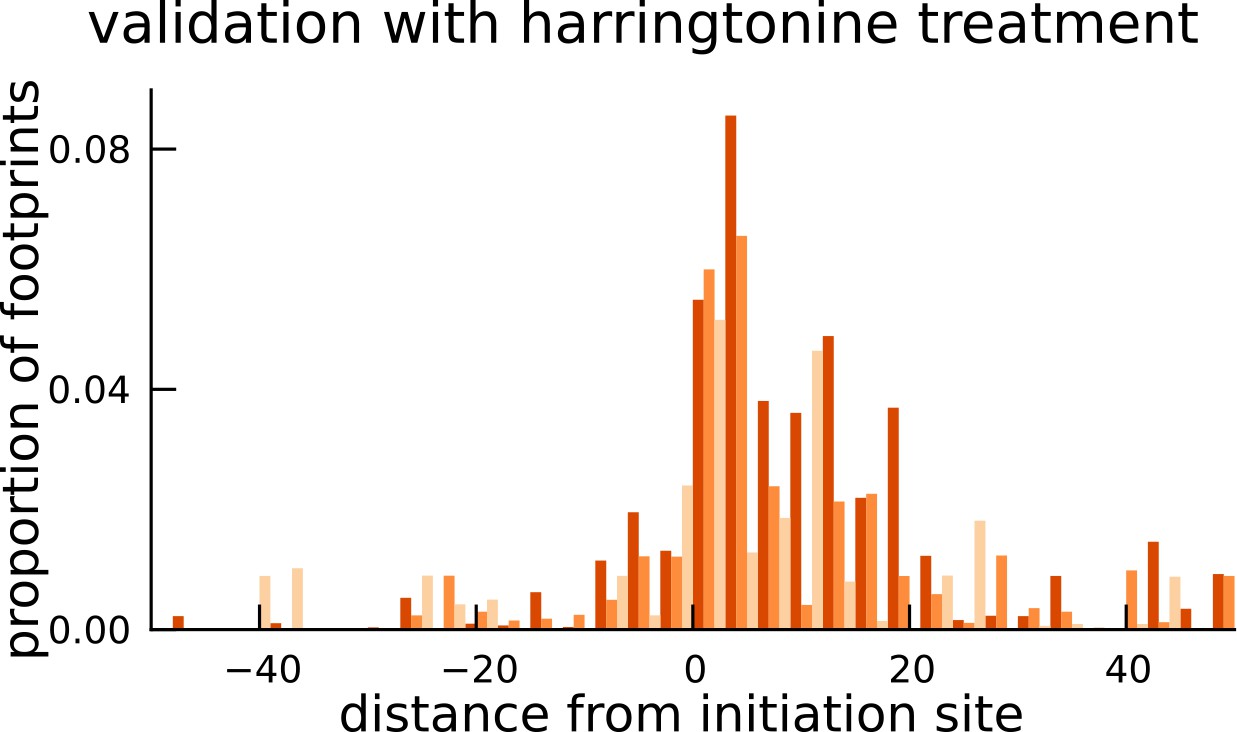
Validation of translated sequences identified in pseudogenes.
Enrichment of harringtonine-arrested ribosome occupancy at the inferred translation initiation sites validates our inferred mCDS in pseudogenes.
