Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome
- University of California, Santa Barbara, United States
- University of Utah School of Medicine, United States
- Southern Illinois University, United States
- J. Craig Venter Institute, United States
- Eli and Edythe L. Broad Institute of Harvard and MIT, United States
- Institute of Molecular Biotechnology, Austria
- The Johns Hopkins University School of Medicine, United States
- MRC Laboratory of Molecular Biology, United Kingdom
- Cornell University, United States
- Chinese Academy of Sciences, China
Figures
Figure 1 with 1 supplement
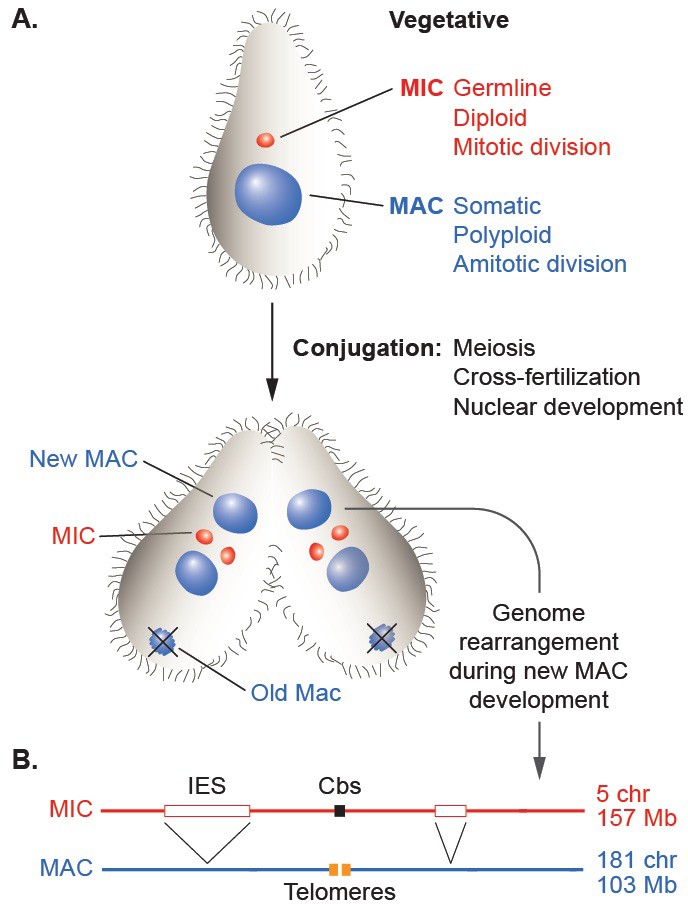
Nuclear dualism and genome rearrangement in Tetrahymena.
(A) Schematic of two stages of Tetrahymena life cycle showing major characteristics of micronuclei (MIC; red) and macronuclei (MAC; blue) and nuclear events of conjugation. (B) Main events of programmed genome rearrangement. A portion of the MIC genome is shown in red, with internal eliminated sequences (IES) shown as open boxes and the Cbs sequence in black. The corresponding MAC regions (blue) lack the IESs, with the flanking MAC-destined sequences (MDSs) joined (represented by ^ symbols). Breakage and addition of telomeres (orange boxes) has occurred at the former site of the Cbs.
Figure 1—figure supplement 1
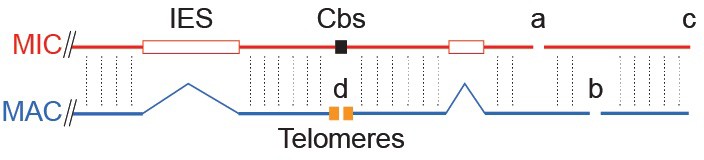
Tiling method used to extend scaffolds into super-assemblies.
The same genomic region as in Figure 1B is represented as portions of the MIC and MAC genome sequence assemblies. Cross-sequence alignment is represented by vertical dotted lines. Inter-scaffold assembly gaps are indicated by letters a, b, and c, for gaps in the MIC, MAC and both assemblies, respectively. The ends of MAC chromosome scaffolds terminating in telomere repeats are indicated by the letter d. When the ends of two MIC scaffolds align to adjacent regions of the same MAC scaffold (letter a), we may infer that these ends are adjacent in the MIC genome (unlike certain ciliates with highly 'scrambled' MIC genomes, T. thermophila's MAC chromosomes are nearly always colinear with their MIC counterparts). The same principle applies to joining MAC scaffolds and chromosomes (letters b and d). This process could partially be carried out computationally, but human judgments (performed independently in two labs) were necessary in many cases to resolve alternative joining paths resulting from repetitive sequences or apparent MIC genome mis-assemblies (see Supplementary file 1C). Unfortunately, both assemblies often 'break' in the same region (letter c), leaving no 'bridge' to the next scaffold in either. This occurred most often in regions abundant in smaller scaffolds, which we later found tend to be located near the middle of MIC chromosomes. Combining tiling and other data, as described in the main text, we constructed ‘best approximation’ MIC chromosome super-assemblies that incorporate 97% of the MIC genome assembly.
Figure 2 with 1 supplement
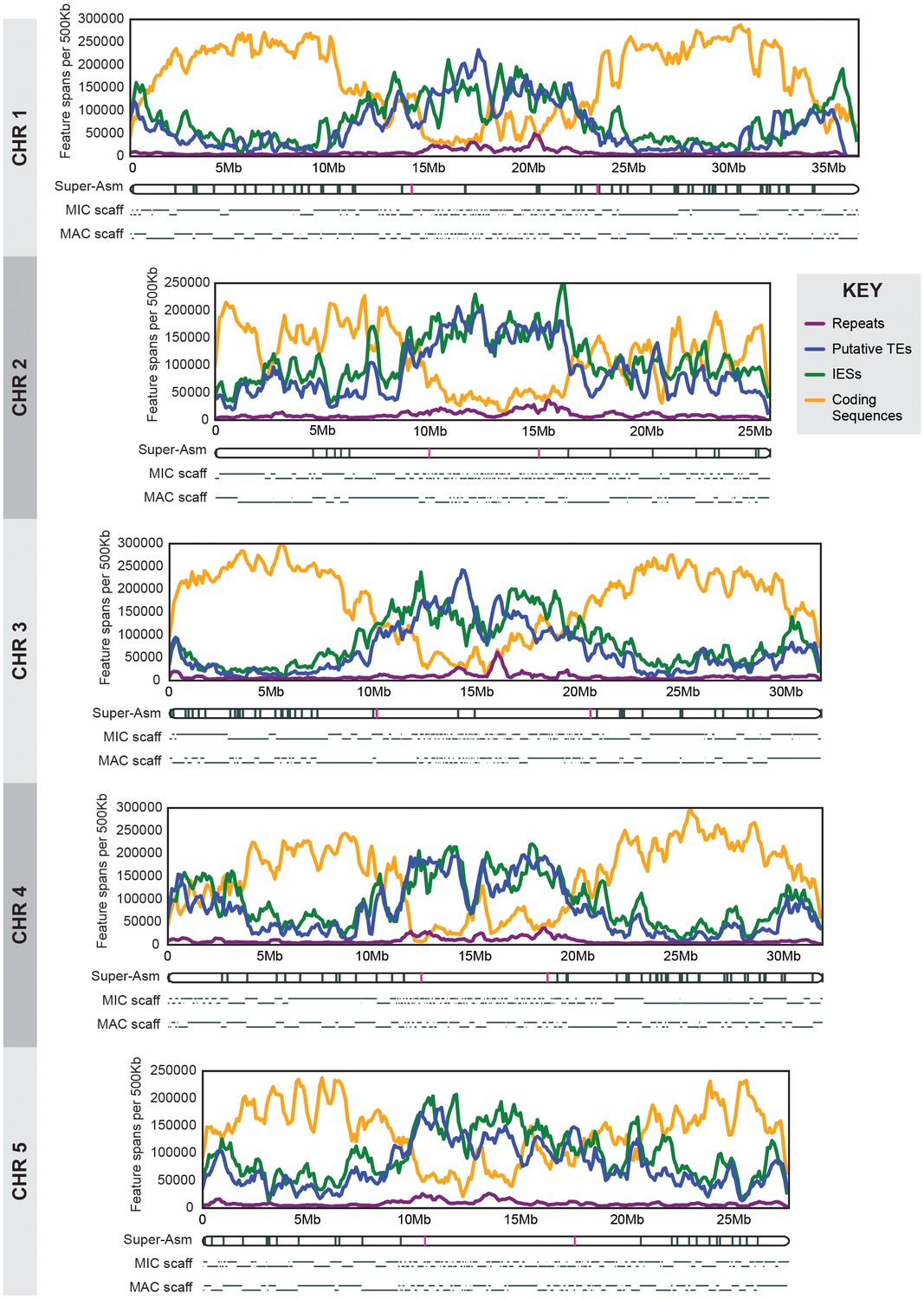
MIC chromosome landscapes.
