Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2
- Stockholm University, Sweden
- MRC Laboratory of Molecular Biology, United Kingdom
- Swedish e-Science Research Center, KTH Royal Institute of Technology, Sweden
Figures
Figure 1
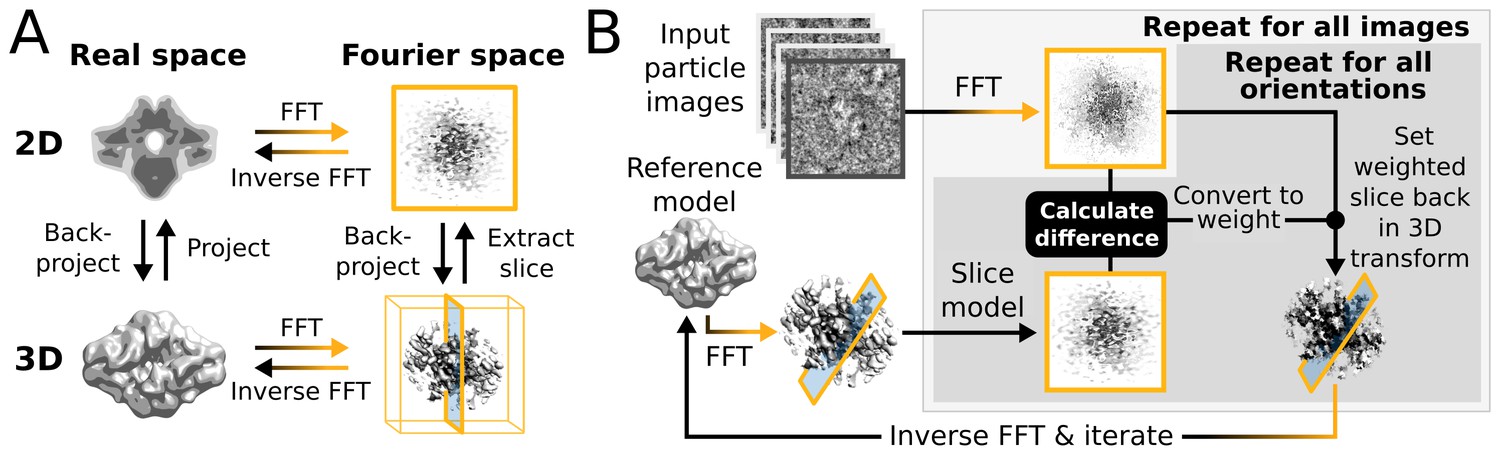
High level flowchart of RELION.
(A) Operations and the real vs. Fourier spaces used during (B) image reconstruction in RELION. Micrograph input and model setup use the CPU, while most subsequent processing steps have been adapted for accelerator hardware. The highlighted orientation-dependent difference calculation is by far the most demanding task, and fully accelerated. Taking 2D slices out of (and setting them back into) the reference transforms has also been accelerated at high gain. The inverse FFT operation has not yet been accelerated, but uses the CPU.
Figure 2
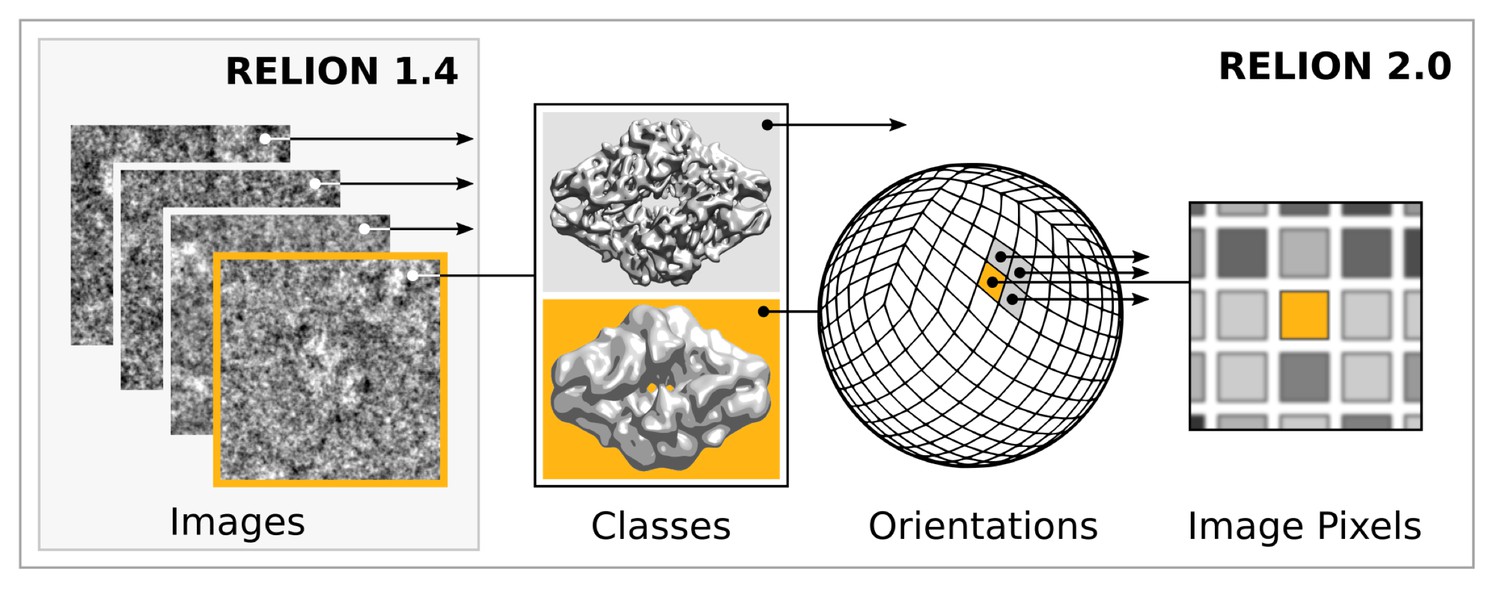
Extensive task-level parallelism for accelerators.
