Labeling proteins inside living cells using external fluorophores for microscopy
- University of Illinois at Urbana-Champaign, United States
Figures
Figure 1 with 1 supplement
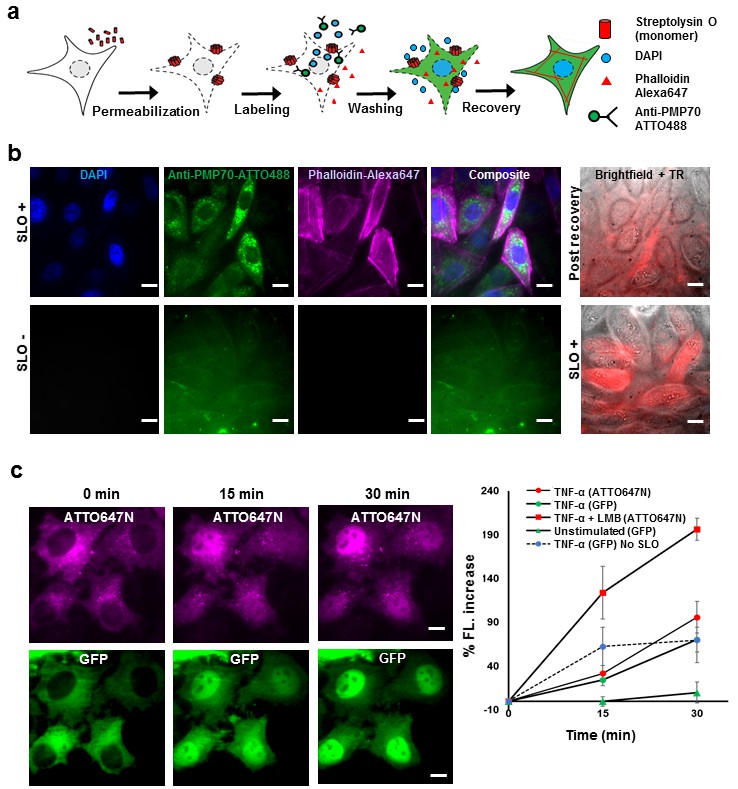
Fluorescent labeling of proteins inside the living cell with high efficiency.
(a) Schematic of permeabilization, delivery, for reversible permeabilization using streptolysin O (SLO) on live CHO-K1 cells. Cells are exposed to SLO for 7–10 min before incubating with the three different fluorescent probes (DAPI, phalloidin-Alexa647, and anti-PMP70-ATTO488) on ice for 5 min. Recovery was initiated by adding 10% FBS DMEM supplemented with nucleotide ATP, GTP, and glucose. (b) Specific labeling of cellular structures by SLO delivered probes. Nucleus, peroxisome, and actin filaments are labeled by DAPI, Anti-PMP70-ATTO488, and phalloidin-Alexa647 respectively. The cells are then incubated with Texas red- 20 kDa dextran to check whether the membrane has recovered. The negative control (SLO-) shows no labeling of cellular structure. The negative control cells are then permeabilized by SLO to show the fluorescence of Texas Red in permeabilized cells. (DAPI is impermeant to CHO-K1 cells.) (c) p65 translocating from cytosol to nucleus after SLO permeabilization in HeLa cells. TNF-α and Leptomycin B (LMB) were used to stimulate nucleus import of the p65-GFP protein labeled by GBP-ATTO647N. The nucleus fluorescence of both GFP and ATTO647N channels increased 15 and 30 min after stimulation as shown in time series fluorescence images (Left). The percent increase of nucleus fluorescence was quantified (Right). (Red square = ATTO647N intensity in the nuclear after TNF-α and LMB treatment, Green circle = GFP intensity after TNF-α treatment [N = 4], Red circle =ATTO647N Intensity after TNF-α treatment [N = 4]. Blue circle with dashed line is the GFP intensity after TNF-α treatment for cells that have never been permeabilized by SLO [N = 4]. Green triangle = GFP intensity without TNF-α and LMB treatments [N = 3]). Scale bar denotes 10 μm.