For each chromosome, the top panel shows the density of several genomic features, measured as number of base pairs (span) per 500 kb sliding window (100 kb slide increment). Purple = simple sequence repetitive DNA (note that exclusion of those simple sequence repeats that overlap with TEs has minimal effect on the distribution pattern). Blue = putative TEs. Green = high-confidence IESs. Orange = protein-coding sequences. The corresponding chromosome-length super-assembly (Super-Asm) is shown immediately below, each Cbs indicated by a vertical tick. Red ticks indicate Cbs’s flanking putative centromeres (see main text and Figure 2—figure supplement 1). In the 'MIC-scaff' schematic, the scaffolds comprising each MIC chromosome super-assembly are depicted as horizontal lines (alternating in vertical position to delineate each from its neighbors). The ‘MAC-scaff’ schematic indicates the positions of MAC scaffolds (many of which are complete, fully sequenced MAC chromosomes) derived from the corresponding regions of the MIC chromosome. Note that, because IESs are absent from MAC scaffolds, their lengths are actually shorter, but for simplicity of viewing, these lengths have been stretched so that MAC-scaff endpoints line up with their corresponding positions in the MIC. Chromosomes are stacked so that their centers align vertically.
Figure 2—figure supplement 1
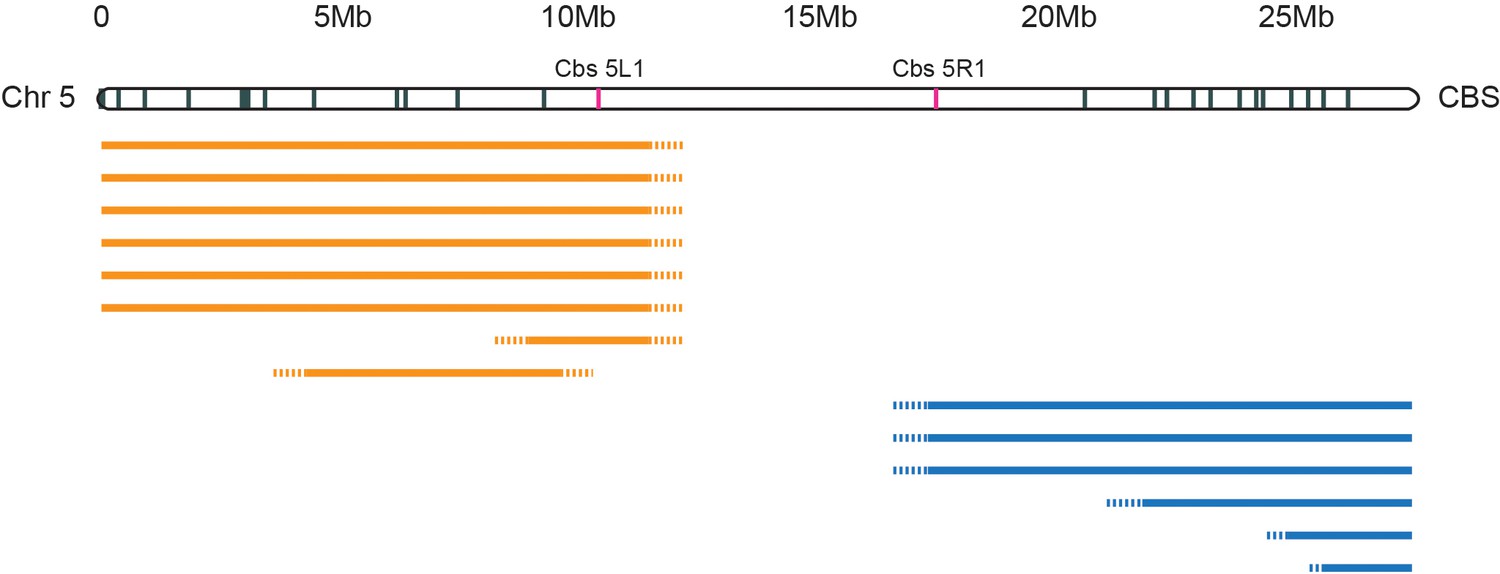
Deletion mapping of Tetrahymena centromeres.
In a separate study, Cassidy-Hanley et al. isolated a collection of strains carrying partial or complete MIC chromosomal deletions. Such deletions are viable because their expressed MAC contains the complete genome. MIC chromosomes are transcriptionally silent, but deletions that mitotically destabilize a chromosome by compromising centromere function would not be recovered. We mapped the extent of the deletions relative to Cbs’s spread along the length of each MIC chromosome. The figure shows mapping of Chromosome 5 deletions as a representative example. The extents of left arm deletions are indicated by orange lines; right arm deletions by blue lines. Six independent deletions removed all the Cbs’s on the left arm while three others removed all the Cbs’s on the right arm (the precise endpoints have not yet been mapped, as indicated by the dotted line termini). Five arm-specific, smaller deletions were also mapped, as shown. Only whole chromosome deletions were recovered that removed both Cbs 5L1 and Cbs 5R1, marked in red, as in Figure 2. We infer that sequences essential for centromere function lie between these two Cbs’s.
Figure 3
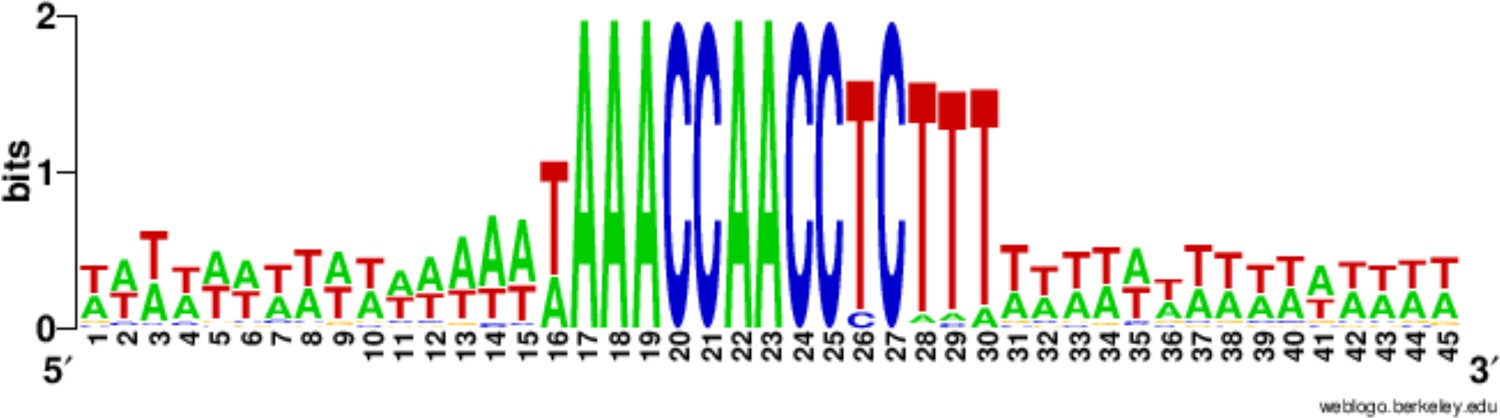
Conservation of the 15 bp chromosome breakage sequence.
Nucleotide conservation was calculated at every position, as described in (Hamilton et al., 2006a), for the 225 Cbs’s and their 15 bp flanking sequences, aligned on the C-rich Cbs strand. The Cbs element occupies positions 16 to 30. At any given position in the logo plot, two bits represent maximum conservation (only one nucleotide occupies that position), and 0 bits corresponds to no conservation (all four nucleotides are equally frequent).
