Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1
- Massachusetts General Hospital, United States
- Harvard Medical School, United States
Figures
Figure 1 with 1 supplement

ER-associated degradation factors and the aspartic protease DDI-1 are required for responses to proteasome disruption.
(a) rpt-3::gfp expression following disruption of proteasome function by rpt-5(RNAi) in various mutant backgrounds. Scale bars 100 µm. (b) Table showing growth vs. arrest phenotypes of various mutants in the presence of bortezomib or upon rpn-10 RNAi. For RNAi experiments, L1 animals were incubated for 3 days on indicated RNAi plates, and scored for developmental arrest (+; normal development, Lva; larval arrest). For bortezomib experiments, ~15 L1 animals were incubated for 4 days in liquid cultures containing varying concentrations of bortezomib, and scored for developmental progression. The number (n) of replicate bortezomib experiments performed for each genotype is shown on the right. Each colored rectangle is divided into equal parts to show results from each replicate.
Figure 1—figure supplement 1
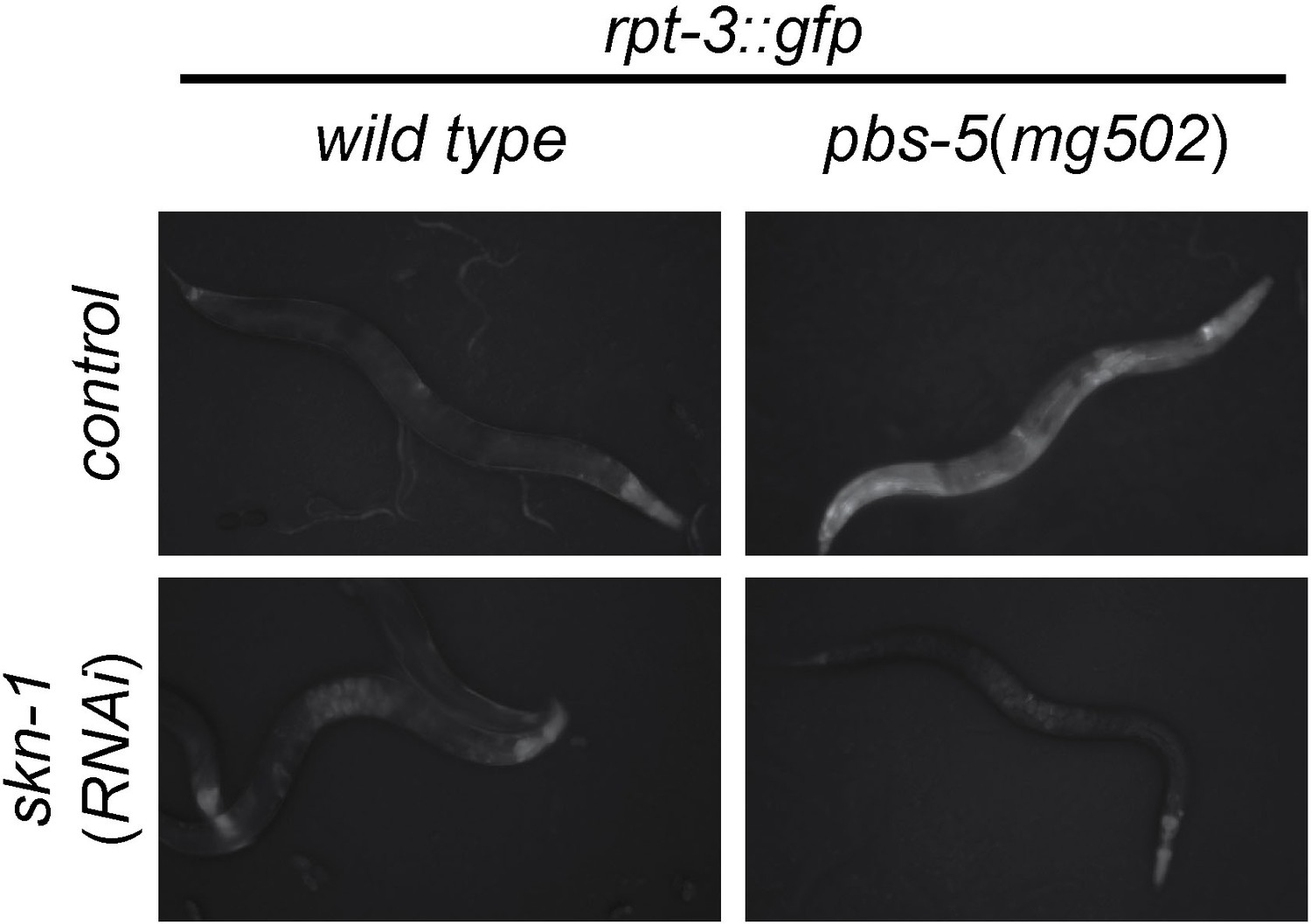
skn-1-dependent activation of rpt-3::gfp in pbs-5(mg502) mutants.
Fluorescence images show rpt-3::gfp expression in animals grown for 3 days at 20°C. Scale bar 100 µm.