While previously relion only exploited parallelism over images (left), in the new implementation classes and all orientations of each class are expressed as tasks that can be scheduled independently on the accelerator hardware (e.g. GPUs). Even individual pixels for each orientation can be calculated in parallel, which makes the algorithm highly suited for GPUs.
Figure 3
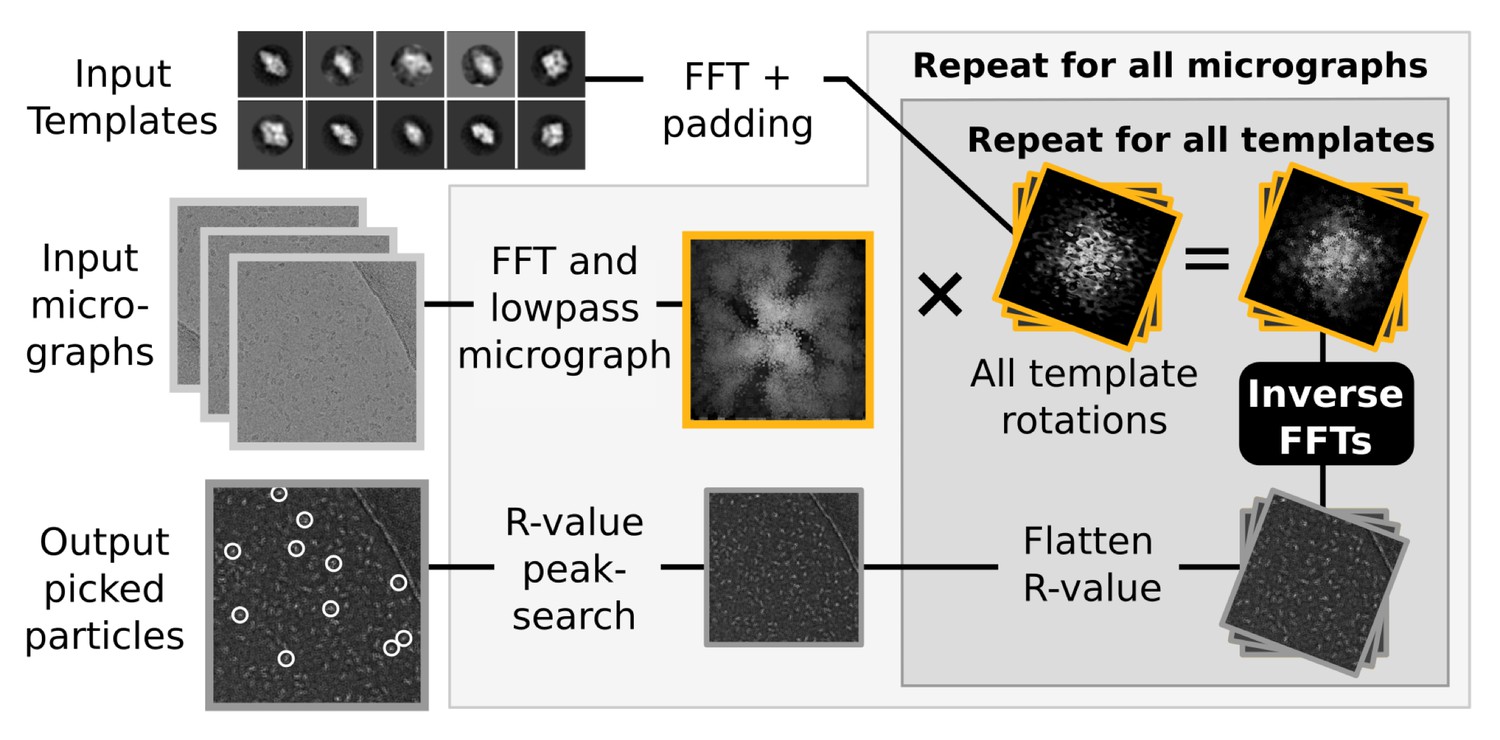
Semi-automated particle picking in RELION-2.
The low-pass filter applied to micrographs is a novel feature in RELION, aimed at reducing the size and execution time of the highlighted inverse FFTs, which accounts for most of the computational work. In addition to the inverse FFTs, all template- and rotation-dependent parallel steps have also been accelerated on GPUs.