Figure 4
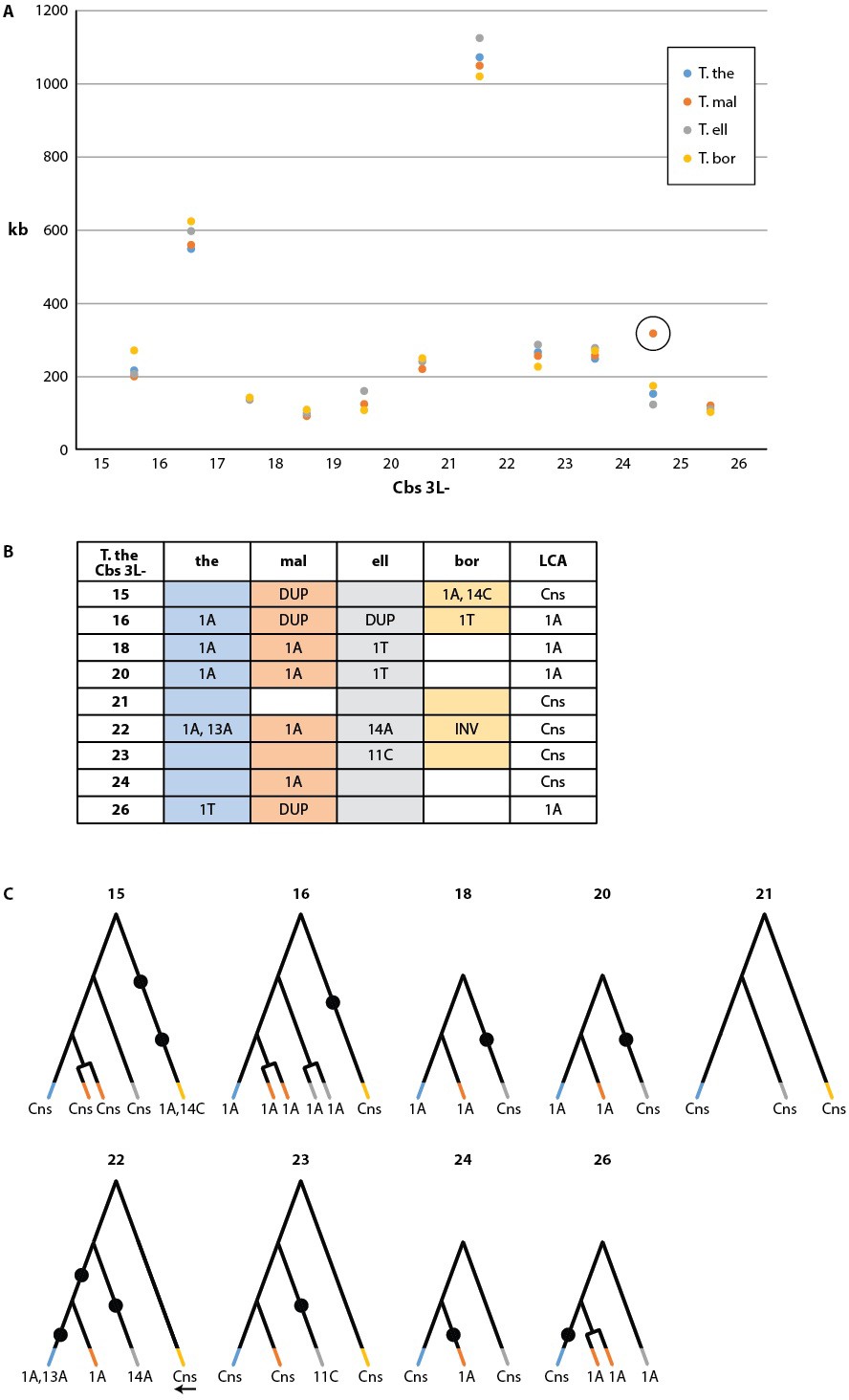
Conservation of chromosome breakage sites and Cbs in four Tetrahymena species.
(A) Conservation of MAC chromosome lengths: X-axis: Cbs 3L-15 to 26 (evenly spaced). Y-axis: Length of the MAC scaffolds in each species whose ends are defined by the flanking Cbs’s. Circle: an extra Cbs site in T. malaccensis creates two MAC chromosomes in this region; length = sum of the two MAC chromosome lengths. (B) Summary of Cbs sequence data at nine chromosome breakage sites; filled in box = sequence available; if no text = single, consensus Cbs in same orientation as T. thermophila; Cbs sequence variants, duplications (DUP) and inversion (INV) indicated; final column = possible last common ancestor (LCA) Cbs, requiring a minimum number of mutations in the clade. (C) Inferred possible descent from Cbs of LCA at each of the nine chromosome breakage sites. Branch tips: Cbs consensus (Cns) or variant in T.the, T.mal., T.ell., and T.bor, in that order (colors consistent with parts A and B; missing branch = unsequenced Cbs). Terminally split branch = local Cbs duplication. Dots indicate minimal number of mutational events; placed in the longest branches when there is a choice. Reverse arrow (T. bor. 3L-22) indicates Cbs inversion.
Figure 5 with 4 supplements
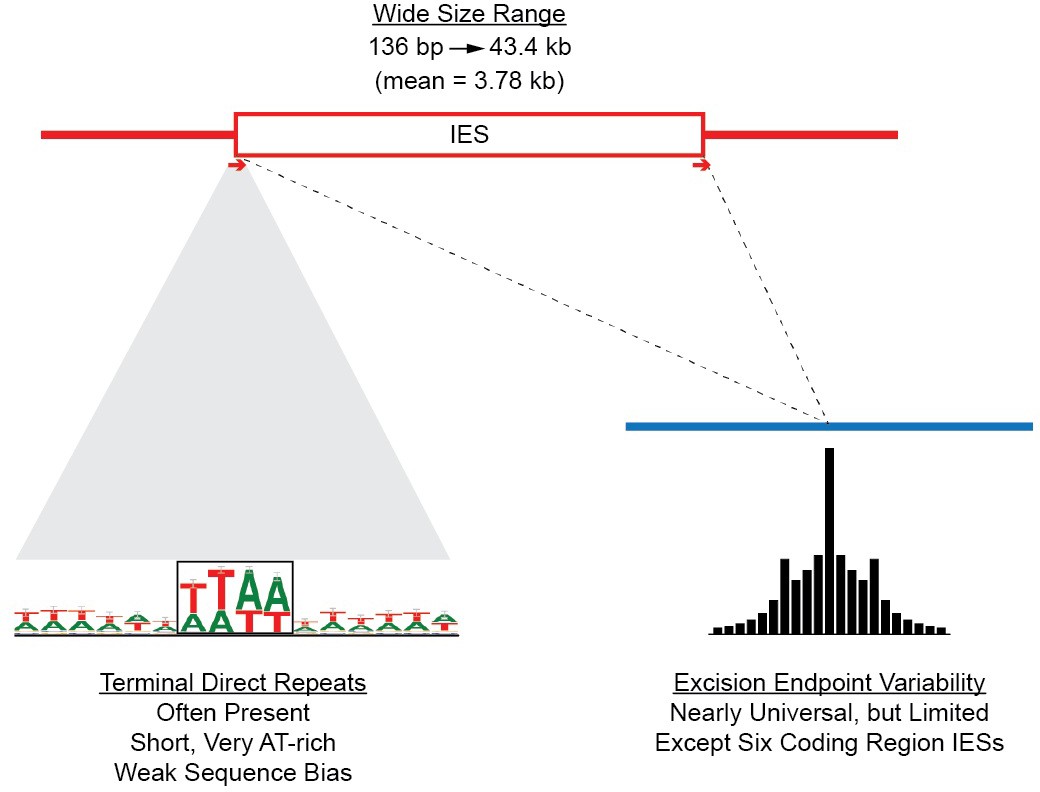
Summary of IES structural features.
Red lines = MIC DNA. Blue lines = MAC DNA. A representative IES is indicated by the open red box. IESs were identified as described in Figure 5—figure supplement 1. Their size distribution is shown in Figure 5—figure supplement 2. The excision endpoint found in the SB210 MAC genome is indicated by the slanted lines converging to the right. Sequences from a large progeny pool representing multiple, independent excision events show most progeny share the parental endpoint, but variation within a limited range is common, as shown in detail in Figure 5—figure supplement 3. The left terminal junction sequences is shown blown up below and to the left. Short Terminal Direct Repeats (TDRs) are often found; they are generally very AT-rich and have a slight sequence pattern bias. A 4 bp TDR sequence logo is shown as an example. More detailed characterization of endpoint TDRs is presented in Figure 5—figure supplement 4.
Figure 5—figure supplement 1

Read alignment methods used for IES dentification.
(A) MAC Sanger sequencing reads (blue arrowed bars) align to MAC scaffolds (thick blue bar) along entire length, but their alignment to MIC scaffold (thick red bar) is interrupted by IES (black bar). (B) Alignment of Illumina MIC reads (red arrowed bars) to margins of IESs is uninterrupted in MIC scaffolds, but broken at 'residual' IES locations in MAC genome. (C) Short direct repeats (grey) at IES/MDS junctions in MIC lead to overlapping read alignment to MAC scaffolds.
Figure 5—figure supplement 2
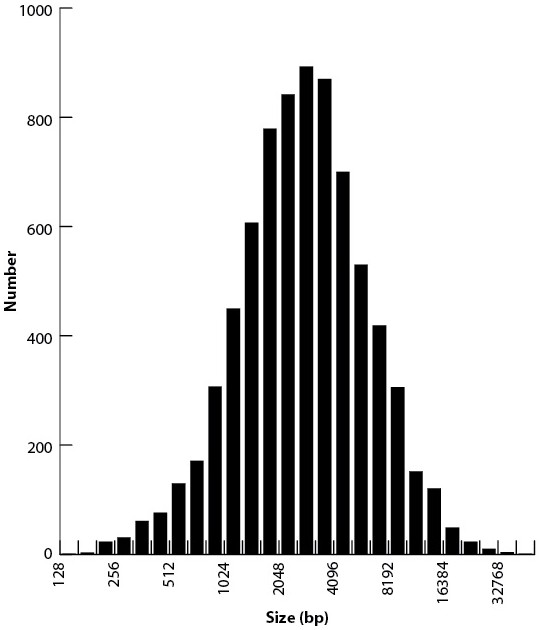
Size distribution of 7551 high-confidence IESs.
Note that the x-axis is log base two transformed.