Figure 2 with 1 supplement
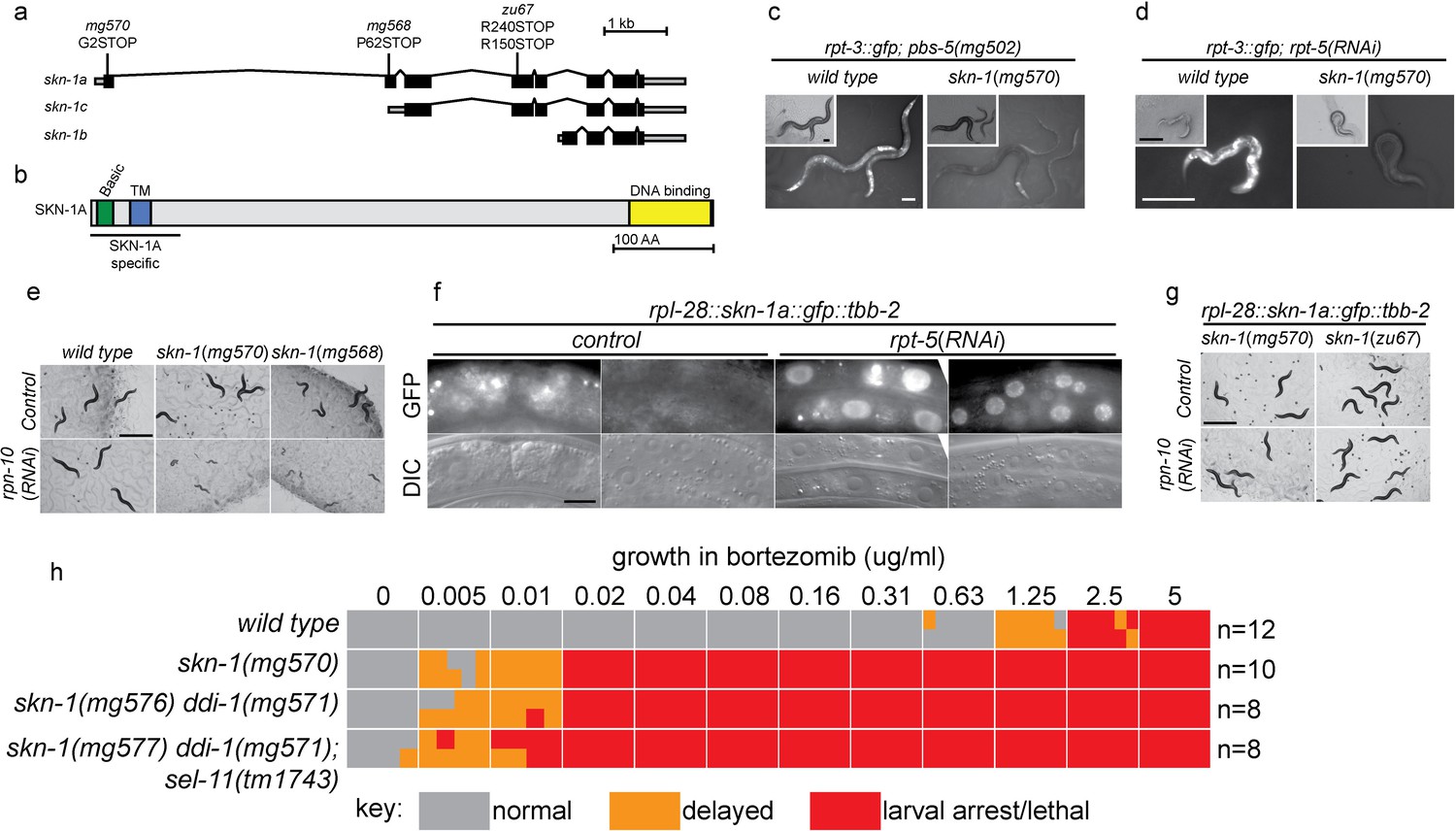
SKN-1A, a transmembrane-domain-containing isoform of SKN-1 mediates transcriptional responses to proteasome disruption.
(a,b) Schematic of the (a) skn-1 locus and (b) SKN-1A protein. In (a), the CRISPR-induced skn-1a-specific mutations are indicated. (c,d) rpt-3::gfp induction in wild type and isoform-specific skn-1 mutants in (c) the pbs-5(mg502) mutant, or (d) rpt-5(RNAi). Scale bars 100 µm. (e) Developmental arrest of isoform-specific skn-1 mutants exposed to mild proteasome disruption by rpn-10(RNAi) but not on control RNAi. Scale bar 1 mm. (f) Expression and localization of functional SKN-1A::GFP fusion protein after proteasome disruption by rpt-5(RNAi). Apparent GFP signal in control treated animals is background auto-fluorescence. Scale bar 10 µm. (g) No developmental arrest of skn-1 mutants carrying an isoform-specific skn-1a::gfp transgene, and exposed to mild proteasome disruption by rpn-10(RNAi). Scale bar 1 mm. (h) Table showing growth vs. arrest phenotypes of skn-1a mutants in in the presence of bortezomib. All skn-1a alleles are identical in their effect on skn-1a coding sequence (G2STOP). Experiments performed identically to those shown in Figure 1b, and data for the wild type from Figure 1 are shown for reference.