Figure 1—figure supplement 1
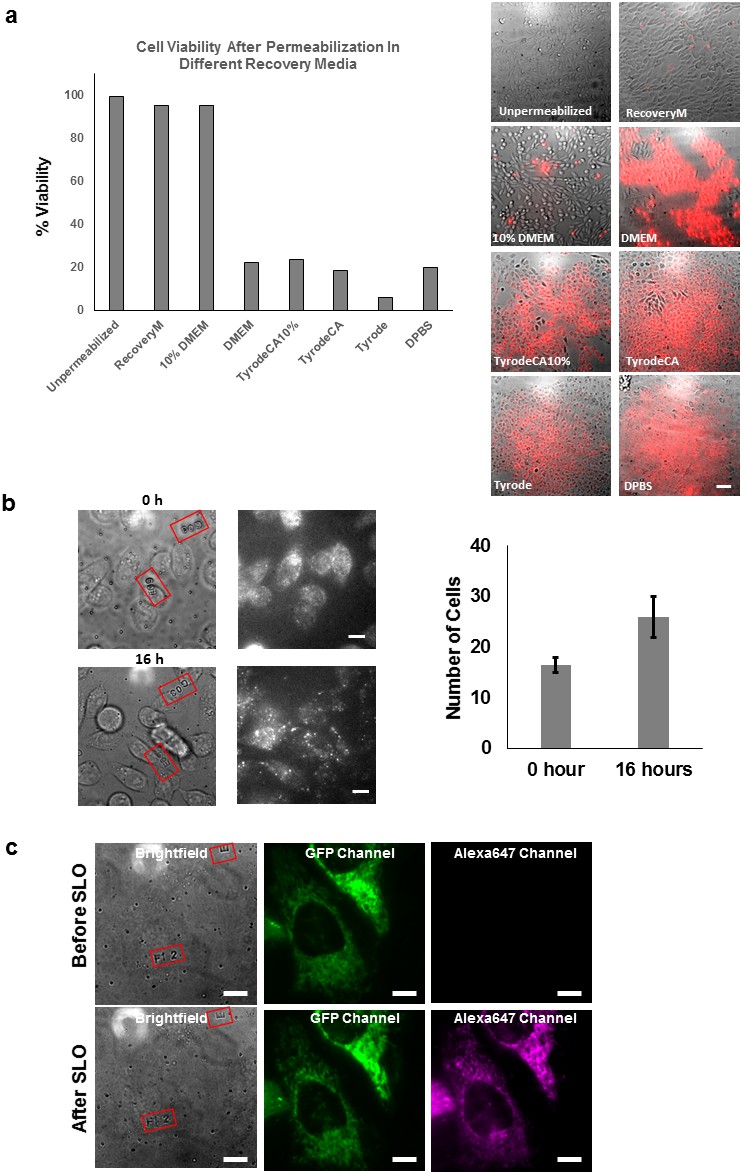
Propidium iodide exclusion and cell division assay.
(a) Propidium iodide assay performed on CHO-K1 cell after SLO permeabilization and recovery. The CHO-K1 cells were recovered in different buffers after permeabilization. Only recovery media and 10% FBS DMEM successfully excluded the cells from PI staining. Only recovery media and 10% FBS DMEM successfully resealed the cells to prevent PI staining. The recovery media is not significantly better than 10% DMEM in terms of repairing the cell membrane. We observed that there is an initial loss of some metabolites from inside the permeabilized cells. This is evident from the fact that a larger proportion of molecular motors were immobile along the microtubules in severely permeabilized cells (Figure 3—figure supplement 3b). The effect from the lost metabolites can be rescued by using our recovery media which is supplemented with essential metabolites (ATP, GTP and glucose), but not with 10% FBS DMEM. Therefore, we emphasize the importance of considering replenishing metabolites for biological system that one is interested in. Scale bar denotes 50 μm. (b) CHO-K1 cells cultured on lettered fiduciary marker for 16 hr after delivering GBP-ATTO647N probes. The letter and number inside the red square assists us in locating the same area. The number of cells are counted in the region initially and after 16 hr. The number of cells increased by nearly 60% despite SLO treatment, suggesting cells were able to undergo cell division [N=2 regions of interest]. (c) HeLa cells expressing Halotag-GFP mitochondria membrane tag were imaged before and after delivery of Alexa647-halotag ligand. The morphology of mitochondria did not appear to differ after the permeabilization and labeling procedure. Scale bar denotes 10 µm.