Figure 4
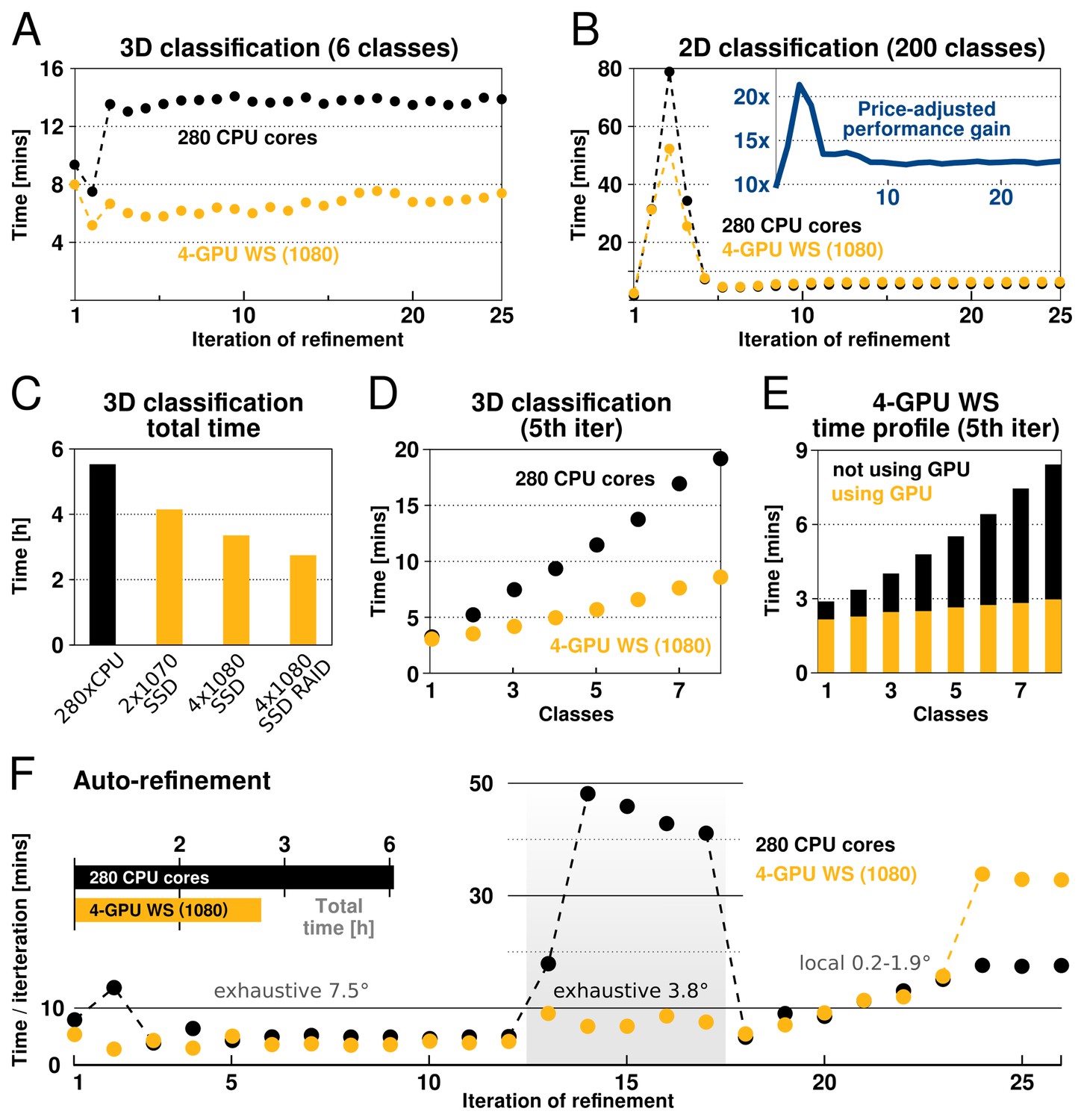
RELION-2 enables desktop classification and refinement using GPUs.
EMPIAR (Iudin et al., 2016) entry 10028 was used to assess performance, using refinements of 105 k ribosomal particles in 3602-pixel images. (A) A quad-GPU workstation easily outperforms even a large cluster job in 3D classification. (B) In 2D classification, the GPU desktop performs slightly better in the first few iteration and then provides performance equivalent to the 280 CPU cores. (C) Total time for 25 iterations of 3D classification for a few different hardware configurations. (D) Additional classes are processed at reduced cost compared to CPU-only execution, due to faster execution and increased capacity for latency hiding. (E) With increasing number of classes, the time spent in non-accelerated vs accelerated execution increases. (F) The workstation also beats the cluster for single-class refinement to high resolution, despite the generally lower degree of parallelism. This is particularly striking for the finer exhaustive sampling at 3.8 due to the GPU’s ability to parallelise the drastically increased number of tasks.
Figure 5 with 1 supplement
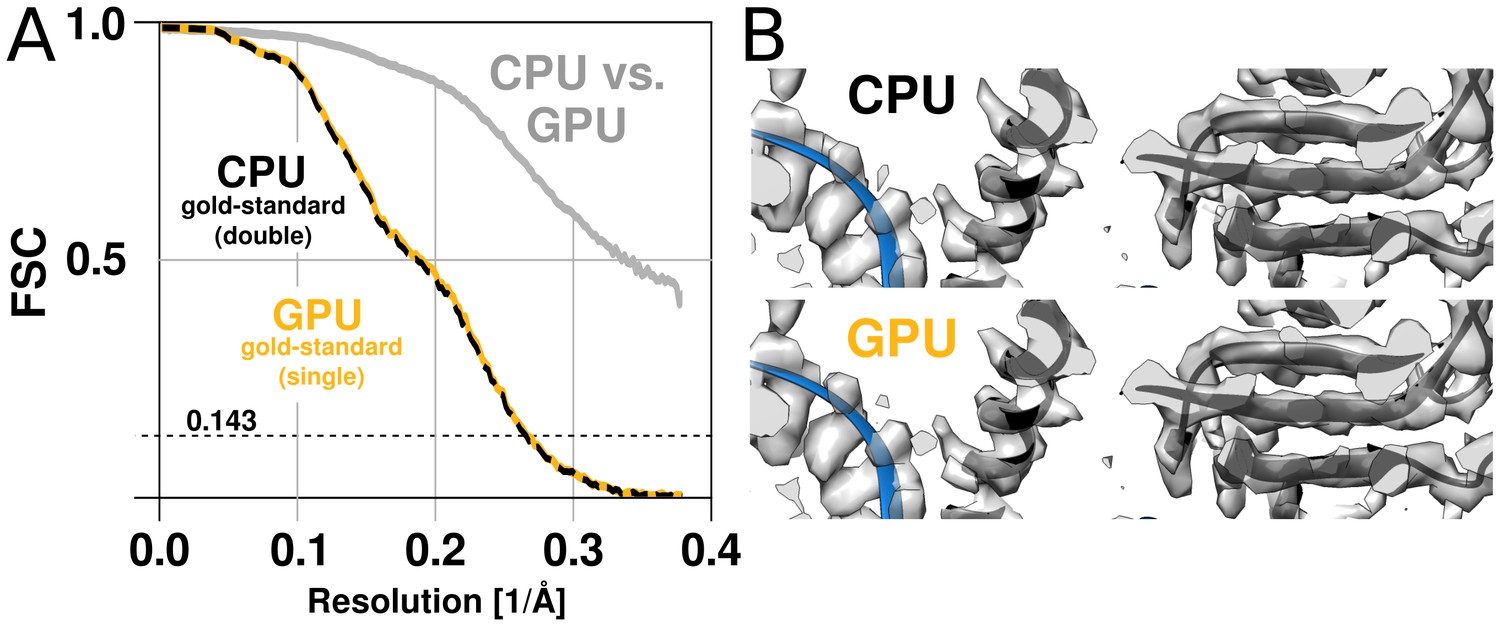
The GPU reconstruction is qualitatively identical to the CPU version.