Figure 2—figure supplement 1
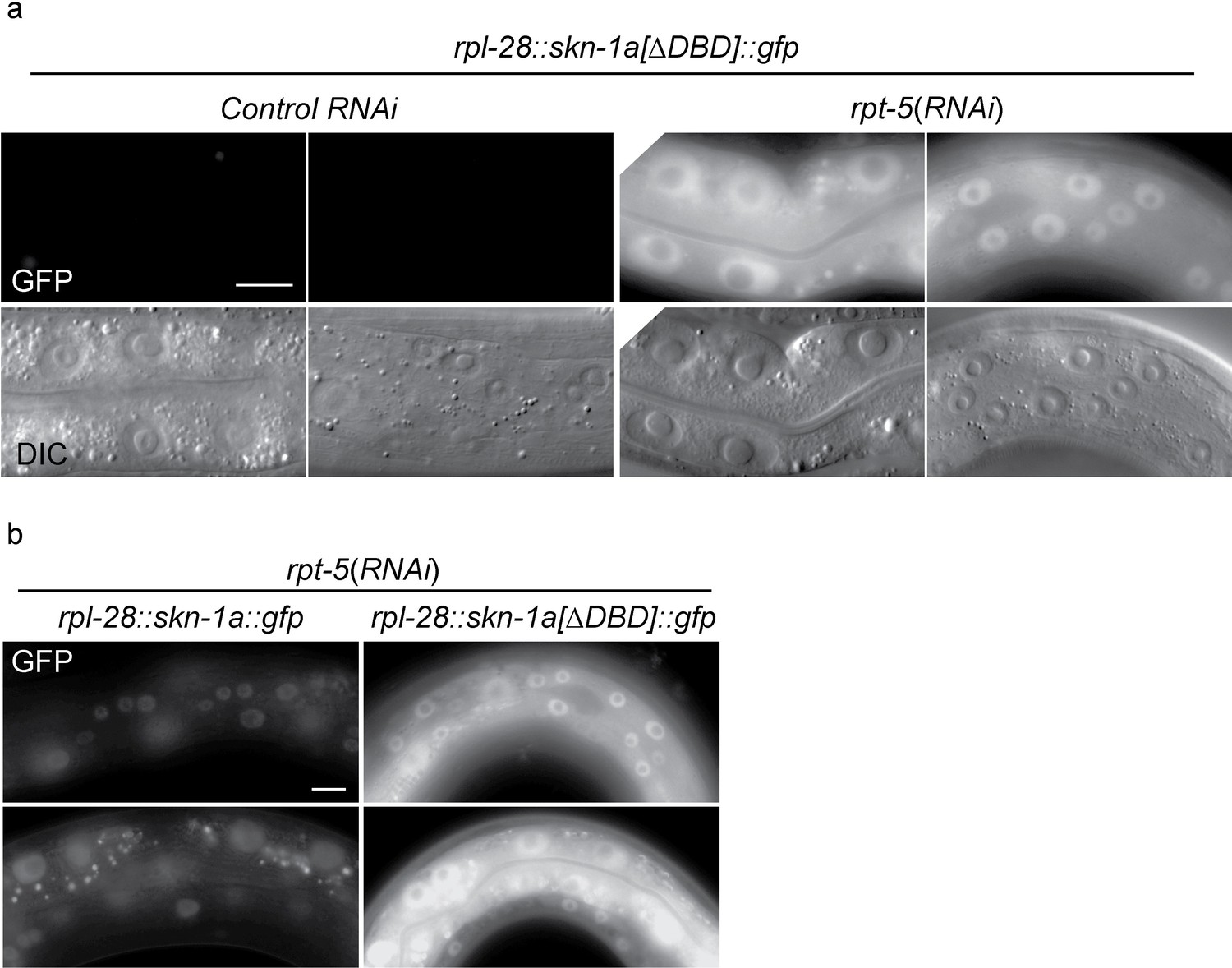
SKN-1A[∆DBD]::GFP is stabilized upon proteasome disruption.
(a) Fluorescence images comparing expression and localization of SKN-1A[∆DBD]::GFP under control conditions and following rpt-5(RNAi). SKN-1A[∆DBD]::GFP is not detected in the absence of proteasome disruption, but accumulates to high levels in both nucleus and cytoplasm following proteasome disruption. Scale bar shows 10 µm. (b) fluorescence images comparing levels of SKN-1A::GFP (left panels) and SKN-1A[∆DBD]::GFP (right panels) upon proteasome disruption. SKN-1A[∆DBD]::GFP reaches much higher levels than the full length fusion protein. Scale bar shows 10 µm. All images show animals raised for two days on the indicated RNAi.
Figure 3
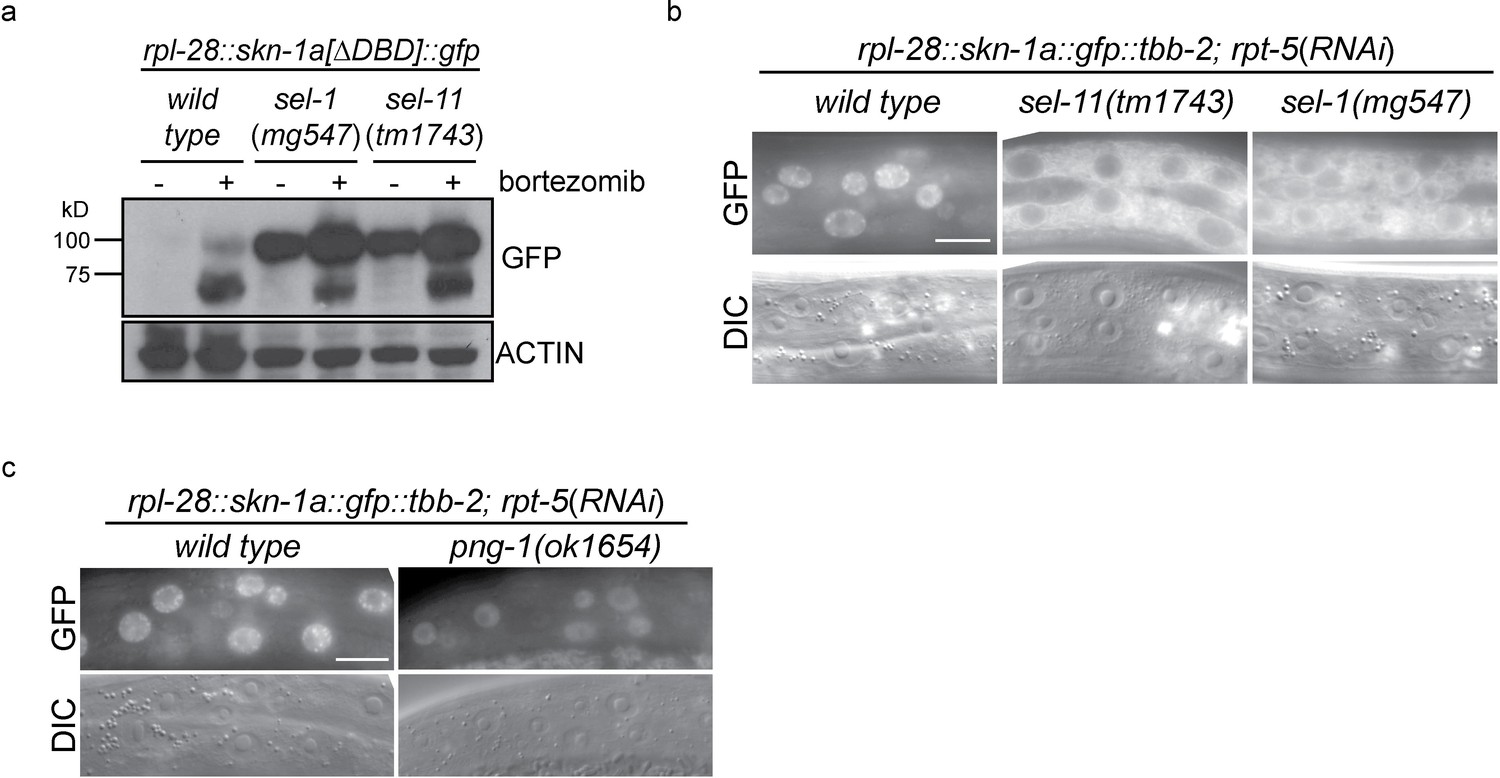
ERAD is required for constitutive SKN-1A degradation, and for activation of SKN-1A upon proteasome disruption.