Figure 5 with 1 supplement
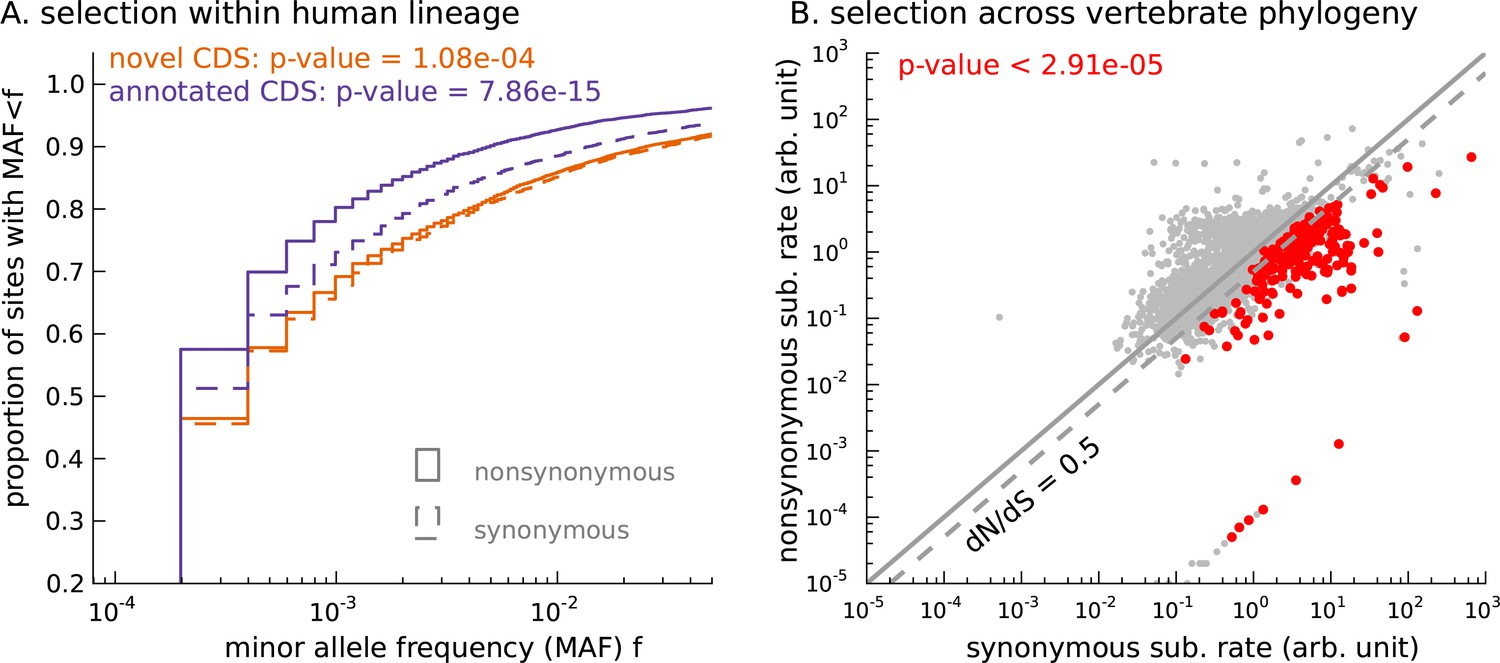
Novel translated sequences show significant signatures of coding function.
(A) Genetic variants that are nonsynonymous with respect to the inferred mCDS segregate at significantly lower frequencies in human populations than synonymous variants. The novel regions are under weaker selective constraint compared to known CDS. (The numbers of variants in each class are matched between novel and annotated CDS.) (B) Scatter plot comparing the substitution rate at inferred synonymous variants versus inferred nonsynonymous variants for each novel mCDS, computed using multiple sequence alignments across 100 vertebrate species. Highlighted in