Figure 2 with 1 supplement
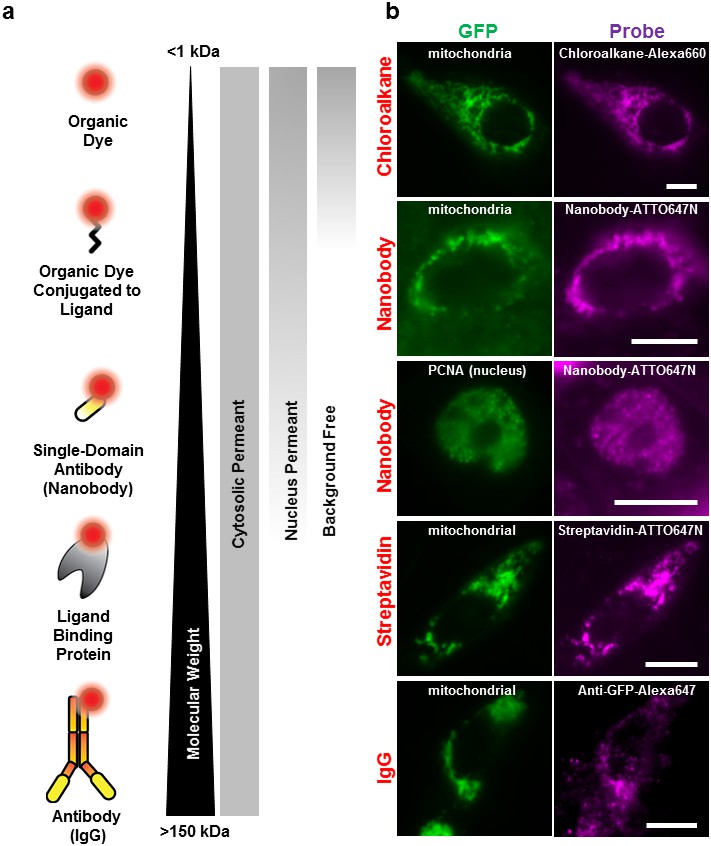
Labeling cellular proteins with a variety of probes.
(a) Delivery of different sized fluorescent probes for specific labeling of intracellular proteins. The drawings depicting the fluorescent probes are not to scale. (b) Cells are labeled by various SLO delivered probes (magenta) to compare with their intracellular or nucleus target, which is tagged with GFP (green). Chloroalkane labeling was performed on U2OS cells, and the rest were done on CHO-K1 cells. All the probes loaded, up to the ~150 kDa IgG, were successfully delivered by SLO and labeled target proteins. However, only small proteins such as GBP (14 kDa) were able to access the nuclear target. Scale bar denotes 10 μm.
Figure 2—figure supplement 1
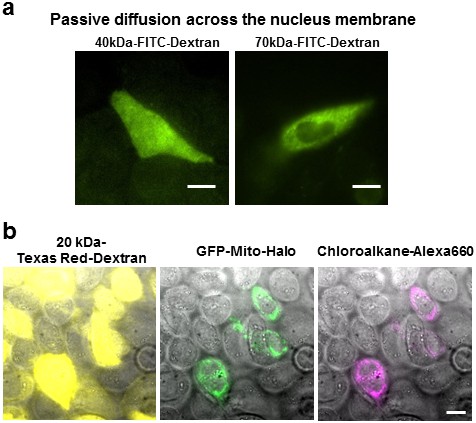
The effect of fluorescent probe sizes on retention and nucleus permeability.
(a) Size cut-off for nucleus accessibility determined using different sized FITC-dextran. We tested the molecular weight cutoff for nucleus entry with fluorescein dextran on CHO-K1 cells. We observed homogenous FITC fluorescence using 40 kDa-dextran, whereas 70 kDa-dextran the fluorescence was excluded in the nucleus. Scale bar denotes 10 μm. (b) The Texas Red Dextran image (yellow) shows permeabilized cells displaying a haze of fluorescence across the whole cell. On the other hand, the chloroalkane-Alexa660 being <2 kDa only stains the cells expressing the GFP-halo protein; no fluorescence was observed in other permeabilized cells. Free chloroalkane-Alexa660 were washed out during our procedure.