(a) Western blot showing expression and post-translational processing of SKN-1A[∆DBD]::GFP in ERAD mutant animals, treated with either solvent control (DMSO) or 5 ug/ml bortezomib. SKN-1A[∆DBD]::GFP is only detected in wild-type animals upon bortezomib exposure; a major band at ~70 kD and a minor band at ~90 kD are detected. In ERAD defective mutants, the ~90 kD band is strongly detected under all conditions, and the ~70 kD band appears only following bortezomib treatment. Actin is used as a loading control. (b) Expression and localization of SKN-1A::GFP in wild type and sel-1 and sel-11 ERAD defective mutants after proteasome disruption by rpt-5(RNAi). In ERAD defective mutants, SKN-1A::GFP fails to localize to the nucleus. Scale bar 10 µm. (c) Expression and localization of SKN-1A::GFP in wild type and png-1 mutants after proteasome disruption by rpt-5(RNAi). In png-1 mutants, SKN-1A::GFP is able to localize to the nucleus, although at reduced levels compared to the wild-type. Scale bar 10 µm.
Figure 4
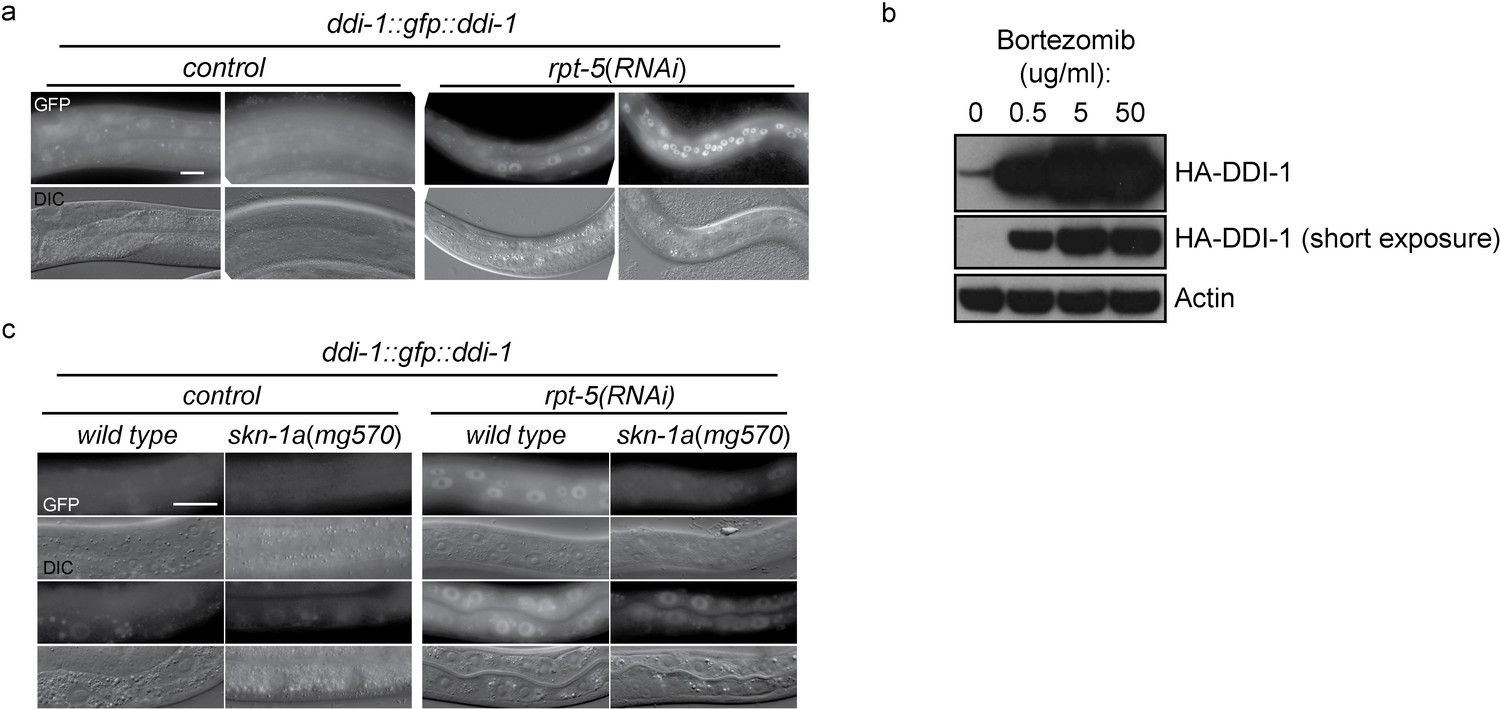
DD1-1 is upregulated upon proteasome disruption.