(A) A high-resolution refinement of the Plasmodium falciparum 80S ribosome using single precision GPU arithmetic achieves a gold-standard Fourier shell correlation (FSC) indistinguishable from double precision CPU-only refinement (previously deposited as EMD-2660). The FSC of full reconstructions comparing the two methods shows their agreement far exceeds the recoverable signal (grey), and as shown in Figure 5—figure supplement 1 the variation in angle assignments match the differences between CPU runs with different random seeds. (B) Partial snapshots of the final reconstruction following post-processing, superimposed on PDB ID 3J79 (Wong et al., 2014).
Figure 5—figure supplement 1
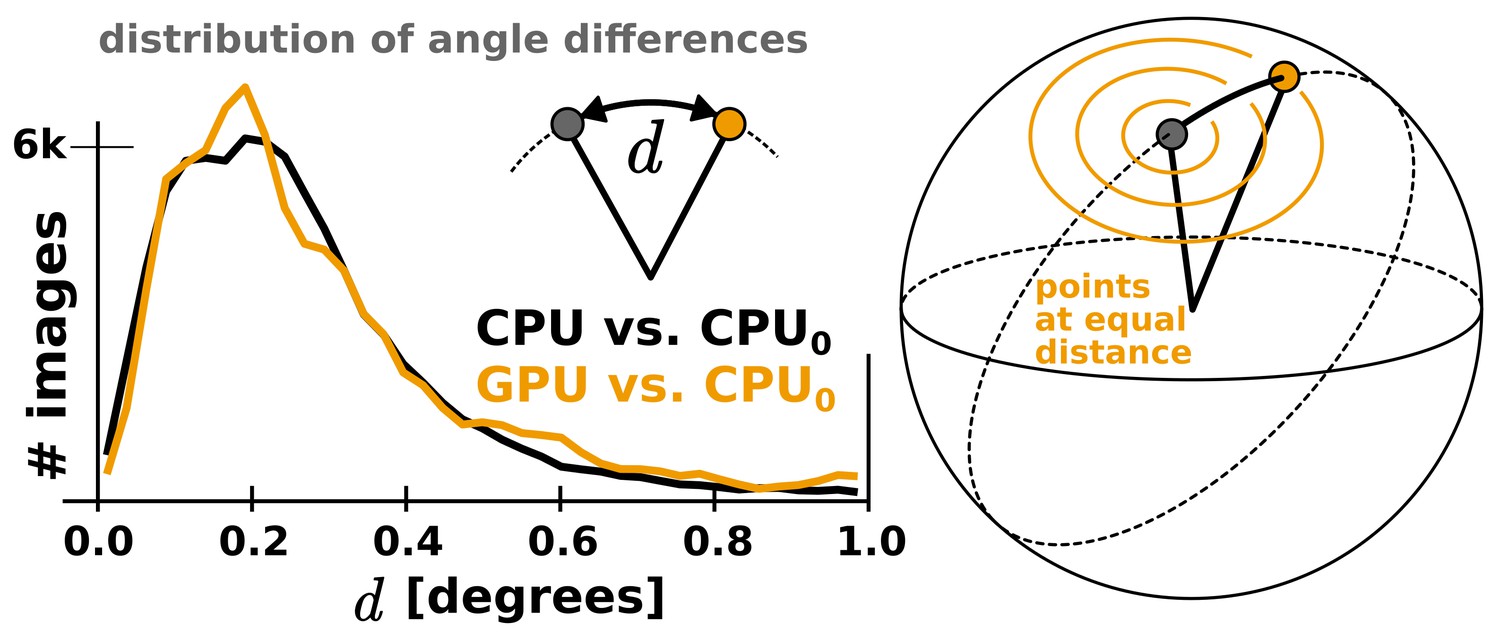
The CPU and GPU implementations provide qualitatively identical distributions of image orientations.
For two CPU runs with different random seeds, 81% of images fall within 1, and for a GPU vs. CPU run 82%. Note that the probability of observing small angles vanishes since the number of potentially available points is proportional to the sine of the angle, which approaches zero for identical orientations. Both distributions were aligned against the reference refinement by fitting reconstructed models.