red are 232 novel mCDS identified to be under significant long-term purifying selection after Bonferroni correction (testing for dN/dS < 1;), indicating conserved coding function for these sequences.
Figure 5—figure supplement 1
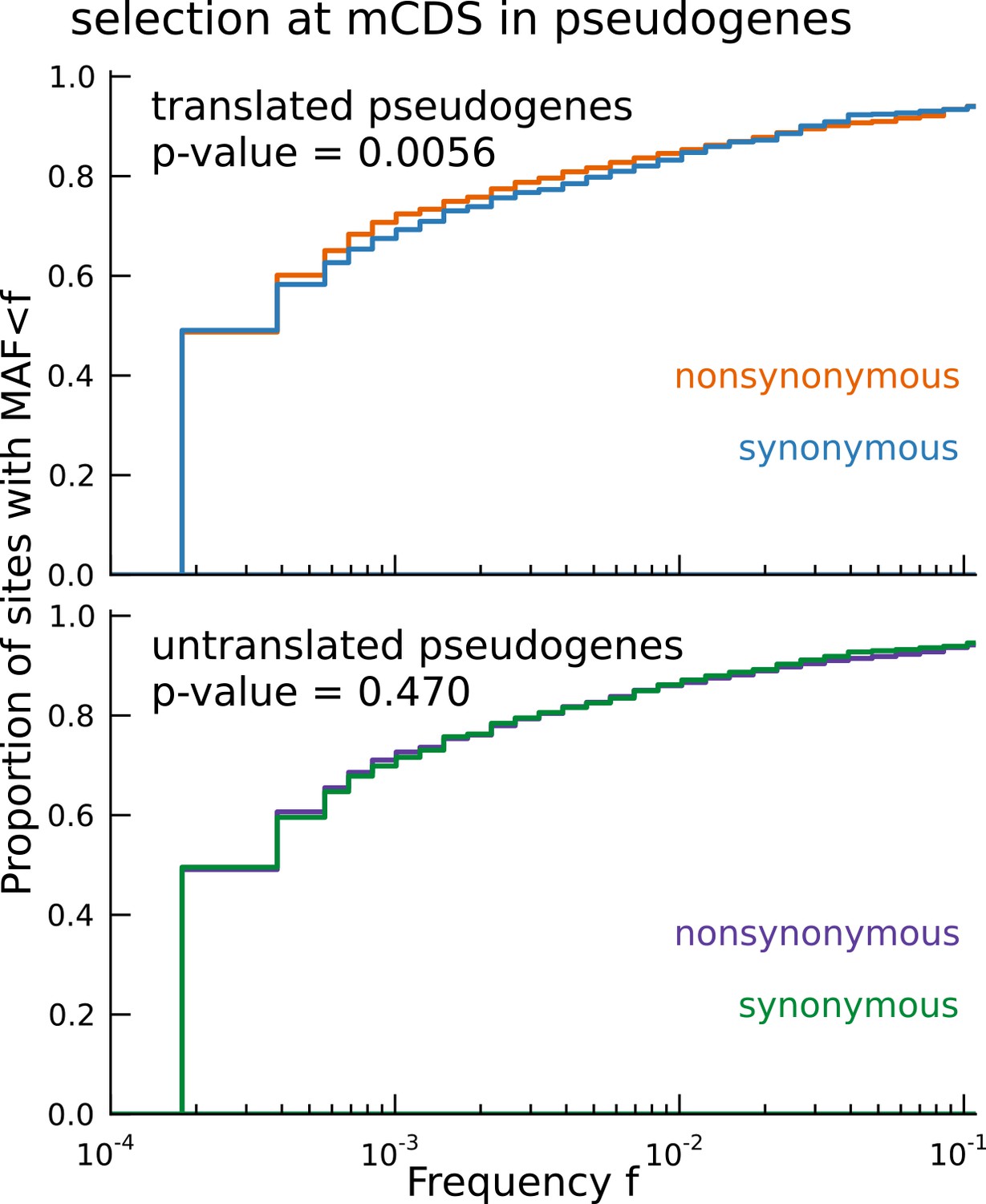
Signature of coding function in translated sequences identified in pseudogenes.
Nonsynonymous variants (orange) segregate at significantly lower frequencies than synonymous variants (blue) in those pseudogenes predicted to have a translated CDS. Pseudogenes with ribosome occupancy, but predicted to have no translated CDS, do not show any significant difference between the site frequency spectra of synonymous and nonsynonymous variants.