(a) A functional GFP::DDI-1 fusion protein is strongly induced and localizes to the nucleus upon proteasome disruption by rpt-5(RNAi). Scale bar 20 µm. (b) Western blot showing induction of HA-tagged endogenous DDI-1 upon proteasome disruption by bortezomib. Actin is used as a loading control. (c) Induction of GFP::DDI-1 upon proteasome disruption by rpt-5(RNAi) is lost in skn-1a mutants. Scale bar 20 µm.
Figure 5 with 1 supplement
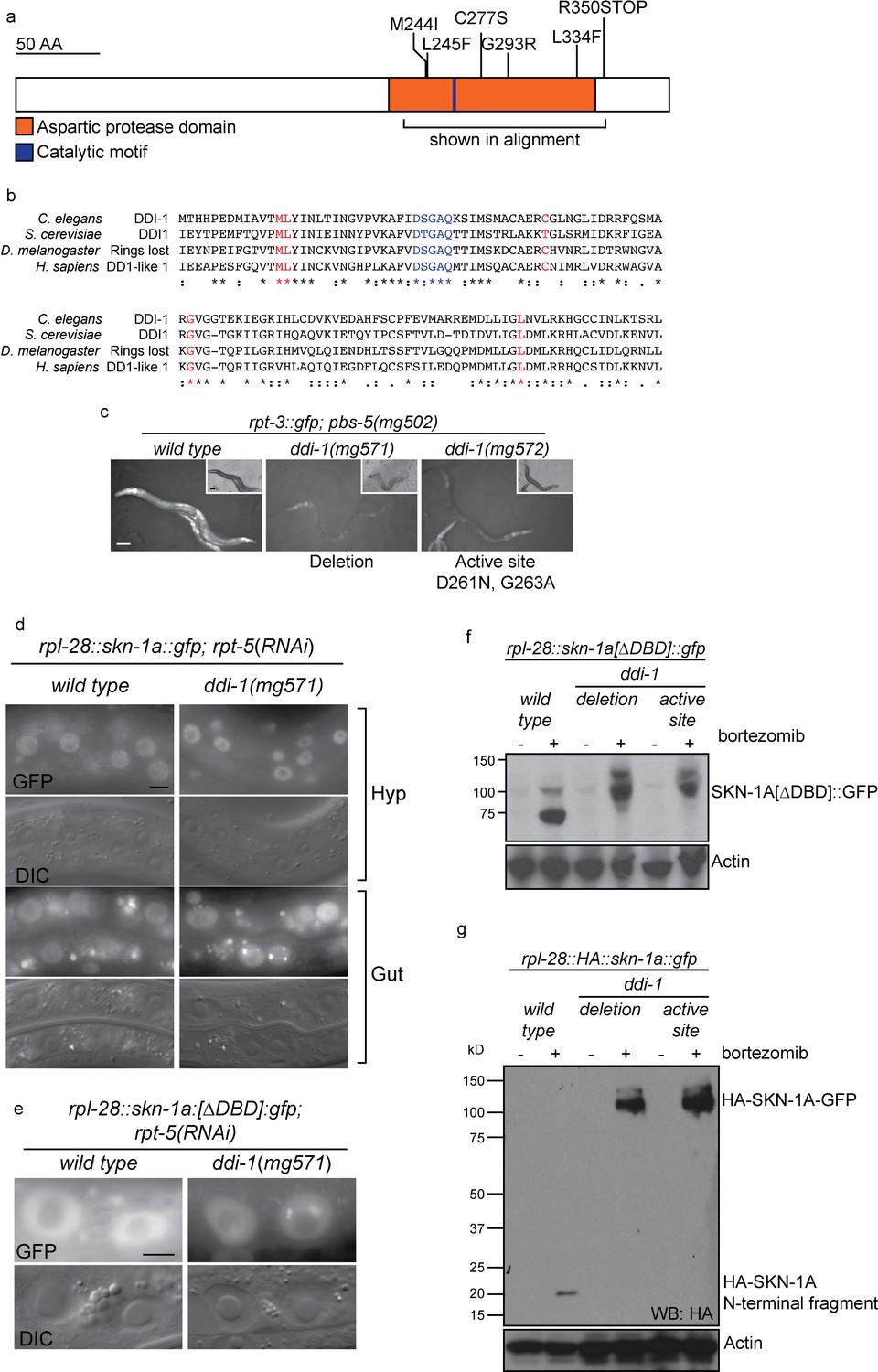
The DDI-1 aspartic protease is required for proteolytic activation of SKN-1A.