Figure 6
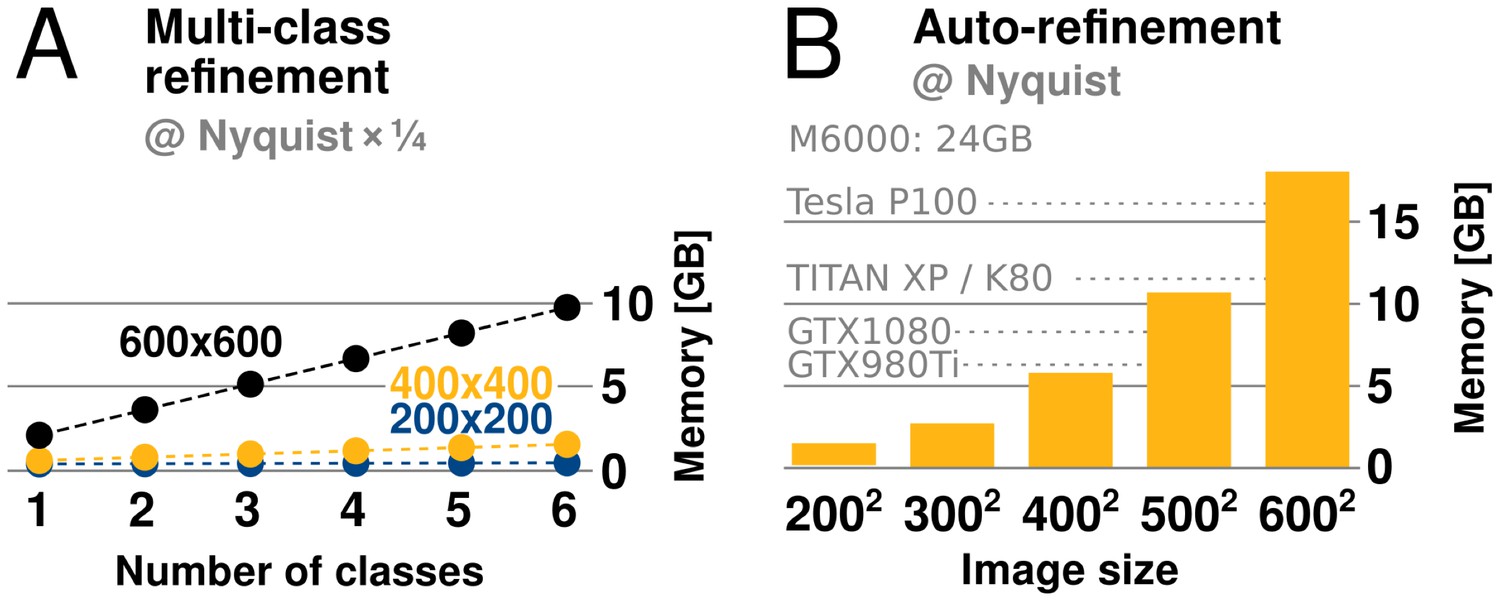
GPU memory requirements.
(A) The required GPU memory scales linearly with the number of classes. (B) The maximum required GPU memory occurs for single-class refinement to the Nyquist frequency, which increases rapidly with the image size. Horizontal grey lines indicate avaliable GPU memory on different cards.
Figure 7
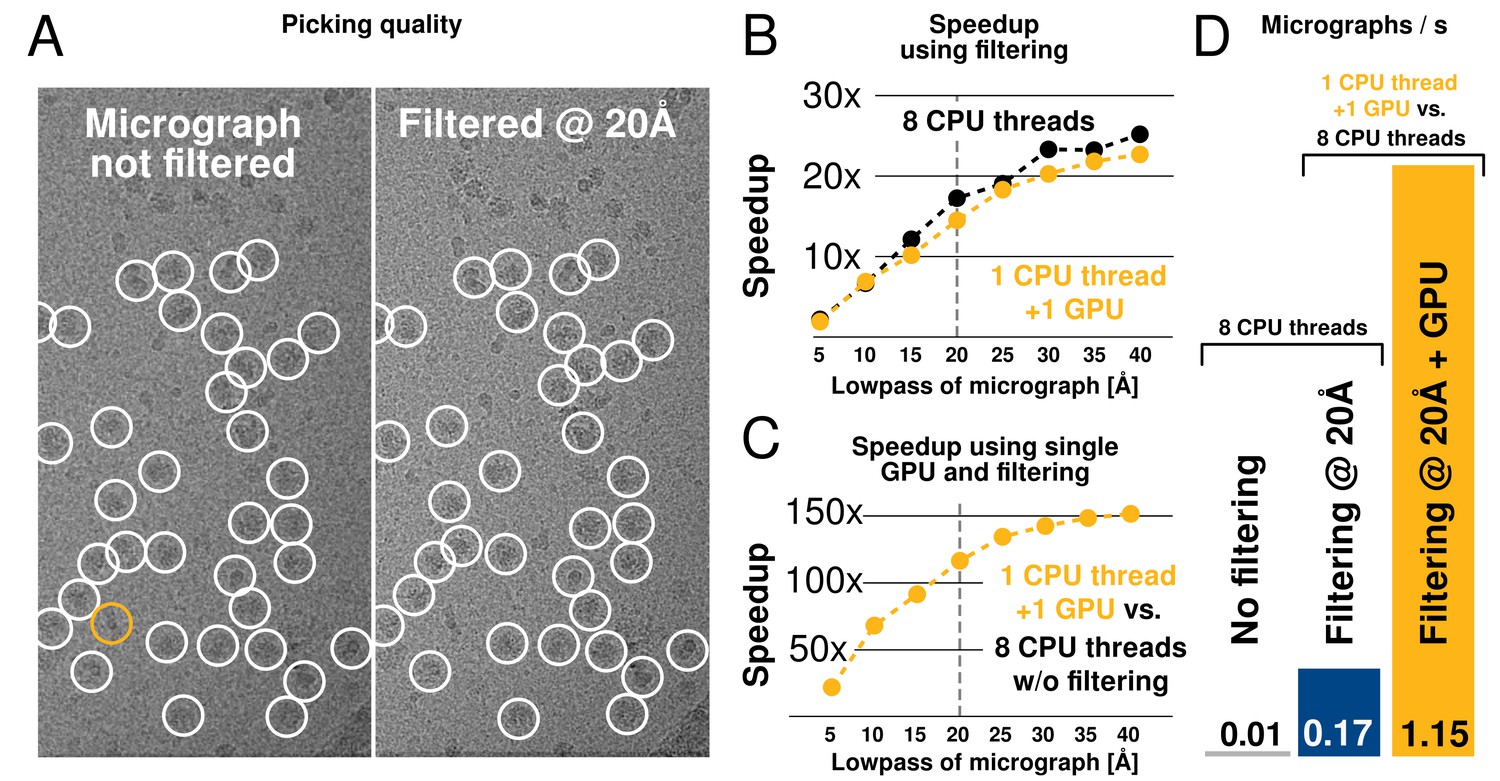
Low-pass filtering and acceleration of particle picking.