Figure 5—figure supplement 3
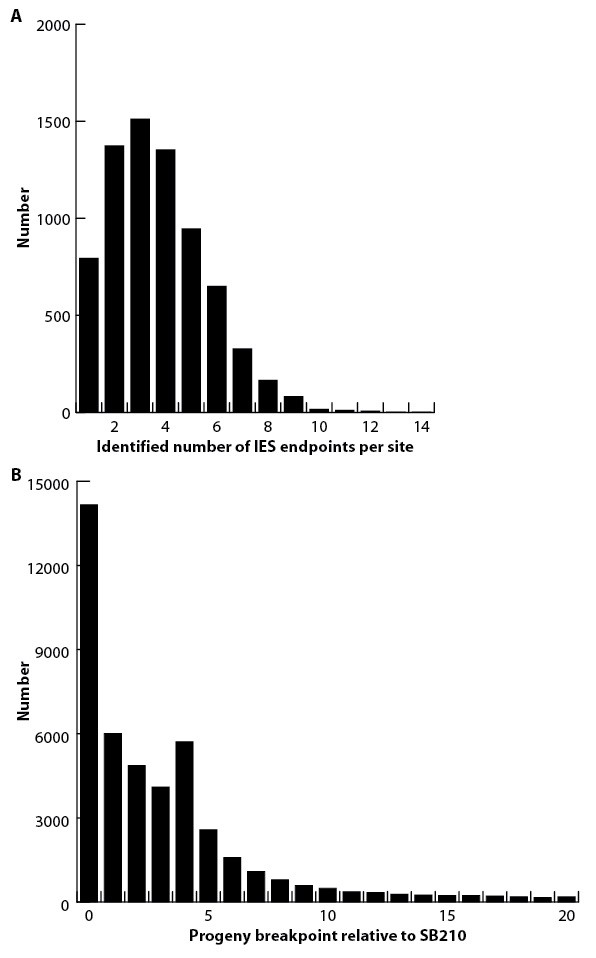
IES excision variability.
(A) The number of variant excision endpoints detected within the progeny pool at each IES site (calculated using excision sites for which data are available for both SB210 and progeny). It is difficult to reliably quantify the degree of endpoint variation because the values depend on several factors, including the number of progeny cells used in DNA purification, the depth of sequencing coverage, the method of mapping endpoints, and the criteria by which endpoints are validated. For this study, endpoints were mapped by the 'split read alignment' method (Figure 5—figure supplement 1A). For validation, at least three identical, independent read alignments were required. Number of progeny cells and sequencing coverage are described in 'Materials and methods'. (B) The positions of progeny pool read alignment breakpoints were mapped relative to the SB210 read alignment breakpoint reference (distances in either direction were added together). The greatest number of progeny breakpoints is identical to the SB210 reference (point 0) Nearly identical results were observed in comparison to SB1969 (data not shown). The frequency of alternative breakpoints generally decreases with increasing distance from the reference, with the exception of a small peak at a distance of 4 bp.
Figure 5—figure supplement 4
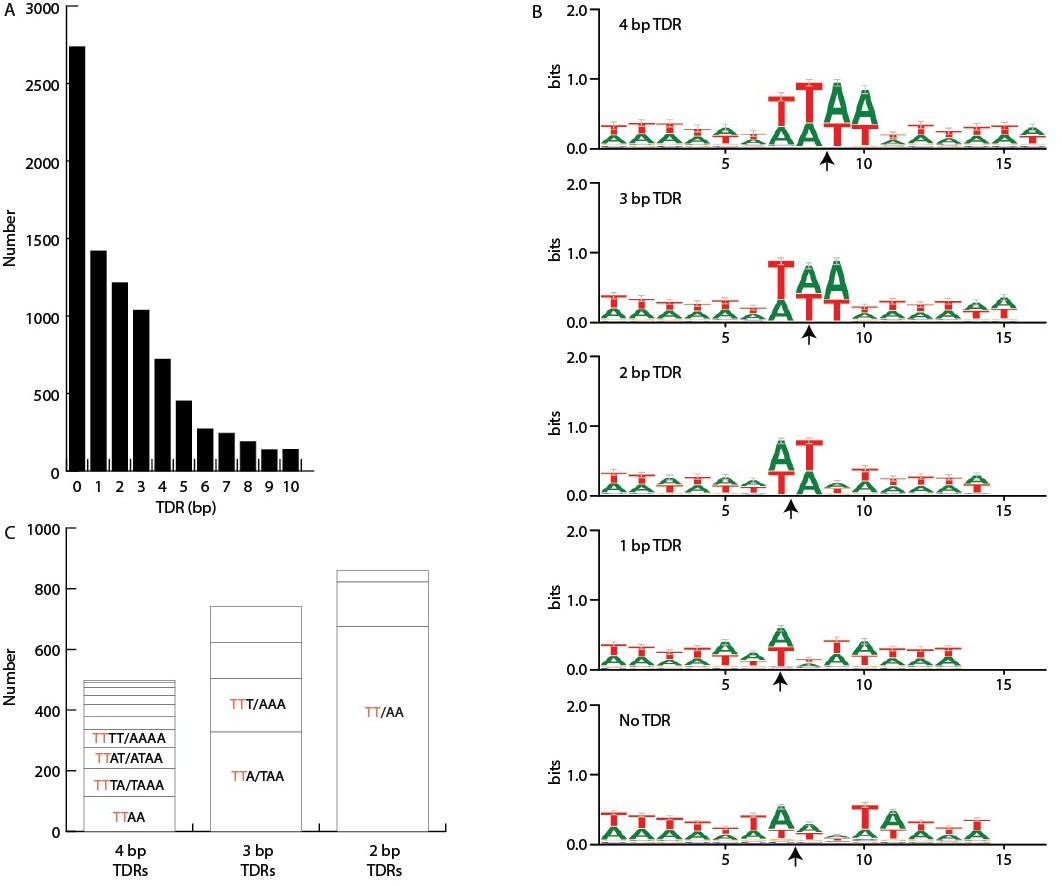
IES/MDS junctions.
Short terminal direct repeats (TDRs) at IES termini were identified by examining alignments between the MIC and MAC genomes at these termini to identify alignment overlaps (i.e. short MAC sequences at precisely the site of excision that align to both precise ends of the IES). These results were confirmed by alignment of MIC sequencing reads to the MAC genome assembly (see Figure 5—figure supplement 1C). (A) TDR length. Numbers of junctions with TDRs of the indicated lengths, showing that IESs with no TDR constitute the largest class. (B) A+T richness. For each of the five TDR classes between 0 and 4 bp, the direct repeat (or two flanking bases, in the case of no overlap) plus six bases on either side were extracted from the MIC genome sequence and aligned (MAC-destined sequence to the left; MIC-limited sequence to the right). Each arrow indicates the center of the TDR (or, in the case of No TDR, the junction point). Sequence logos derived from the alignments show that the TDRs are more AT-rich than surrounding sequence. Bases within the four, three, and two base direct repeats are approximately 97% AT overall and the one base direct repeats are 92% AT, whereas the two bases flanking the 'zero overlap' junctions are 80% AT, similar to the adjacent sequence composition. (C) Sequence pattern bias. In addition to overall AT-richness, the sequence patterns of the TDRs are not entirely random. We compared the frequency of each of the possible TDRs between 2 and 4 bp in length that consist of only As and Ts. Reverse complementary sequences were found to have approximately equal frequencies, as expected because the orientation of the sequenced strand is random, and they were grouped together. This makes for 10 groupings of 4 bp TDRs, 4 groupings of 3 bp TDRs, and 3 groupings of 2 bp TDRs. As shown in this panel, the frequencies of each grouping are unequal; the most common are: 4-mer TTAA (palindromic), 3-mer TTA/TAA, and 2-mer TT/AA (the latter two are pairs of reverse complementary sequences). Furthermore, it is notable that the four most common groupings of 4-mers all contain one member with a 5' TT dinucleotide (red font) and together account for two thirds of the total 4-mers. Likewise, the two (out of four total) 3-mer groupings containing a 5' TT dinucleotide account for two thirds of 3-mers, and the single TT/AA 2-mer grouping accounts for over three quarters of all 2-mers. These findings suggest that IES junctions have a slight bias in favor of beginning with TT and an extended weak consensus of 5'-TT(A)(A)−3', the most common 2, 3, and 4 bp TDRs (but far from the majority) including successively more of the consensus, from left to right.