(a) Schematic of the DDI-1 protein showing residues affected by EMS-induced loss of function alleles. (b) Multiple alignment of the aspartic protease domain of DDI-1. Red text indicates residues affected by mutations that disrupt rpt-3::gfp activation, blue text indicates the DS/TGAQ catalytic motif. (c) rpt-3::gfp induction in the pbs-5(mg502) mutant background (left panel), compared to animals carrying a ddi-1 deletion (middle panel), or a point mutation affecting the catalytic motif (right panel). Scale bars 100 µm. (d) Localization of SKN-1A::GFP in wild-type and ddi-1 mutant animals following disruption of proteasome function by rpt-5(RNAi). SKN-1A::GFP nuclear localization is intact in the ddi-1 mutant. In some gut cells of ddi-1 mutant animals SKN-1A::GFP localizes to puncta that are not detected in the wild type. Scale bars 10 µm. (e) Localization of SKN-1A[∆DBD]::GFP in gut nuclei of wild-type and ddi-1 mutant animals. Nuclear foci of SKN-1A[∆DBD]::GFP are found in ddi-1 mutants, but not wild type. (f) Western blot showing expression and processing of SKN-1A[∆DBD]::GFP in ddi-1 mutant animals, treated with either solvent control (DMSO) or 5 ug/ml bortezomib, and blotted for GFP. In the ddi-1 mutant animals, the major band detected is ~30 kD larger than in the wild type. (g) Western blot showing expression and processing of HA::SKN-1A:GFP in ddi-1 mutants animals, treated with either solvent control (DMSO) or 5 ug/ml bortezomib, and blotted for HA. In the wild type, a ~20 kD band is detected in animals exposed to bortezomib. In ddi-1 mutants this low molecular weight fragment is absent, and a ~110 kD band is detected. In (f) and (g) ddi-1 mutations were ddi-1(mg571)[deletion] or ddi-1(mg572)[active site] and actin is used as a loading control.
Figure 5—figure supplement 1
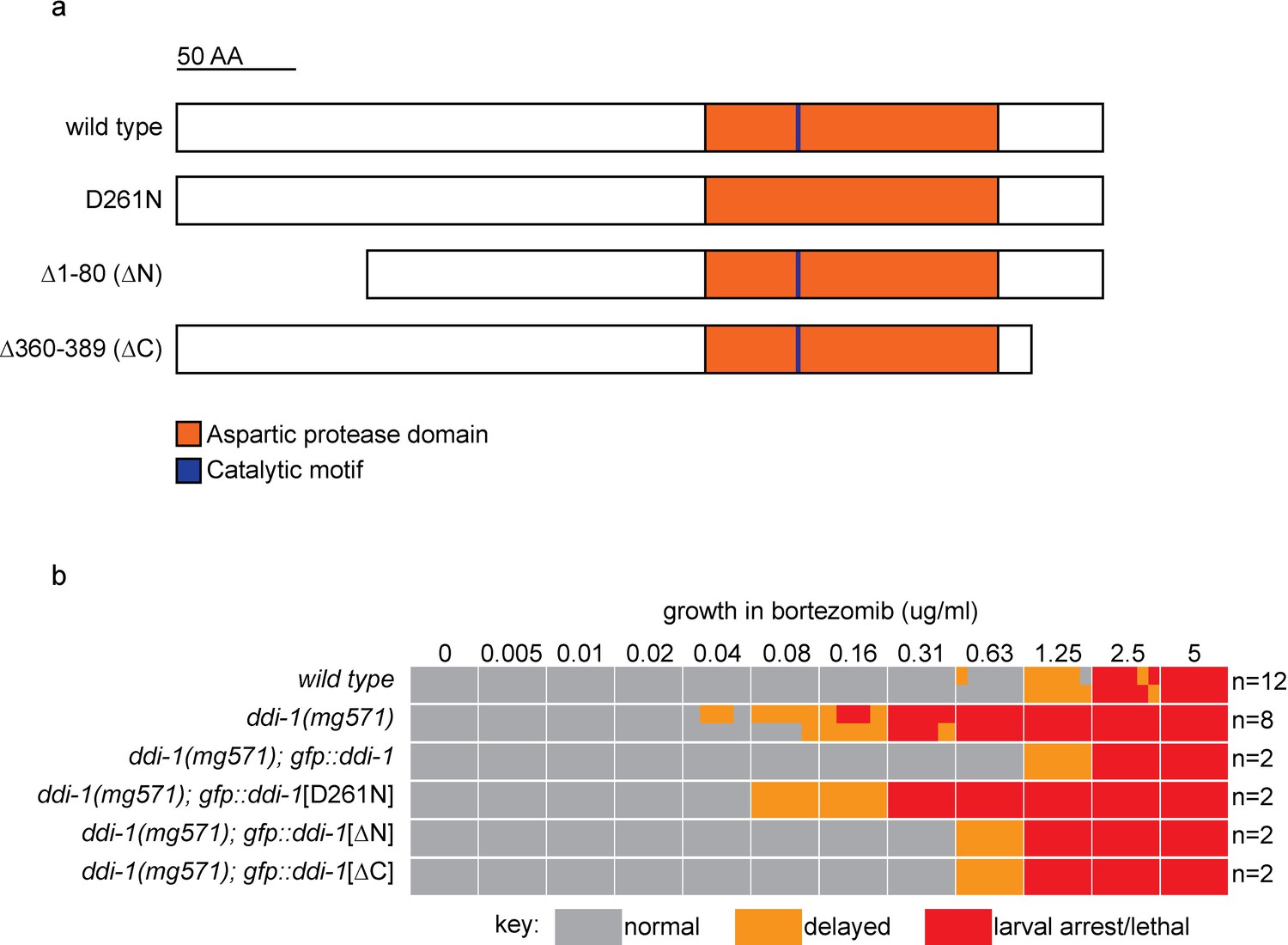
The aspartic protease, but not the N- or C-terminal domains of DDI-1 are essential for resistance to bortezomib.
(a) Schematic showing wild type and mutant DDI-1 proteins tested. Each protein was expressed under the ddi-1 promoter and fused to GFP at the N-terminus. (b) Table showing growth vs. arrest phenotypes of ddi-1(mg571) mutants expressing wild-type or mutant GFP::DDI-1 in the presence of bortezomib. ~15 L1 animals were incubated for 4 days in liquid cultures containing presence of varying concentrations of bortezomib (as shown in Figure 1), and scored for developmental arrest. The number (n) of replicate bortezomib experiments performed for each genotype is shown on the right, for each condition the corresponding colored rectangle is divided into n equal parts and shows the result from each replicate. Data for the wild type and ddi-1(mg571) from Figure 1 are shown for reference.