(A) Ribosomal particles were auto-picked from representative -pixel micrographs collected at 1.62 Å/pixel using four template classes, showing near-identical picking with and without low-pass filtering to 20 Å. The only differing particle is indicated in orange, and likely does not depict a ribosomal particle. (B–C) Despite near-identical particle selection, performance is dramatically improved. (D) Filtering alone provides almost 20-fold performance improvement on any hardware compared to previsos versions of relion, and when combined with GPU-accelerated particle picking the resulting performance gain is more than two orders of magnitude using only a single GPU (GTX 1080).
Figure 8
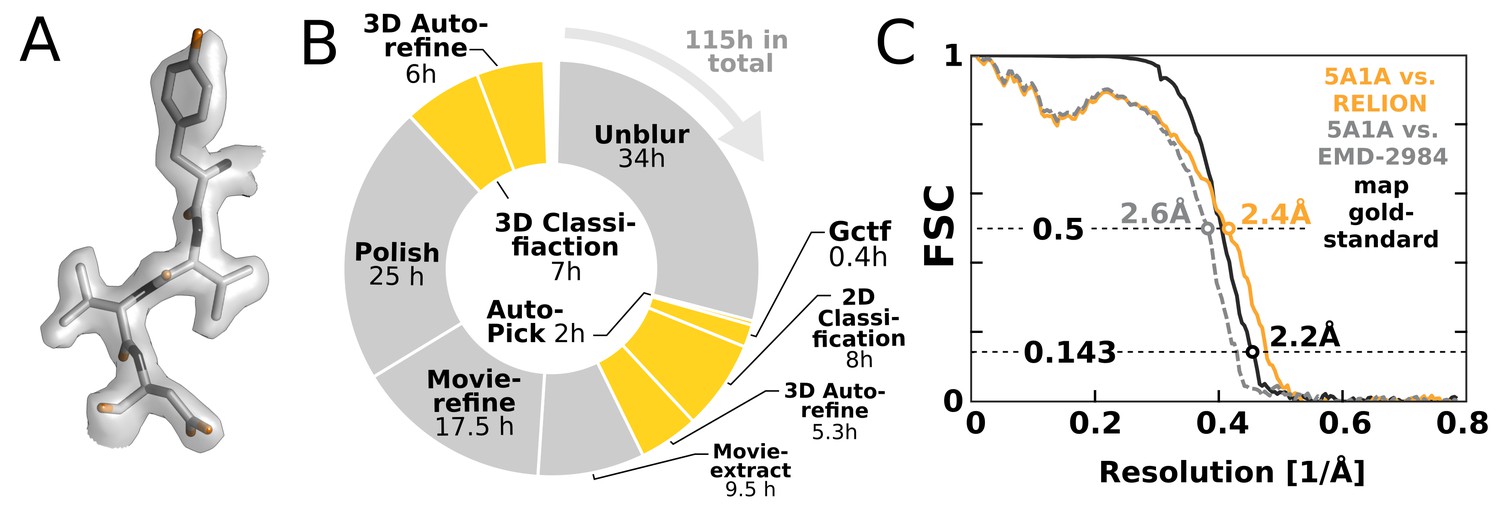
High-resolution structure determination on a single desktop.
(A) The resulting 2.2 Å map (deposited as EMD-4116) shows excellent high-resolution density throughout the complex. (B) The most time-consuming steps in the image processing workflow. GPU-accelerated steps are indicated in orange. The total time of image processing was less than that of downloading the data. (C) The resolution estimate is based on the gold-standard FSC after correcting for the convolution effects of a soft solvent mask (black). The FSC between the relion map and the atomic model in PDB ID 5A1A is shown in orange. The FSC between EMD-2984 and the same atomic model is shown for comparison (dashed gray).