Figure 6 with 2 supplements
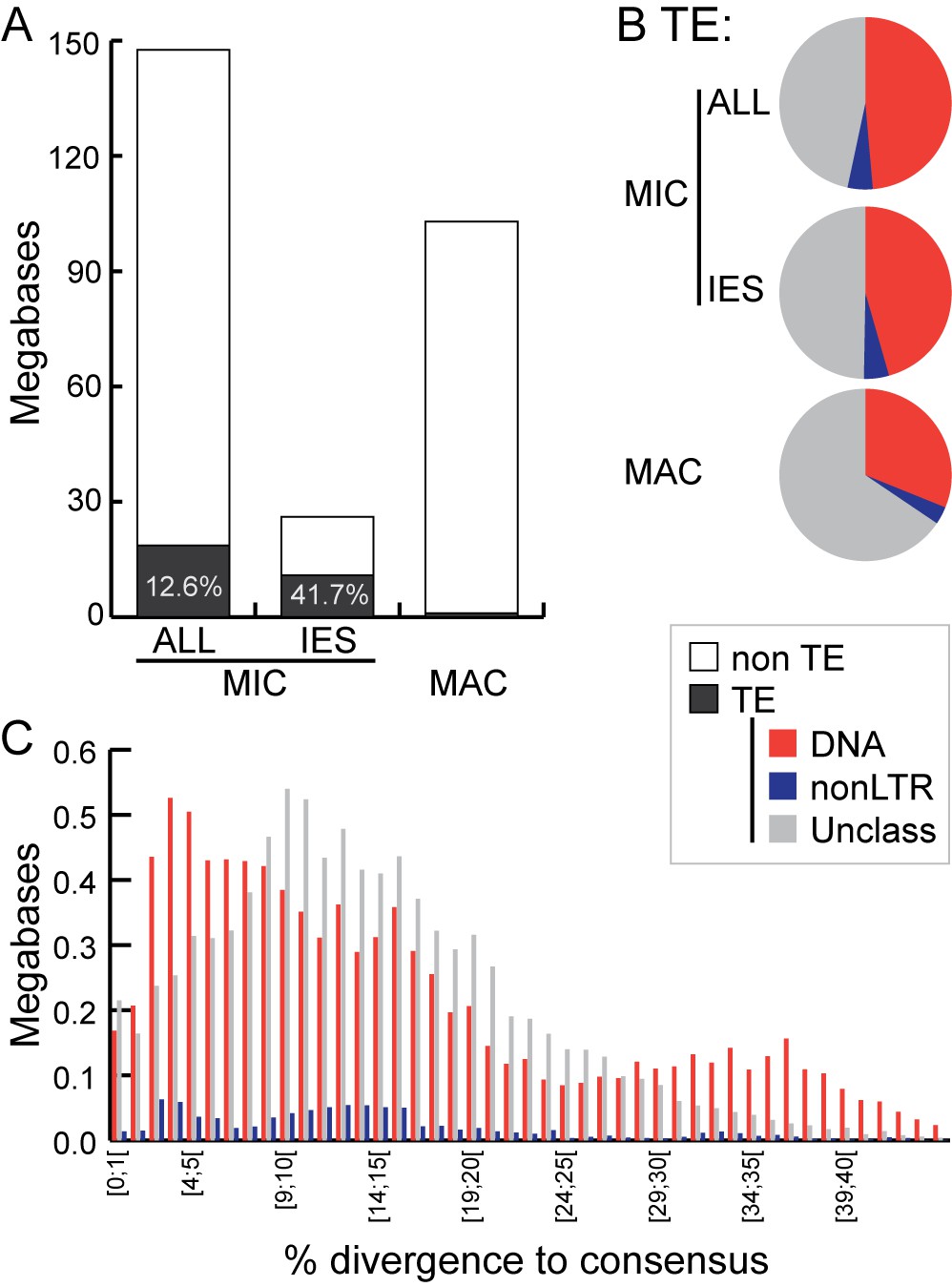
Transposable element landscape.
(A) Proportion of DNA annotated as TEs (black) or unannotated (white) using RepeatMasker (Smit et al., 2015) and a custom putative TE library (see text). MAC putative TE content is about 1 Mb, potentially corresponding to a mixture of TE sequences retained in the MAC assembly and repeats not corresponding to TEs still in the library. (B) Proportion of putative TEs by class for MIC (ALL and high-confidence IESs) and MAC. In MIC(ALL), the most abundant elements (besides unclassified) correspond to DNA TEs (‘cut-and-paste’, Mavericks and Tlr elements). More than half of the MIC(ALL) non-LTR elements could be annotated as LINE1 elements. (C) Evolutionary view of putative TEs in the MIC. For each class, amounts of DNA are shown as a function of the percentage of divergence to the consensus (by bins of 1%), as a proxy for age: the older the TE invasion, the more copies will have accumulated mutations (higher percentage of divergence, right of the graph). Conversely, sequences corresponding to youngest elements show little divergence (left of the graph).
-
Figure 6—source data 1
Tetrahymena putative TE library.
Consensus sequences of putative TEs of the Tetrahymena germline genome (fasta format): combination of de novo repeat library and TEs from the literature. See 'Materials and methods', 'Repeat Analysis'.
- https://doi.org/10.7554/eLife.19090.018
-
Figure 6—source data 2
Details of putative TEs contribution to the MIC chromosome super-assemblies.
Detailed metrics for each putative TE contribution to Tetrahymena MIC chromosomes super-assemblies, such as numbers of fragments, total amount, average divergence to consensus (proxy for age), etc. Obtained by parsing the RepeatMasker output (.out file), using https://github.com/4ureliek/Parsing-RepeatMasker-Outputs/blob/master/parseRM_simple.pl.
- https://doi.org/10.7554/eLife.19090.019
-
Figure 6—source data 3
Putative TE annotation of high-confidence 7551 IESs.
Raw RepeatMasker output of the high confidence set of 7551 IESs, with two additional columns: the IES identifier (as in Supplementary file 3A) and the total putative TE content of each IES.
- https://doi.org/10.7554/eLife.19090.020
Figure 6—figure supplement 1
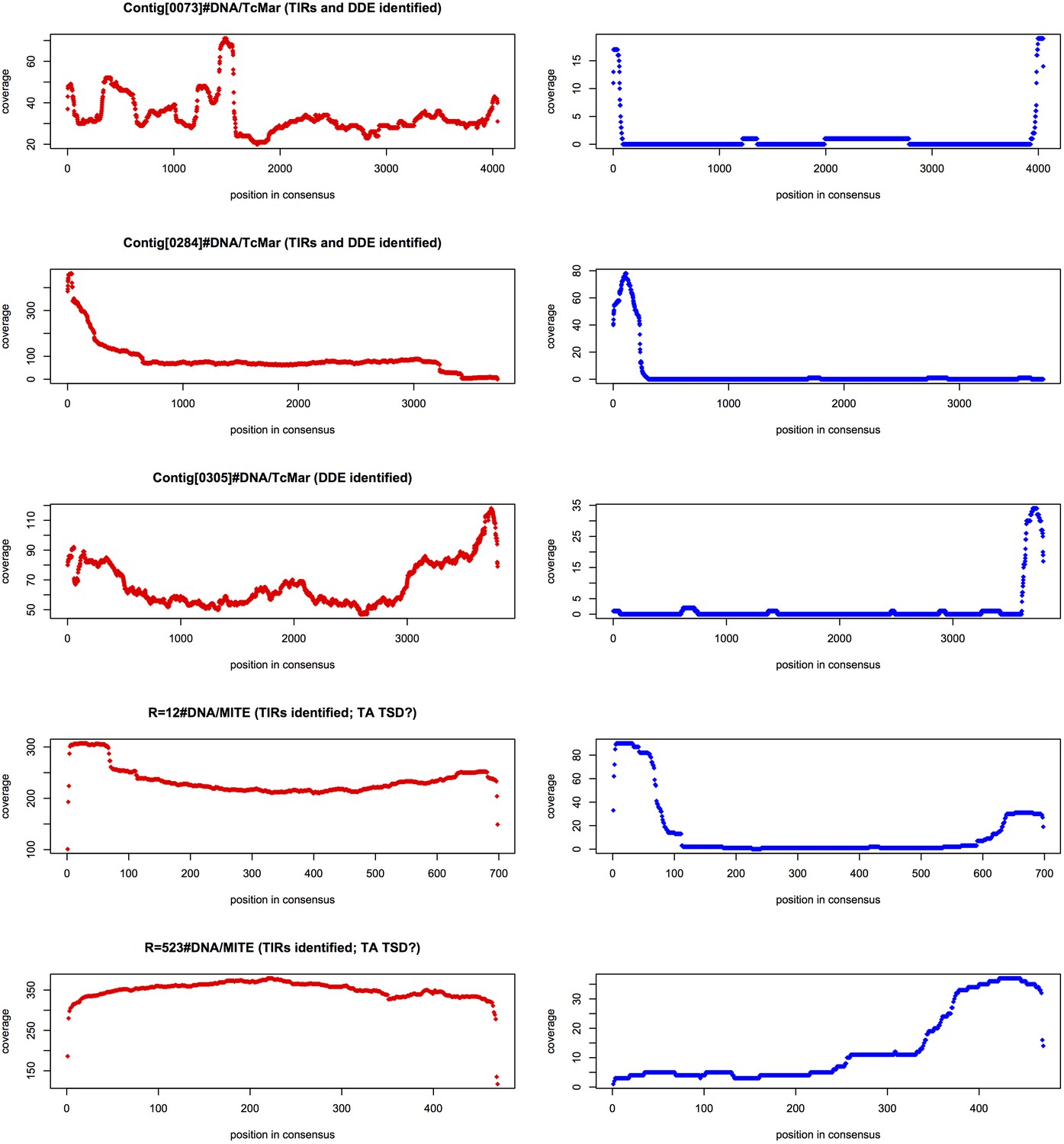
MAC retention of TE termini.