Figure 3 with 3 supplements
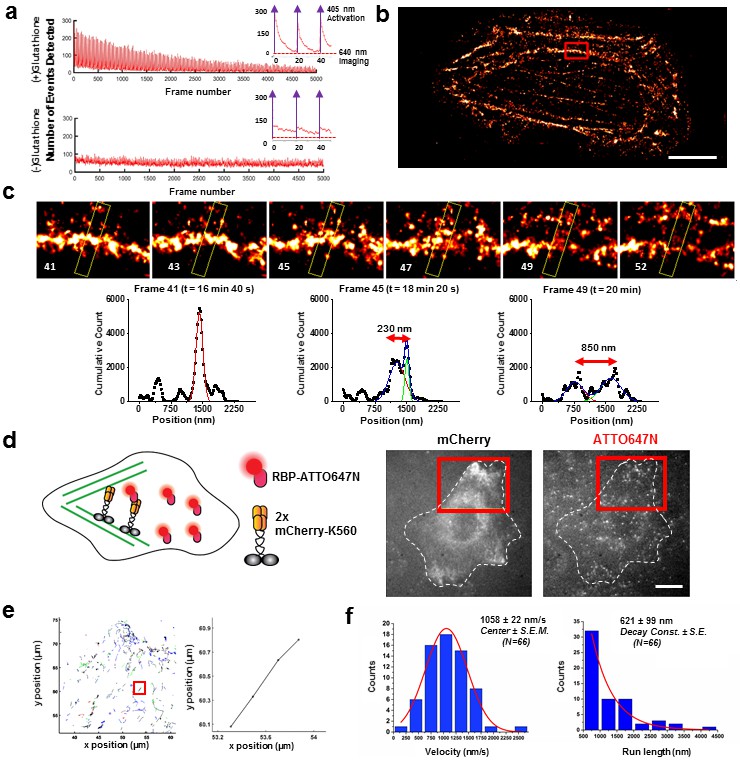
Applications of live cell protein labeling using SLO permeabilization.
(a) The number of detected single molecule activation events per frame with and without the addition of glutathione in the recovery media. With the addition of 4 mM glutathione in the recovery media after SLO permeabilization, there are a greater number of activation events detected per frame, as well as a fewer number of detected events during illumination, suggesting that the added glutathione assists in turning off the fluorophore more efficiently. The insets show the first 50 frames of the images, where with each pulses of 405 nm laser new waves of single molecules are activated. (b) A dynamic dSTORM movie was acquired to examine large-scale actin dynamics. Each frame of the movie is composed of 500 frames, with a 250 frames moving average time window. The movie was recorded for a total of 20,000 frames at 100 ms exposure time, for a total acquisition time of over 30 min. Scale bar denotes 5 μm (c) A series of zoomed in images focusing on a small section of actins in (b) (yellow rectangle). A thick actin bundle can be seen to split into two thinner actin filaments. The actin filaments grew 230 nm apart after 1 min 40 s, and then 850 nm apart after another 1 min 40 s (Video 1). (d) U2OS cells expressing 2x-mCherry-kinesin (K560) and labeled by RBP-ATTO647N. After just 3 hr of expression, the cell is filled with over-expressed mCherry-kinesin, and it is not possible to track individual protein. Single kinesin molecules can easily be tracked by sparsely labeling mCherry-kinesin with Red fluorescent protein Binding Protein (RBP) conjugated to ATTO647N. (e) Tracking individual ATTO647N-RBP labeled kinesin. Majority of kinesin trajectory in the selected area can be seen to travel uni-directionally in a short distance (<5 μm). The trajectory enclosed in the red rectangle is an example trace of detected kinesin trajectory. (f) To quantify the kinesin behavior in vivo, velocity and run length of labeled kinesin were measured. The kinesins are moving at an average velocity of 1058 ± 22 nm/s (Center ± SEM), and the average run length is 621 ± 99 nm (Decay constant ± SE). The measurements were taken on a heated stage at 30°C.
Figure 3—figure supplement 1

Activation of Alexa647 in fixed and live cells in the presence and absence of Oxyrase.
(a) Representative images of Alexa647 de-activation. U2OS cells were fixed and labeled with Alexa647 to confirm the condition for photoactivation. The cells were fixed in 4% paraformaldehyde and permeabilized in 0.2% Triton X-100. After staining, the Alexa647 probes were deactivated using 640 nm laser. The fluorescence intensity enclosed in area depicted by the yellow line was analyzed and plotted in panel b. (b) Activation of Alexa647 in the presence and absence of Oxyrase. The experiment was performed in the presence and absence of 20 µL of Oxyrase stock with 20 mM of sodium lactate. Pulses of 405 nm lasers are applied to the either fixed or live cells with stained actin, and the intensity of the enclosed region as a function of frame number is plotted. If the probe reactivated, spikes of intensity increase would be observed. No reactivation was observed without addition of Oxyrase on fixed cell. Activation of Alexa647 on actin in fixed cells and in live cells in the presence of Oxyrase.