Figure 6 with 1 supplement
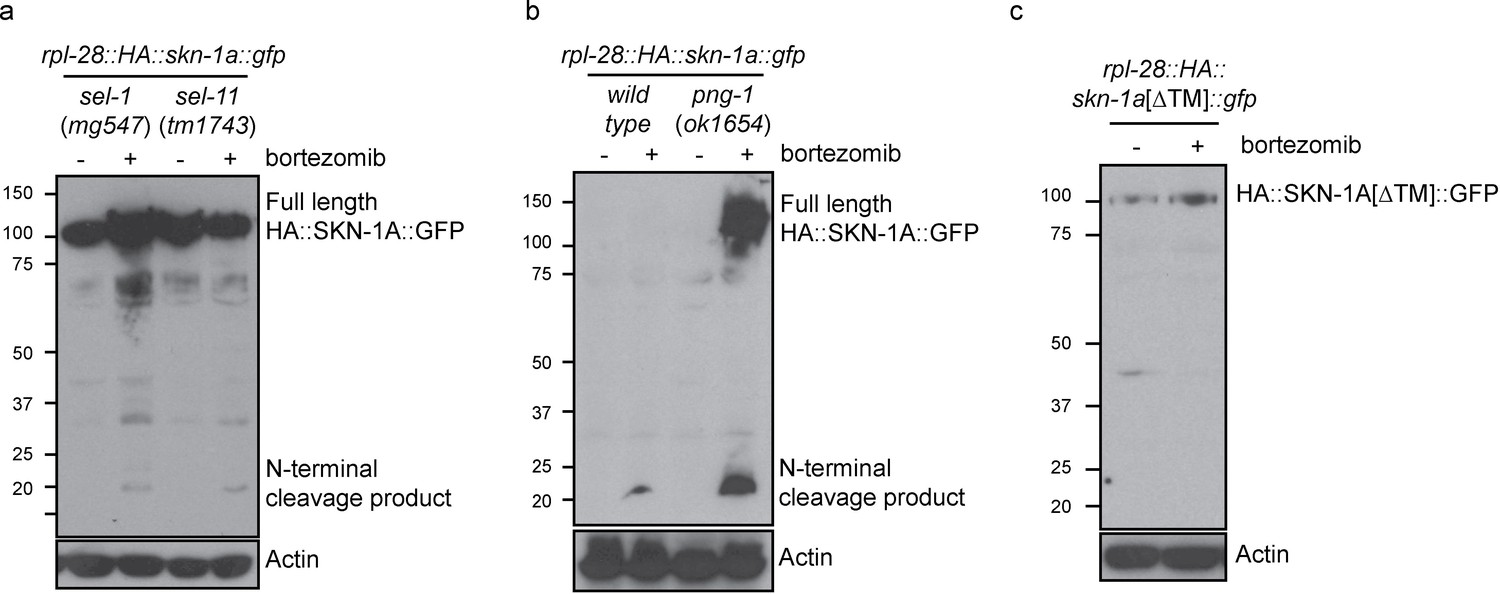
Proteolytic processing of SKN-1A occurs downstream of ER trafficking.
(a) Western blot showing expression and processing of HA::SKN-1A::GFP upon proteasome disruption by bortezomib in sel-1 and sel-11 mutant animals. ERAD-defective mutants accumulate similar levels of uncleaved (~110 kD) HA::SKN-1A::GFP in both absence and presence of bortezomib. A relatively small amount of the ~20 kD cleavage product accumulates only upon exposure to bortezomib. (b) Western blots comparing expression and processing of HA::SKN-1A::GFP upon proteasome disruption by bortezomib in wild-type and png-1 mutant animals. Both cleaved (~20 kD) and uncleaved (smear ~100-150 kD) HA::SKN-1A::GFP accumulates upon bortezomib treatment in png-1 mutants. (c) Western blot showing expression and processing of HA::SKN-1A[∆TM]::GFP upon proteasome disruption by bortezomib, in otherwise wild-type animals. Uncleaved (~100 kD) HA::SKN-1A[∆TM]::GFP is detected at similar levels under both conditions suggesting the protein is not subject to proteasomal degradation, and a low molecular weight cleavage product is not detected under either condition. For each experiment, mixed stage cultures were treated with either solvent control (-), or 5 ug/ml bortezomib (+) prior to collection for SDS-PAGE. HA::SKN-1A::GFP/ HA::SKN-1A[∆TM]::GFP is detected by anti-HA antibodies, Actin is used as a loading control.