https://doi.org/10.7554/eLife.19090.021
Figure 6—figure supplement 2
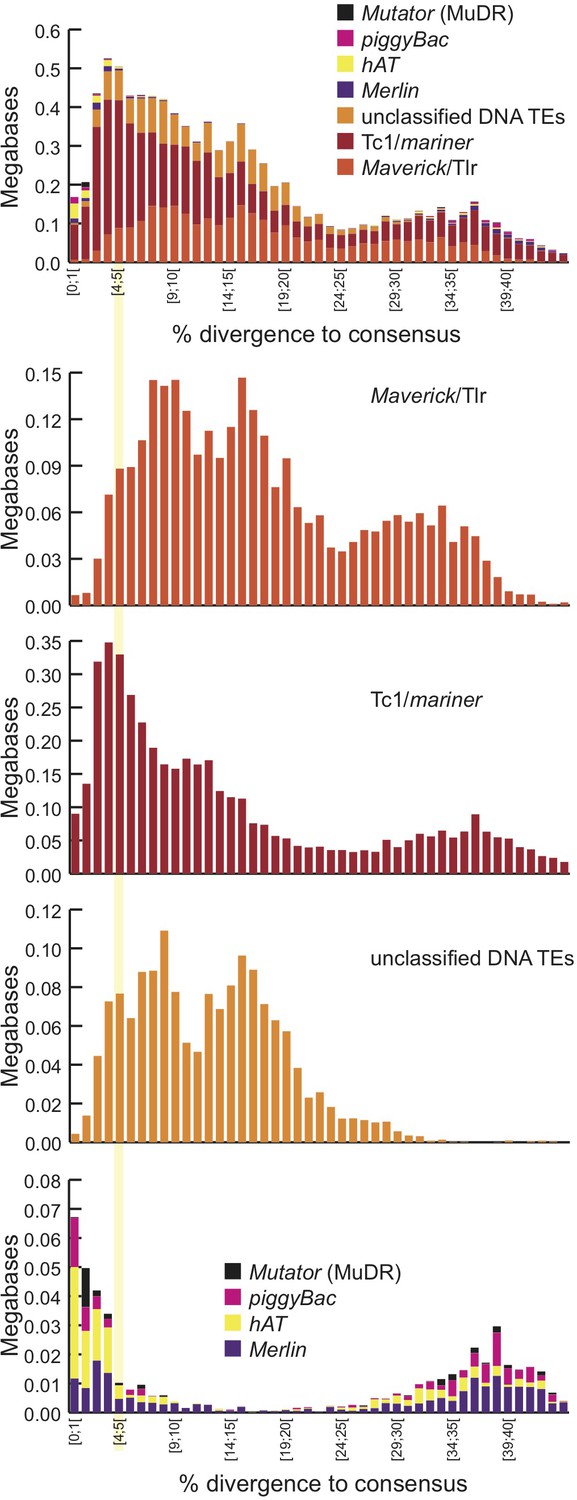
Landscape details of DNA TEs.
https://doi.org/10.7554/eLife.19090.022
Figure 7
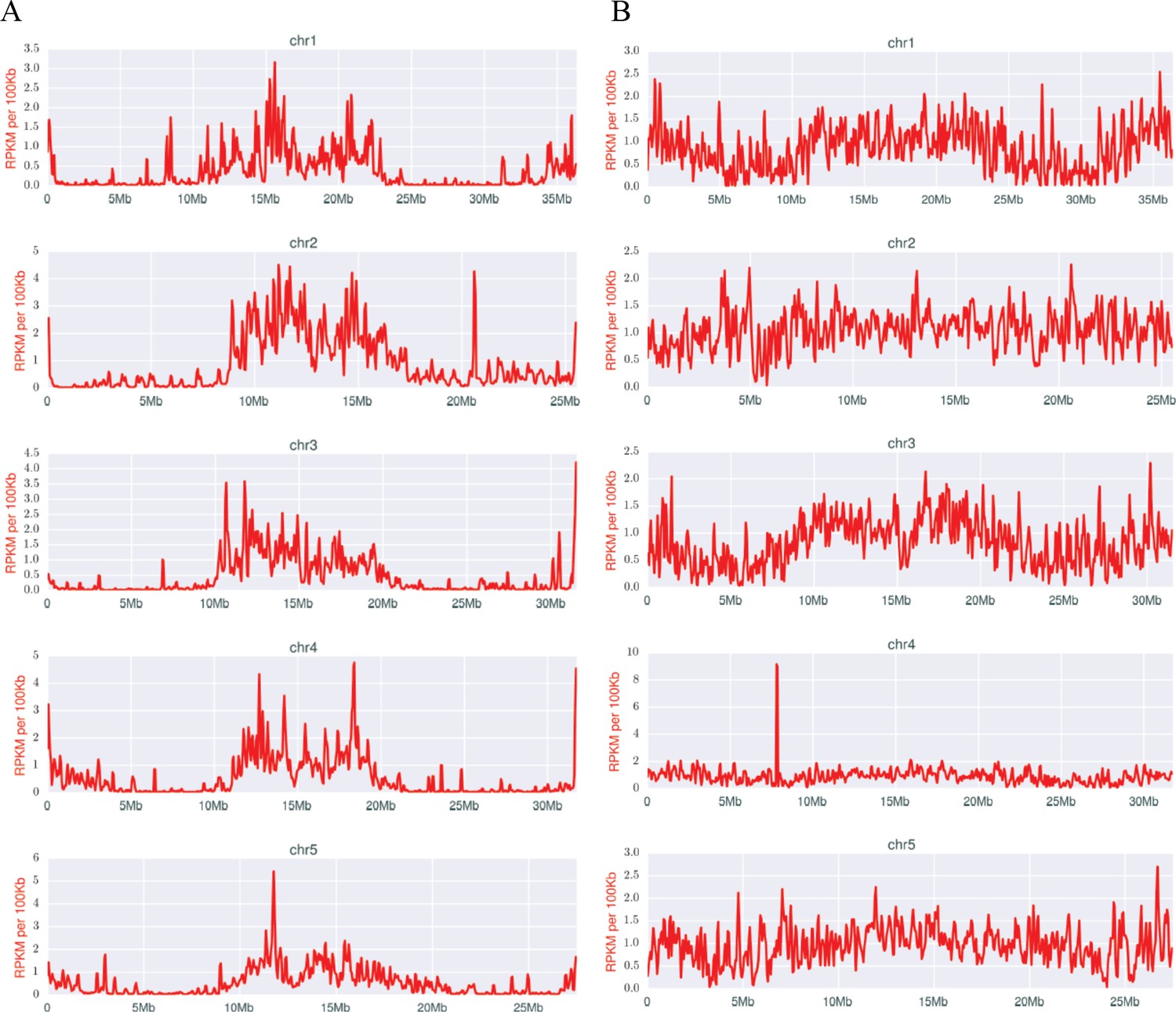
Densities of early (A) and late (B) scnRNAs on MIC chromosomes.
X-axis = position on MIC chromosome super-assembly; all graphs normalized to the same length. Early-scnRNAs were co-purified with Twi1p at three hpm and Late-scnRNAs with Twi11p at 10.5 hpm. Normalized numbers (Reads per kb per million reads [RPKM] in 50 kb bins) of sequenced 26–32-nt RNAs that uniquely map to the MIC genome are shown. A few locations on the chromosomal arms where Early- or Late-scnRNAs were extensively mapped (e.g. ~20.6 Mb on Chr2 for Early-scnRNA and ~7.8 Mb on Chr4 for Late-scnRNAs) were examined in detail, but we have failed to detect any obvious unusual sequence features at these loci to account for the observed enrichment.
Figure 8
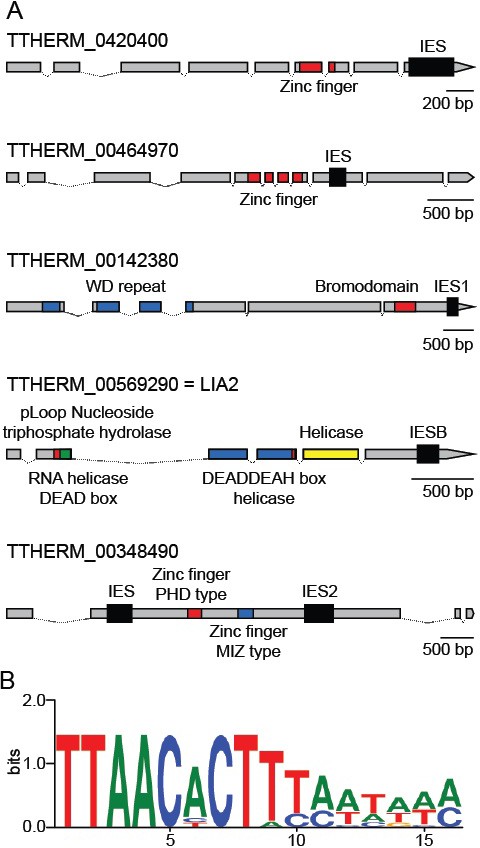
Coding region IESs.