Figure 3—figure supplement 2
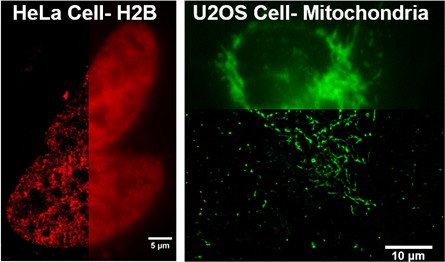
Additional examples of intracellular structures of living cells imaged by dSTORM using cell impermeant chloroalkane-dye that are delivered by using SLO.
On the left is a dSTORM image of Histone 2B protein in the nucleus of HeLa cells. The H2B was labeled by Alexa647-chloroalkane. The image on the right hand side is a dSTORM image of mitochondria labeled with Alexa660-chloroalkane. These two chloroalkane probes were cell impermeant prior to SLO treatment.
Figure 3—figure supplement 3
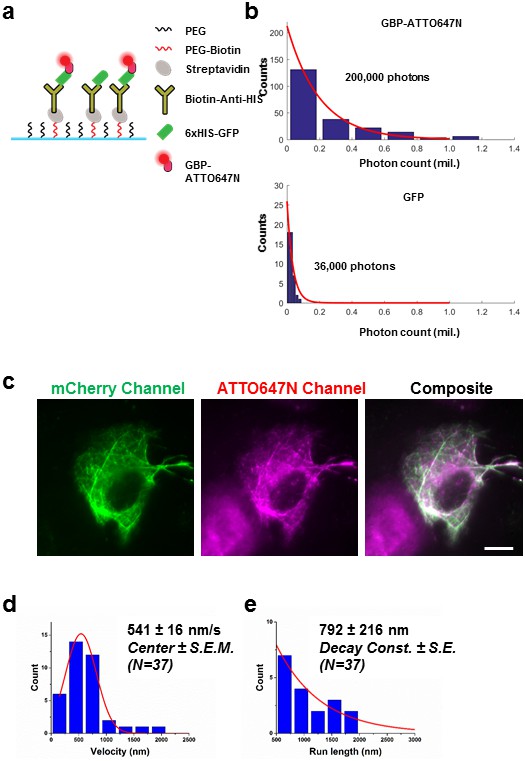
Labeling fluorescent protein with conjugated nanobody for kinesin tracking.
(a) Improving the detection of proteins attached to fluorescent protein by using nanobody labeling. 6xHIS-GFP was immobilized on glass coverslip using the described schematic. ATTO647N-GBP was flowed into the glass coverslip chamber to label the GFP. (b) The photostability of GFP and ATTO647N was evaluated, and an increase of ~6x in total photon output was observed. (c) Demonstration of specific binding of nanobody to mCherry. U2OS cells were first transfected with plasmid encoding 2xmCherry kinesin, and were then permeabilized by SLO. RBP-ATTO647N was added after permeabilization. U2OS cells were recovered in 10% FBS DMEM without ATP, so that in heavily permeabilized cells, the ATP concentration was low after immediately after a 15 min recovery. mCherry Kinesin labeled with RBP-ATTO647N appear to be saturating the microtubule. Specific labeling was demonstrated in composite image. Scale bar denotes 10 μm. (d) Velocity measurement of kinesin in live U2OS cells at room temperature. The peak after Gaussian fitting shows a velocity of 541 ± 16 nm/s (Center ± SEM). e) The average run length of kinesin is found to be 792 ± 216 nm (Decay Const. ± SE).
Videos
Video 1
Dynamic direct super-resolution imaging of actin labeled with Alexa647-phalloidin related to Figure 3.
Real time is displayed at the bottom right hand corner of the movie in min:s.
Video 2
Single molecule detection of individual mCherry kinesin using ATTO647N labeled anti-RFP nanobody.
Real time is displayed at the bottom right hand corner of the movie in min:sec, the movie is played back in 3x the speed compared to real time.
Tables
Table 1
Dilution and incubation time of SLO to use for cell lines used in this study at various confluency.
Cell line | Confluency | Dilution of SLO stock | Incubation time (min) |
|---|---|---|---|
CHO-K1 | 90–100% | 250x | 7 |
CHO-K1 | 75% | 334x | 7 |
U2OS | 80% | 500x | 7 |
U2OS | 50% | 774x | 7 |
U2OS | 30% | 1000x | 7 |
HeLa | 90% | 250x | 10 |
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Labeling proteins inside living cells using external fluorophores for microscopy
eLife 5:e20378.
https://doi.org/10.7554/eLife.20378




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}