Figure 6 with 1 supplement
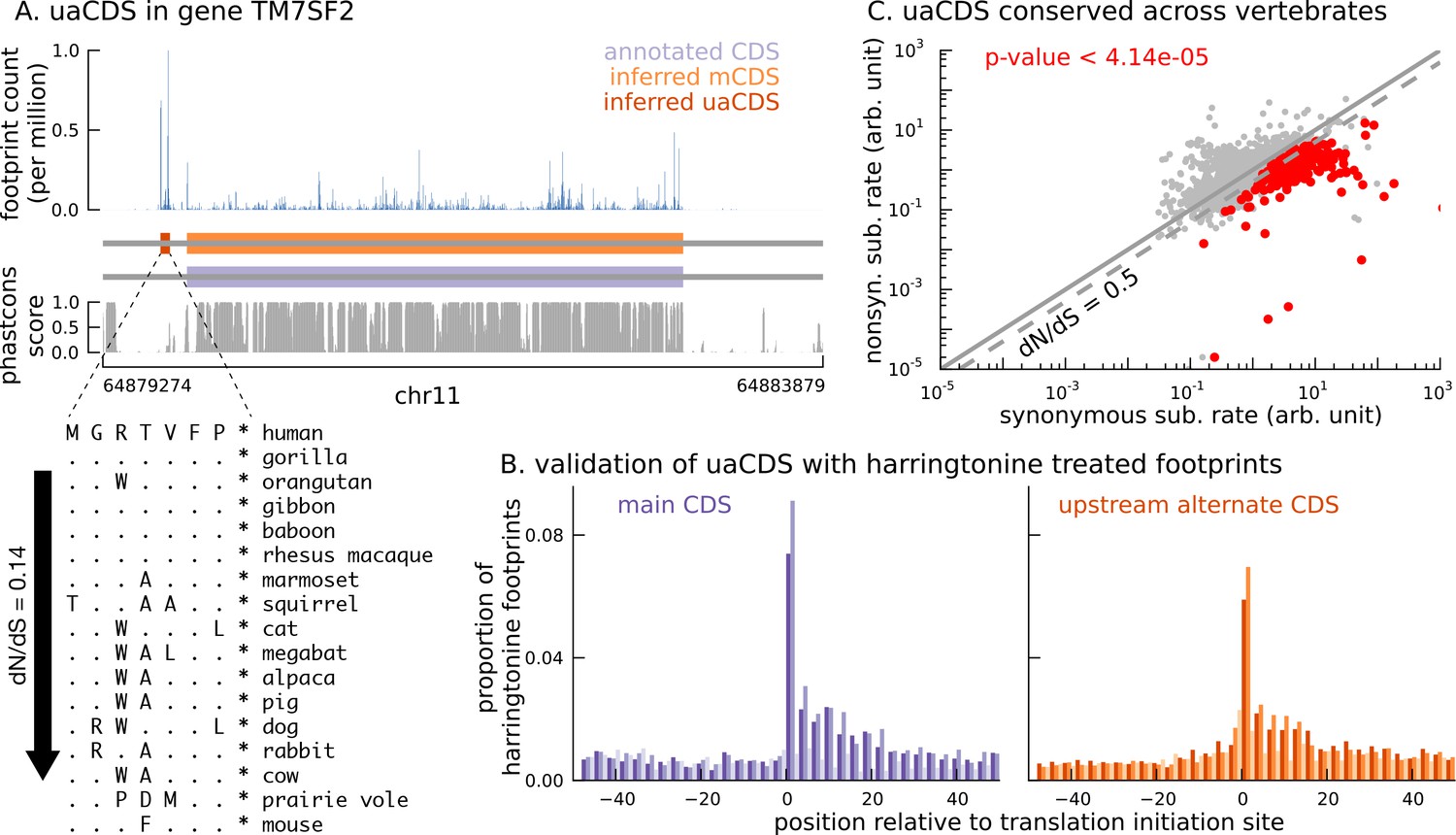
Short translated sequences identified upstream of thousands of translated main coding sequences.
(A) An alternate, novel CDS was identified upstream of the inferred main CDS in gene TM7SF2. As shown in its protein sequence alignment across mammals, the uaCDS (in particular, the start and stop codons) is highly conserved with dN/dS = 0.14. (B) Harringtonine-treated ribosome footprints show strong enrichment at the inferred initiation sites of uaCDS, comparable to the enrichment at the initiation sites of the corresponding mCDS, suggesting that ribosomes do initiate translation of these uaCDS. (C) Using multiple sequence alignment across 100 vertebrate species, 317 uaCDS were identified to have strong, significant long-term conservation.