(A) MIC structures of the five genes containing coding region IESs (thick black boxes). Predicted protein-coding regions indicated by thinner boxes, conserved coding sequence domains by colored boxes, and introns by thin lines. Three coding region IESs previously identified (Arnaiz et al., 2012) are indicated as IESB, IES1, and IES2. (B) Sequence logo generated from the 12 IES/MDS junctions of the six IESs depicted in part A (interior of IES to the right). See also Supplementary file 3D.
Appendix 1—figure 1
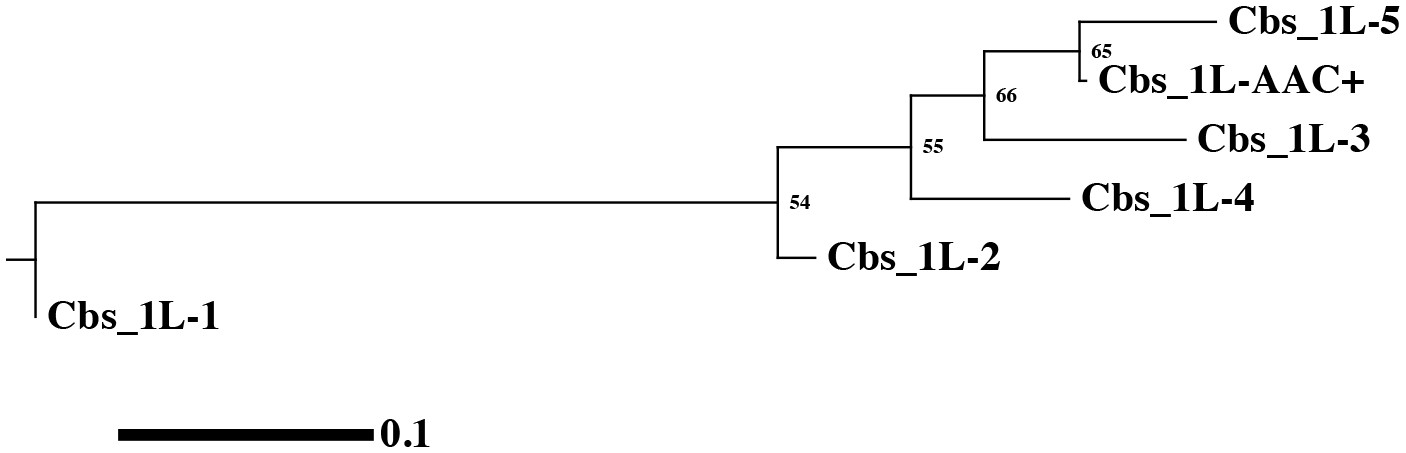
Phylogenetic tree of the 1L-1 clade.
Phylogenetic tree of the 1L-1 clade. The branches show significant statistical support, as indicated by bootstrap percentages.
Appendix 1—figure 2
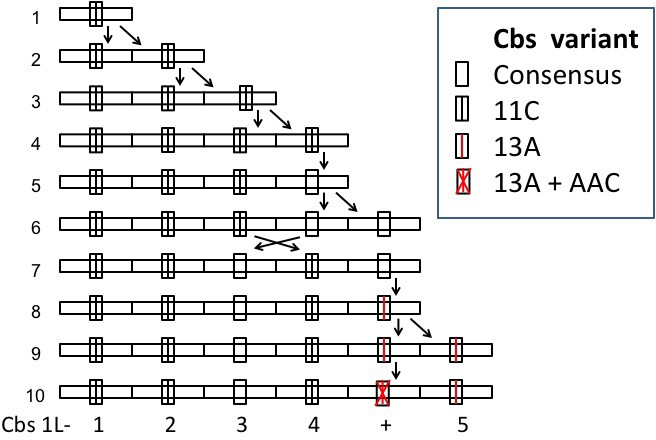
A possible history of the Cbs 1L-1 clade.
Cbs 1L-+ represents the nonfunctional variant with an internal trinucleotide insertion. Line 1: the putative ancestral Cbs and adjacent sequence. Line 10: final (current) state of the 1L-1 clade. Divergent pair of arrows: repeat unit duplication. Crossed arrows: circular permutation of two repeat units. Vertical single arrow: Cbs mutation. Generation of duplications and the circular permutation by unequal crossing-over is diagrammed in Appendix 1—figure 3.
Appendix 1—figure 3
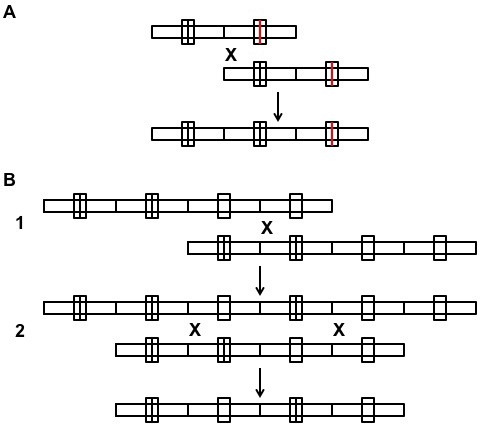
Examples of unequal crossing over.
(A) Repeat unit duplication. (B) Circular permutation of two adjacent repeats. X: unequal crossing over by non-allelic homologous recombination; only the recombinant product of interest is shown. The circular permutation shown involves a series of two independent unequal cross-overs, one of which is a double cross-over; the latter step could alternatively be replaced with two serial single cross-overs (not shown). Another alternative, starting with the original 4-repeat sequence, is a unimolecular unequal cross-over that excises a circle containing the two middle repeats (not shown). Immediate re-insertion of the circle by unequal crossing over at the circle location diametrically opposed to that of the excision site, would accomplish the identical circular permutation more economically.
Appendix 1—figure 4
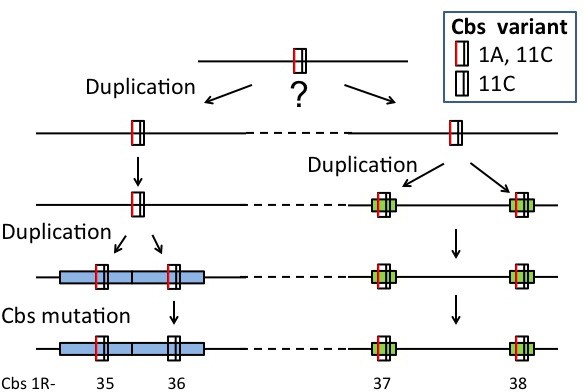
A putative superclade encompassing the Cbs 1R-35 and 1R-37 clades.
The Cbs in these two clades are consecutive (bottom line). The top line represents the putative ancestral Cbs. The lengths of the alignments are 512 bp (blue shading) and 150 bp (green shading) for Cbs 1R-35/36 and Cbs 1R-37/38, respectively.
Appendix 1—figure 5
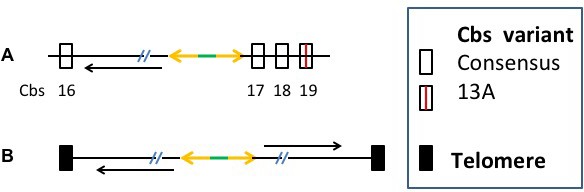
Cbs in the 1L-16 and 17 clades flank the MIC rDNA chromosome-destined DNA.
(A) The ~11 Kb MIC form of the rDNA MAC chromosome-destined DNA. 5’and 3’ ends, as defined with respect to the rRNA coding region, are on the right and left, respectively. Rectangles: flanking Cbs; orange arrows: inverted 42 bp M-repeats; green segment: 28 bp M-repeat non-palindromic spacer. Diagonal slashes: rDNA segments, including the rRNA gene, not shown. (B) The mature, palindromic MAC rDNA chromosome. Black rectangles: Telomeres. Long black arrows in both panels indicate the 5’ to 3’ direction of the coding strand of the rRNA gene.
Appendix 1—figure 6
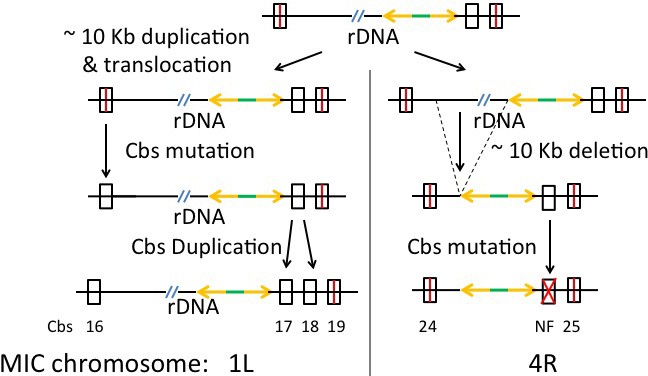
Cbs 1L-16 and 17 Clade: Simplest duplication, translocation and Cbs mutation history.