Appendix 1—figure 1
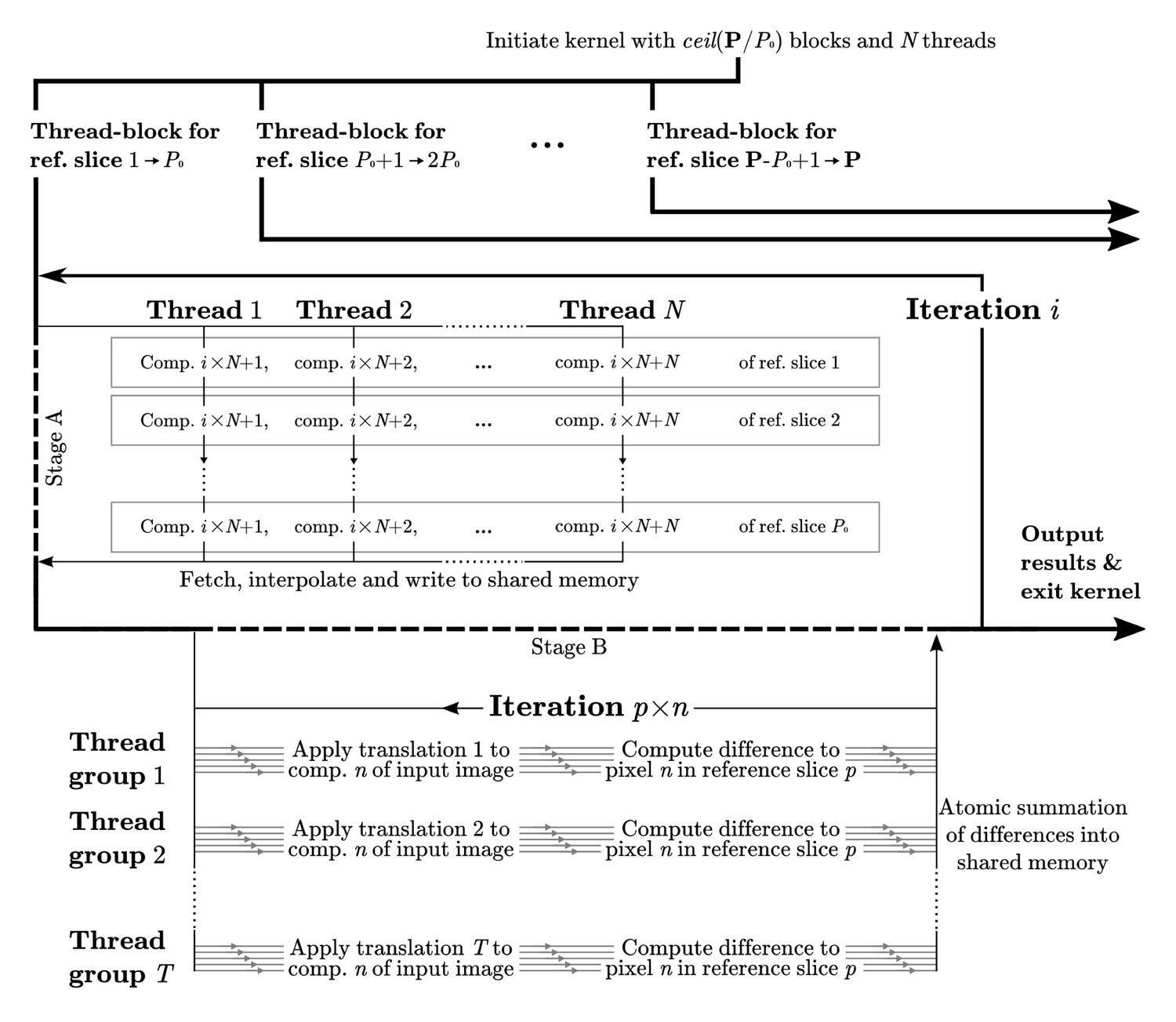
Computational flow in difference calculation kernel.
The kernel is initiated with thread-blocks and threads, where P is the total number of projections. The work flow of a thread-block in each iteration is divided into two stages. In stage A the pixels of reference slices are fetched through texture memory, interpolated, and stored in shared memory. This data is then exhaustively reused in stage B, where groups of threads compute the differences to the corresponding translated image components. Individual threads within a group work with different image components, , of each reference slice, . Collectively all threads iterate through the components of each reference slice, for a total of components for each iteration . The final result is reduced back into shared memory through atomic reduction operations. All image components are covered as goes from 1 to , where C is the total number of Fourier components. A reduced sum of differences for each pair of orientation and translation is written to global memory prior to the kernel exiting.
Appendix 1—figure 2
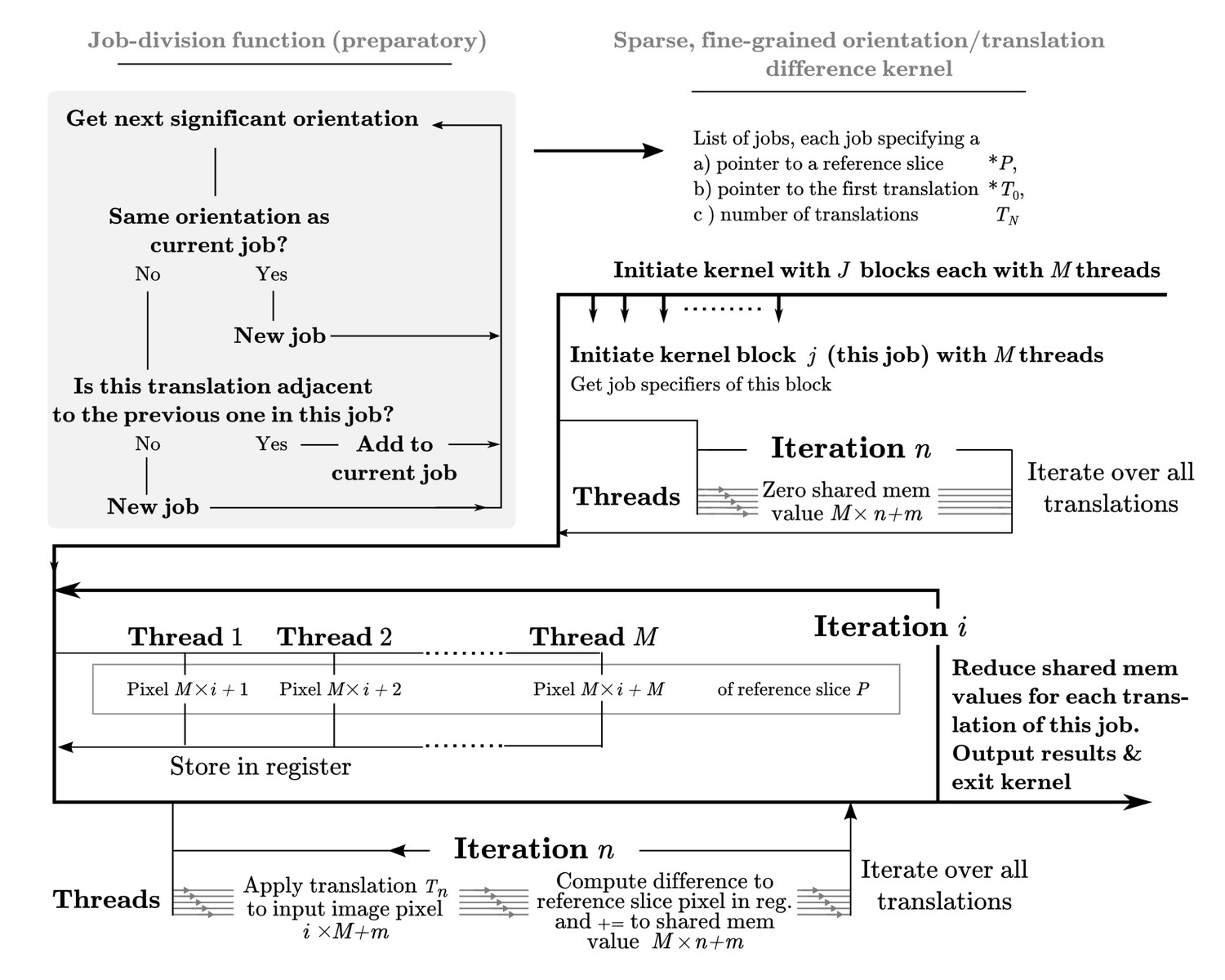
A dedicated kernel function performs the targeted fine-grained examination of the most significantly matching regions during image alignment against a reference model.