Figure 6—figure supplement 1
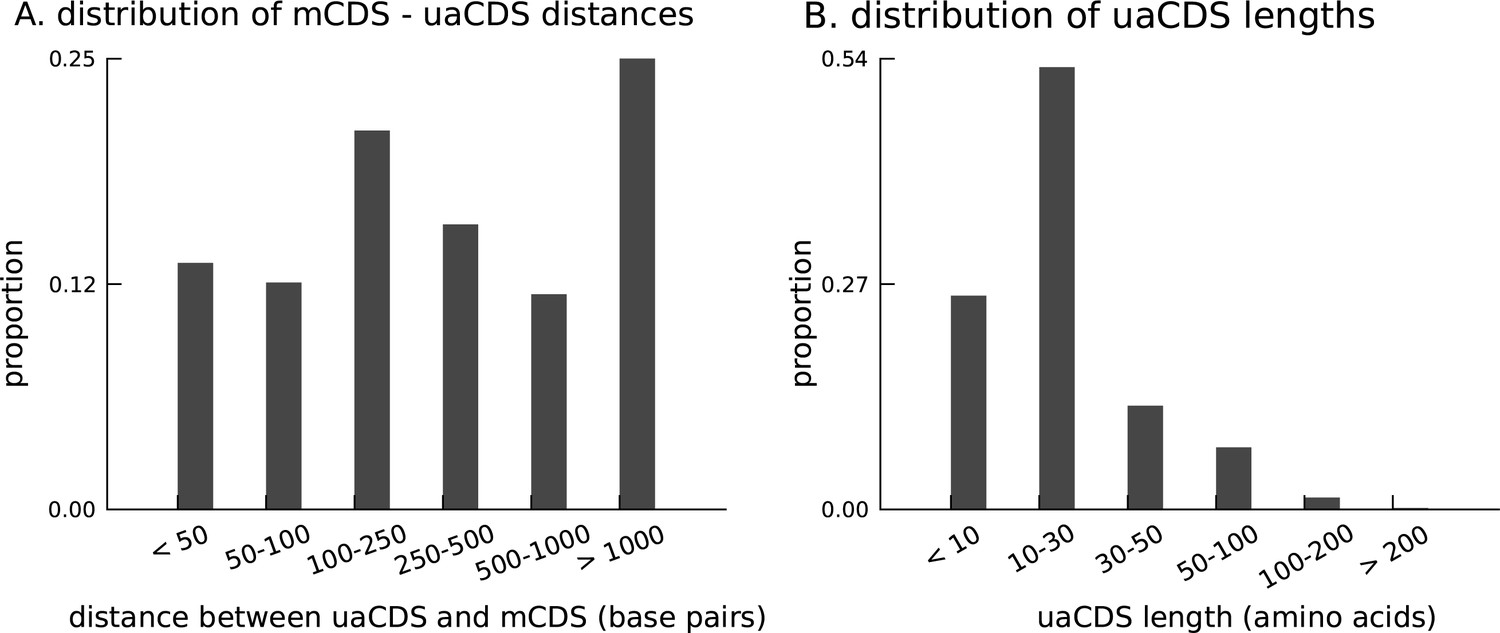
Characteristics of novel uaCDS.
(A) Histogram of the distance between uaCDS and their corresponding mCDS. (B) Histogram of the lengths of uaCDS (median length of 16 amino acids).
Figure 7
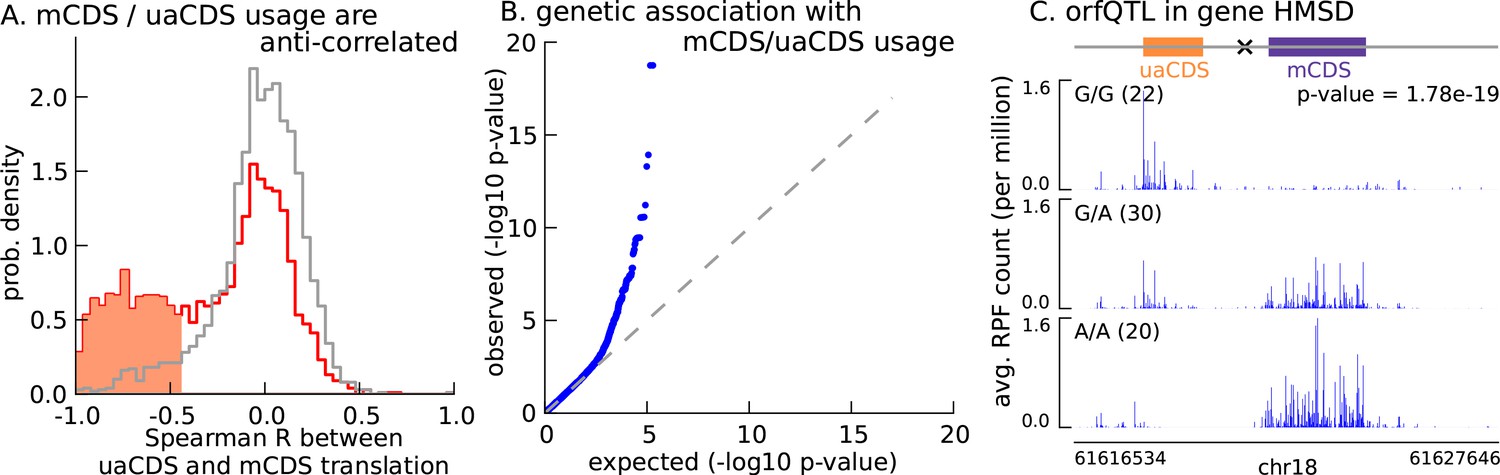
Translation of uaCDS regulates translation of mCDS.