Crossed rectangle labeled NF: Non-functional mutant Cbs; other symbols as in Appendix 1—figure 5. The top line is the putative ancestral rDNA region, in either chromosome 1L or 4R. The bottom line represents the current state of the duplicated/translocated sequences.
Tables
Table 1
MIC centromere regions and centric MAC chromosomes.
MIC chromosome | L-Cbs location (Mb) | R-Cbs location (Mb) | Cen length (Mb) | # super-contigs in Cen | MIC chromosome length (Mb) | Cen midpoint (Mb) | Chromosome midpoint (Mb) |
|---|---|---|---|---|---|---|---|
1 | 13.98 | 23.24 | 9.26 | 87 | 36.32 | 18.61 | 18.16 |
2 | 9.81 | 14.85 | 5.04 | 77 | 25.51 | 12.33 | 12.76 |
3 | 9.98 | 20.32 | 10.34 | 120 | 31.52 | 15.15 | 15.76 |
4 | 12.23 | 18.34 | 6.11 | 74 | 31.72 | 15.29 | 15.86 |
5 | 10.37 | 17.39 | 7.02 | 62 | 27.47 | 13.88 | 13.74 |
Total | 37.77 (24.7%) | 152.54 |
-
L-Cbs and R-Cbs represent the most Cen-proximal Cbs on the left and right chromosome arms, respectively. Centromere locations were established by deletion mapping (see text for details). For chromosomes 2, 4, and 5, the L-1 and R-1 Cbs flank the putative centromere region. The remaining centromeres contain Cbs’s. Cbs 3L-3 and 3R-1 flank the chromosome 3 centromere, while Cbs 1L-6 and Cbs 1R-11 flank the centromere region of chromosome 1. Locations in Mb use the far (telomere) end of the left arm as the origin.
Table 2
Variation within the Cbs family. Pink and gray shading: single- and double-substituted variants, respectively.
Cbs designation | Count | Cbs nucleotide position | Number of substitutions | Total substitutions per subset | ||||
|---|---|---|---|---|---|---|---|---|
1 | 11 | 13 | 14 | 15 | ||||
canonical | 109 | 0 | 0: 109 | |||||
1A | 53 | A | 1 | |||||
11C | 8 | C | 1 | |||||
13A | 7 | A | 1 | |||||
13C | 2 | C | 1 | |||||
14A | 9 | A | 1 | |||||
14C | 4 | C | 1 | |||||
15A | 10 | A | 1 | 1: 93 | ||||
1A,11C | 5 | A | C | 2 | ||||
1A,13A | 2 | A | A | 2 | ||||
1A,13C | 1 | A | C | 2 | ||||
1A,14C | 2 | A | C | 2 | ||||
1A,15A | 8 | A | A | 2 | ||||
11C,13A | 1 | C | A | 2 | ||||
11C,13G | 1 | C | G | 2 | ||||
11C,14C | 1 | C | C | 2 | ||||
11C,15A | 1 | C | A | 2 | ||||
14A,15A | 1 | A | A | 2 | 2: 23 | |||
Total | 225 | 71 | 17 | 14 | 17 | 20 | ||
Table 3
Summary of salient features of T. thermophila Cbs clades.
Cbs clade | Cbs Members 1 | Expect value range | Tandem duplications (repeat size) | Number of NMCs 2 | Inter-chromosomal duplications |
|---|---|---|---|---|---|
1L-1 | 1L-1 to 1L-5 | E-22 to E-48 3 | 144 bp 4 | 4 | |
1L-16 | 1L-16, 4R-24 | E-47 | 0 | 1L-4R 5 | |
1L-17 | 1L-17, 1L-18, 1L-19, 4R-25 | E-18 to E-46 3 | 45 bp | 2 6 | 1L-4R 5 |
1L-20 7 | 1L-20, 3L-14 | E-14 | 0 | 1L-3L | |
1L-28 | 1L-28, 1L-29 | E-18 | 530 bp | 1 | |
1R-1 | 1R-1 to 1R-7, 2L-2, XX-1, XX-3 | E-21 to E-178 | 13.6 Kb | At least 8 | 1R-2L |
1R-35 8 | 1R-35, 1R-36 | E-139 | 469 bp | 1 | |
1R-37 8 | 1R-37, 1R-38 | E-18 | 796 bp | 1 | |
2R-1 | 2R-1, 2R-2 | E-66 | 605 bp | 1 | |
3L-3 | 3L-3, 3L-29 | E-175 | 0 | ||
3L-4 | 3L-4, 4L-2, 4L-3 | E-48 to E-85 | 3.8 Kb | 1 next to 3L-4, 1 between 4L-2 and 4L-3 | 3L-4L |
4R-3 | 4R-3 to 4R-7, 4R-38, XX-2, XX-4 | E-49 to E-171 | 17.5 Kb | At least 6 | |
5L-9 | 5L-9, 5L-10, 5L-11 | E-58 to E-76 | 10.4 Kb | 2 | |
5R-5 | 5R-5, 5R-6 | E-22 3 | 53 bp | 1 6 | |
5R-14 7 | 5R-14, 5R-15 | E-14 | 84 bp | 1 6 |
-
All clades are described in greater detail in Supplementary file 2E.
-
1 Exact MIC supercontig locations of each Cbs are given in Supplementary file 2A.
-
2 The number of predicted non-maintained chromosomes (NMCs) is one less than the number of repeat units.
-
3 Shorter query length – expected values are potentially higher than for most alignments, which are based on a query length of 415 bp, for the same degree of sequence conservation.
-
4 Average repeat unit length is artificially increased because an additional repeat unit containing a mutationally disabled Cbs between Cbs 1L-4 and Cbs 1L-5.
-
5 Simultaneous duplication event; see Supplementary file 2E.
-
6 These NMCs may be too short to be telomerized after chromosome breakage.
-
7 Support for these clades is weaker than for the others; see Supplementary file 2E.
-
8 This pair of adjacent clades may be a single clade.
-
Table 3—source data 1
MIC DNA sequences surrounding Cbs sites.
- https://doi.org/10.7554/eLife.19090.011
Appendix 1—table 1
Match statistics of all-by-all Blastn alignments of Cbs 1L-1 clade members.
Cbs | Len* | 1L-1 | 1L-2 | 1L-3 | 1L-4 | 1L-AAC | 1L-5 |
|---|---|---|---|---|---|---|---|
1L-1 | 94 | −48 | −29 | −22 | −23 | −29 | −25 |
1L-2 | 120 | 83:74%,0% | −62 | −45 | −43 | −43 | −40 |
1L-3 | 122 | 96:80%,5% | 122:89%,3% | −63 | −49 | −45 | −41 |
1L-4 | 118 | 82:69%,0% | 118:89%,0% | 122:92%,3% | −61 | −40 | −39 |
1L-AAC | 121 | 93:80%,4% | 119:89%,2% | 123:89%,5% | 119:87%,2% | −62 | −49 |
1L-5 | 118 | 90:84%,1% | 116:88%,0% | 120:88%,3% | 116:87%,0% | 121:93%,2% | −61 |
-
* Len = length; 1L-AAC - non-functional Cbs variant with AAC insertion.
-
Main diagonal: Self-match E-values (highlighted yellow). Cells above main diagonal: Expected value of non-self matches. Cells below main diagonal: match length: % sequence identity, % gaps.
Additional files
-
Supplementary file 1
Genome assembly and centromere structure.
(A) Genome assembly statistics. (B) T. thermophila superscaffolds assembled by HAPPY physical mapping. (C) Chromosome super-assemblies. (D) Suspected Chimeric MIC Supercontigs. (E) Most centric MAC chromosomes are among the ten longest MAC chromosomes.
- https://doi.org/10.7554/eLife.19090.025
-
Supplementary file 2
Chromosome breakage and NMCs.
(A) The 225 Cbs identified in the T. thermophila MIC genome. (B) The 181 T. thermophila MAC chromosomes: Lengths and flanking Cbs's. (C) Conservation of chromosome breakage sites 3L-15 to 26 relative to flanking genes in all four examined Tetrahymena species. (D) Identity and lengths of Tetrahymena species scaffolds and supercontigs defined by Cbs's homologous to T. thermophila 3L-26 to 15. (E) T. thermophila Cbs clade alignments. (F) Properties of predicted non-maintained chromosomes. (G) Predicted genes on non-maintained chromosomes.
- https://doi.org/10.7554/eLife.19090.026
-
Supplementary file 3
Internally eliminated sequences.
(A) 7551 High Confidence IESs. (B) Summary of the putative TE content in IESs. (C) Interspersed repeat (putative TEs) content. (D) Characteristics of IESs within protein-coding regions.
- https://doi.org/10.7554/eLife.19090.027
-
Supplementary file 4
Cbs segment sequences and alignments.
- https://doi.org/10.7554/eLife.19090.028
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome
eLife 5:e19090.
https://doi.org/10.7554/eLife.19090




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}