The oversampling of each of five fitting dimensions during fine-grained search renders storage of all possible weights intractable, so input and output data are stored with explicit mapping arrays. These are read by the kernel function thread-block, rather than inferred based on block ID. This creates overhead and possible latency of global memory access, which makes this kernel even further separated from the exhaustive kernel represented in Figure 9. Here, a pixel-chunk of a single projection is reused for a number of sequential translations, arranged contiguously if possible. Invoking separate thread blocks for non-contiguous translations allows some implicit indexing of them, which affords better access patterns for SIMD instructions and reduced latency. Due to the sparseness, shared memory can also be used for in-kernel summation of all pixels of each image, which despite some some required explicit thread-level synchronisation increases throughput by avoiding the higher latency of atomic write operations during image summation in the coarse-search kernel.
Appendix 2—figure 1
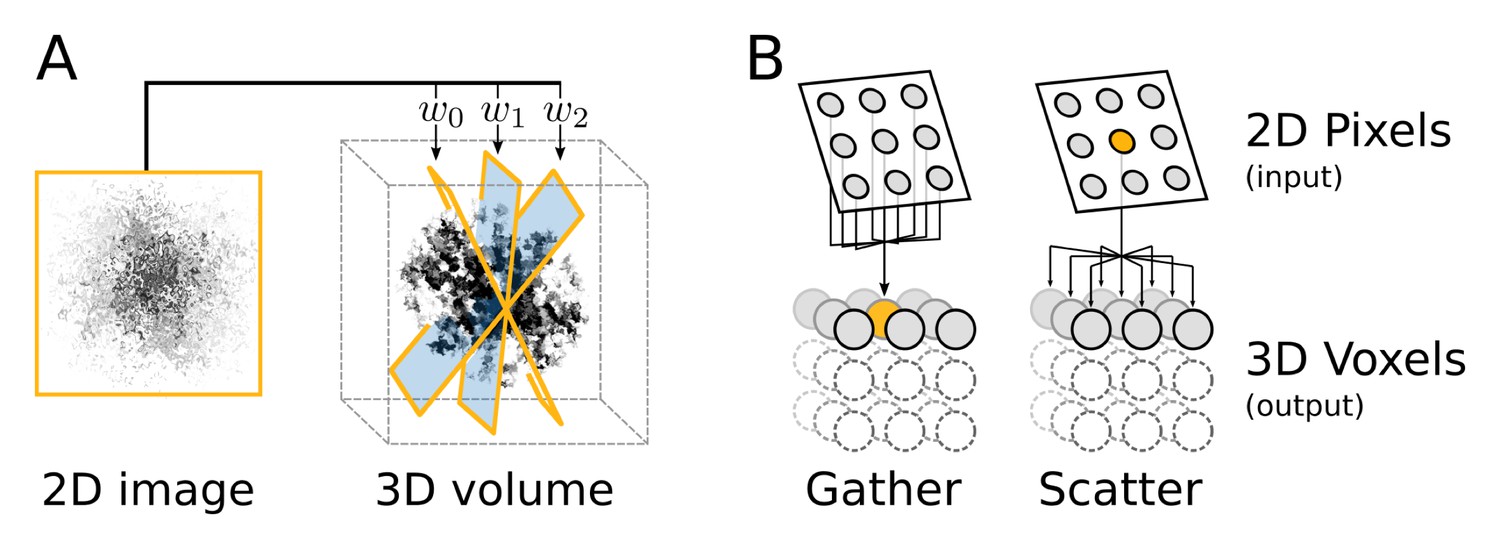
Computational flow of fine-grained search kernel.
(A) Weighted back-projection of a 2D image into three different planes. We explored two memory access approaches (B) for this task, namely gather and scatter. In the gather approach a process (marked with orange) is assigned to individual or groups of 3D voxels. The process read from the input image and updates the data of the assigned voxel(s).
Tables
Table 1
Quality and speed of autopicking for the -galactosidase benchmark. Comparing the CPU version with the GPU version using increasing levels of low-pass filtering yields progressively higher recalls at similar FDRs. The GPU version yields identical results to that of the CPU version, but at a much reduced computational costs. Filtering does not depend on GPU-acceleration, and will perform similarly using only CPUs.
| Code | Filter | # picked | Recall | FDR | Time | Performance in |
|---|---|---|---|---|---|---|
| (Å) | particles | (s/micrograph) | CPU core units | |||
| CPU | none | 54,301 | 0.88 | 0.34 | 1,227 | 1 |
| GPU | none | 54,325 | 0.88 | 0.34 | 10 | 122 |
| GPU | 5 | 55,629 | 0.90 | 0.34 | 5.8 | 211 |
| GPU | 10 | 55,886 | 0.90 | 0.34 | 2.1 | 584 |
| GPU | 15 | 56,450 | 0.92 | 0.33 | 1.6 | 766 |
| GPU | 20 | 57,361 | 0.95 | 0.33 | 1.3 | 943 |
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2
eLife 5:e18722.
https://doi.org/10.7554/eLife.18722




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}