(A) Spearman correlation, across LCLs, between mCDS translation and uaCDS translation (red histogram). Using random (mCDS, uaCDS) pairs, matched for length and pairwise distance, we computed an empirical null distribution of Spearman correlations (gray histogram). At 10% FDR, 917 inferred (uaCDS, mCDS) pairs have significant negative correlation (shaded red region). (B) 365 orfQTLs (genetic variants associated with ORF usage; i.e., whether the mCDS or uaCDS of a transcript is translated) were identified at 10% FDR (41 pairs of mCDS/uaCDS). (C) Illustrating an example of an orfQTL in the histocompatibility minor serpin domain-containing (HMSD) gene (introns removed for better visualization). The most significant variant (marked x) lies within an intron between the mCDS and uaCDS of the transcript.
Author response image 1
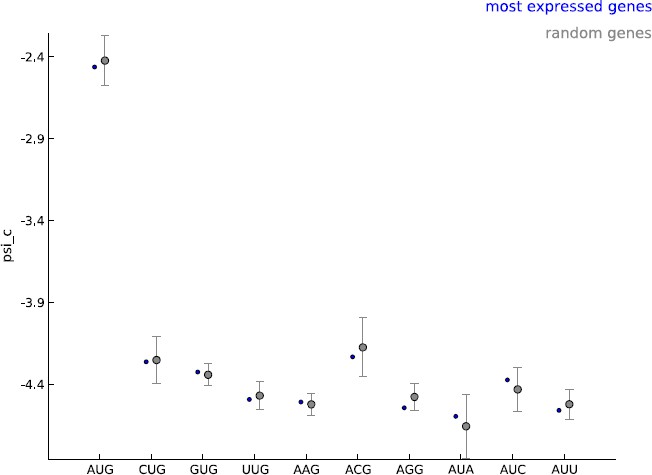
Comparing the estimated values of parameter psi_c, when using the 5000 most expressed genes (blue) and random sets of 5000 genes (gray).
https://doi.org/10.7554/eLife.13328.025
Author response image 2
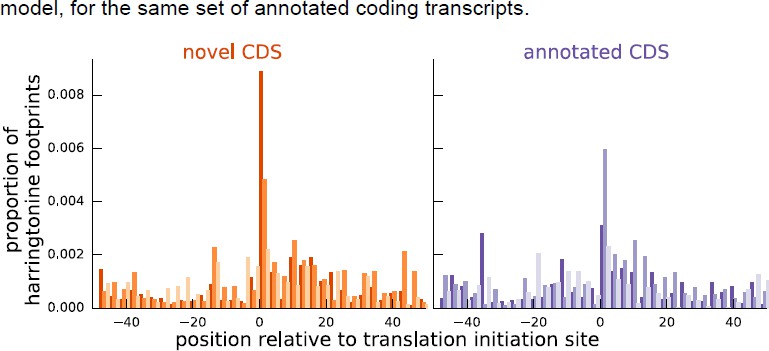
Comparing the proportion of harringtonine-treated ribosome footprints at the inferred initiation sites of novel coding sequences and the annotated initiation sites of annotated coding sequences.
Both plots are an aggregate over the same set of annotated coding genes for which the inferred coding sequence does not match the annotated coding sequence.
Additional files
-
Supplementary file 1
Peptide matches to novel CDS detected using mass spectrometry.
This table lists the 207 novel CDS (161 mCDS and 46 uaCDS) that have at least one mass-spectrum uniquely matching a peptide in the inferred protein sequence, at protein-level 10% FDR.
- https://doi.org/10.7554/eLife.13328.024
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling
eLife 5:e13328.
https://doi.org/10.7554/eLife.13328




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}