Figure 6—figure supplement 1
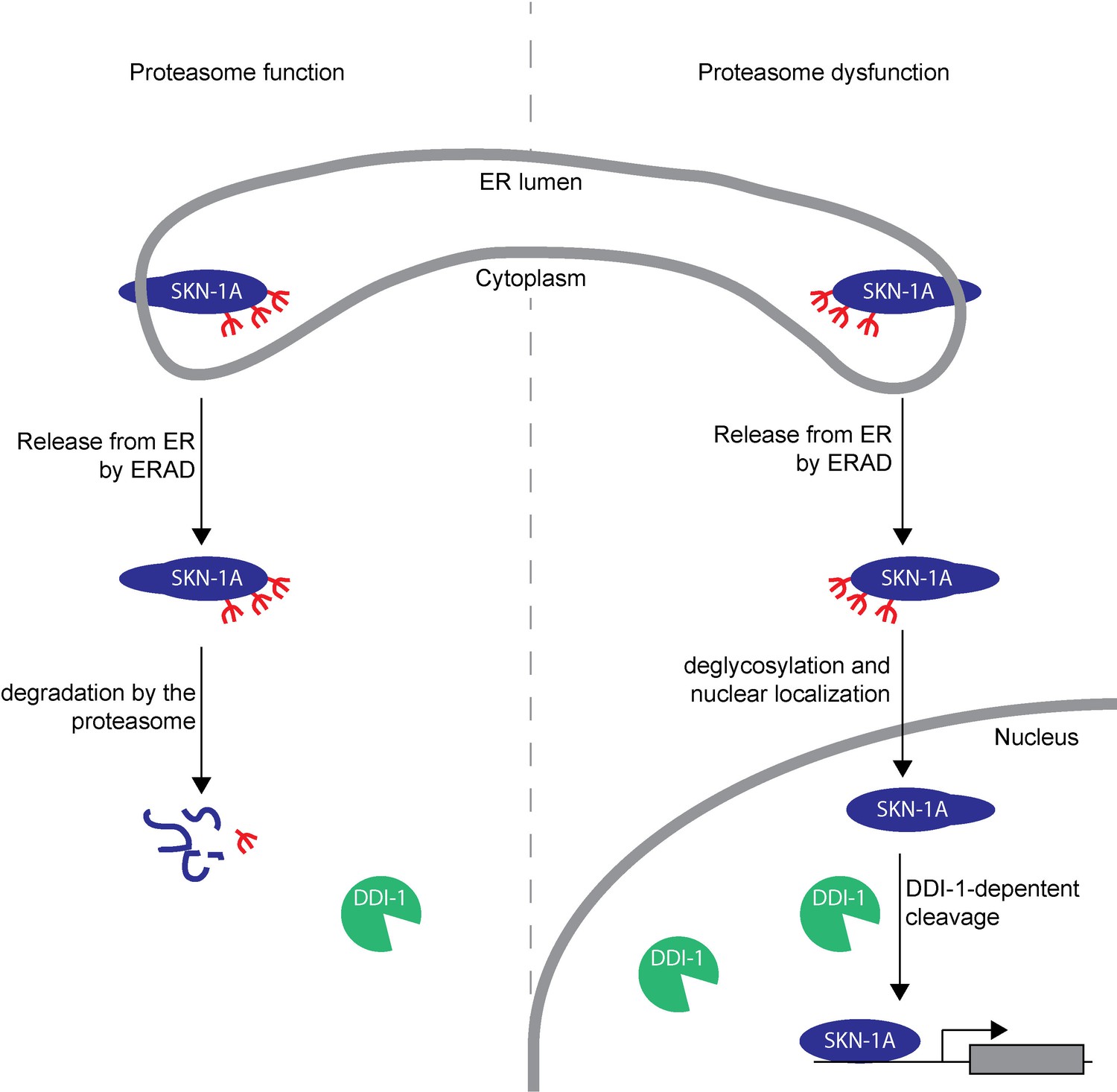
Model showing post-translational processing of SKN-1A by DDI-1.
SKN-1A associates with the ER via its N-terminal transmembrane domain. In the ER, SKN-1A is subject to glycosylation, and possibly other post-translational modifications. ERAD-dependent release of SKN-1A from the ER targets SKN-1A for proteasome-mediated degradation. When the proteasome is functional (shown on the left half of the model), SKN-1A is rapidly degraded by the proteasome. Under these conditions, the DDI-1 protease is expressed at low levels and localizes largely to the cytoplasm. During proteasome dysfunction (shown on the right half of the model), SKN-1A is not efficiently degraded. Stabilized SKN-1A is deglycosylated by PNG-1 (this deglycosylation might take place in the nucleus or cytoplasm) and localizes to the nucleus. Proteasome dysfunction also leads to increased expression and nuclear localization of DDI-1 (this is in part due to regulation of ddi-1 transcription by SKN-1A, but likely involves additional SKN-1A-independent mechanisms). Once in the nucleus, SKN-1A is cleaved by DDI-1 to produce the active truncated form of SKN-1A, allowing upregulation of proteasome subunits and other downstream genes. Cleavage of SKN-1A is disrupted by mutations that affect ERAD, or if SKN-1A lacks its transmembrane domain, suggesting that DDI-1-dependent cleavage occurs downstream of ER-trafficking and may require post-translational modifications to SKN-1A acquired during this process.
Tables
Table 1
EMS-induced mutations that disrupt rpt-3::gfp activation.
| Allele | Affected gene | Effect | Homologues |
|---|---|---|---|
| mg563 | C01G5.6 | L245F | DDI1. Aspartic protease. |
| mg555 | C01G5.6 | C277S | |
| mg544 | C01G5.6 | G293R | |
| mg542 | C01G5.6 | R350STOP | |
| mg543 | C01G5.6 | M244I | |
| mg557 | C01G5.6 | L334F | |
| mg565 | sel-1 | G594E | HRD3/SEL1. ER membrane protein, required for ERAD substrate recognition. |
| mg567 | sel-1 | A522T | |
| mg547 | sel-1 | splice site | |
| mg550 | sel-9 | S140F | EMP24/TMED2. ER membrane protein. |
| mg561 | png-1 | G498R | PNG1/NJLY1. Peptide N-glycanase. Removes N-linked glycans during ERAD. |
| mg564 | png-1 | splice site |
Additional files
-
Supplementary file 1
Supplementary Tables 1-4.xlsx.
Contains 4 Supplementary tables: Table 1. C. elegans strains used in this study. Table 2. CRISPR guide RNA constructs and repair template sequences. Table 3. Constructs used to generate miniMos transgenics. Table 4. Evidence used in identification of EMS-induced mutations that cause failure to activate rpt-3::gfp expression.
- https://doi.org/10.7554/eLife.17721.014
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1
eLife 5:e17721.
https://doi.org/10.7554/eLife.17721




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}