Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans
- Memorial Sloan Kettering Cancer Center, United States
- Weill Cornell Medical College, United States
Figures
Figure 1. Purification and sequencing of Okazaki fragments from C. elegans with 4 supplements
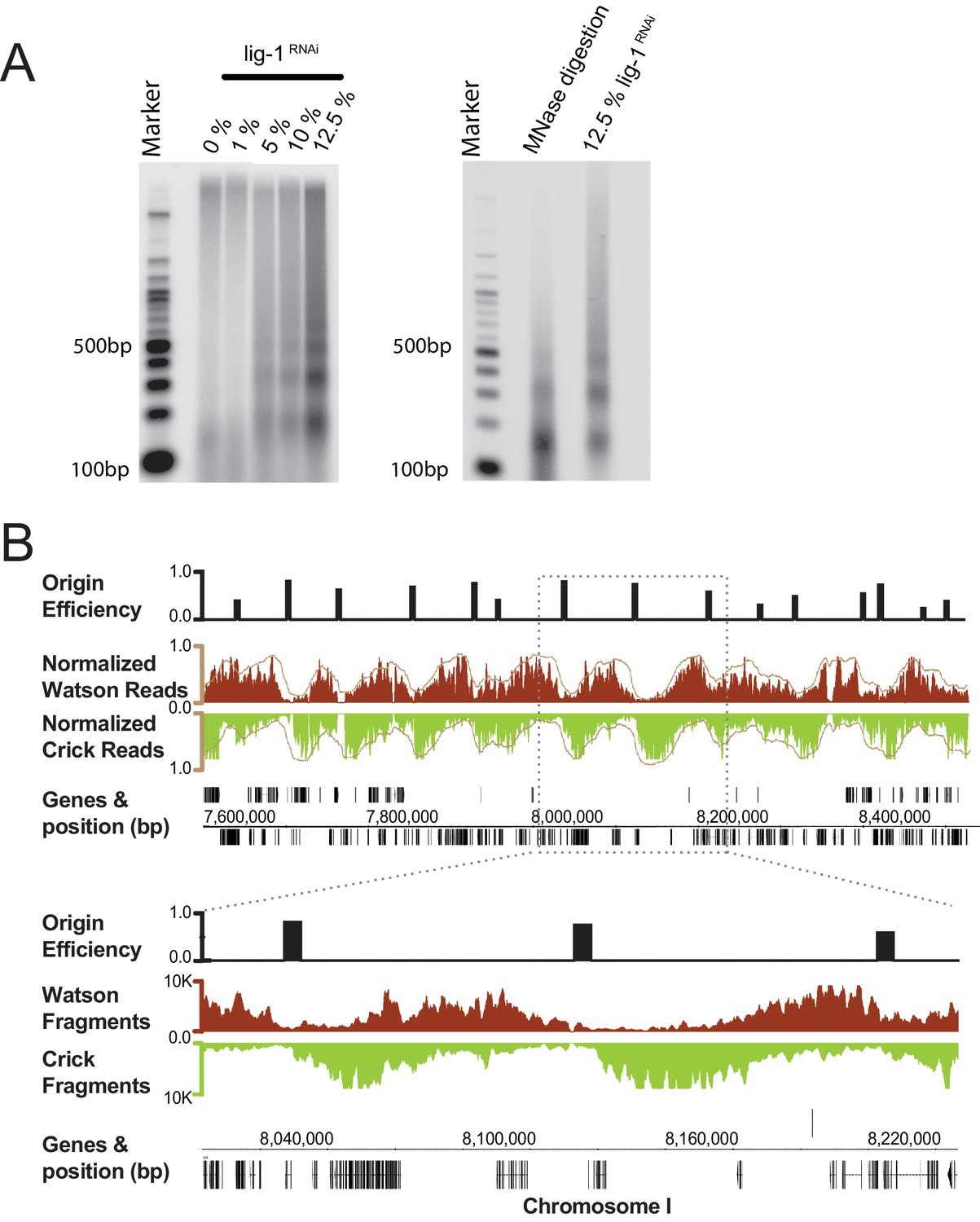
(A) Left panel: increasing DNA ligase I (lig-1) depletion by RNAi results in the generation of small DNA molecules with periodic size distribution. Right panel: comparison of the putative Okazaki fragments with DNA resultant from a Micrococcal Nuclease digestion of chromatin, reveals both species have similar periodicity. (B) Fragments mapped to the C. elegans genome display a characteristic strand bias expected of Okazaki fragments. Regions of high Watson-strand signal (red) that transition to regions of high Crick-strand signal (green) are replication origins; normalized signal is shown in brown. Mapped replication origins are black bars above the Okazaki fragment traces and are shown as 5 kb blocks centered on the origin midpoint to aid visualization. Annotated genes and their coordinates are shown below the fragment traces.
Figure 1—figure supplement 1
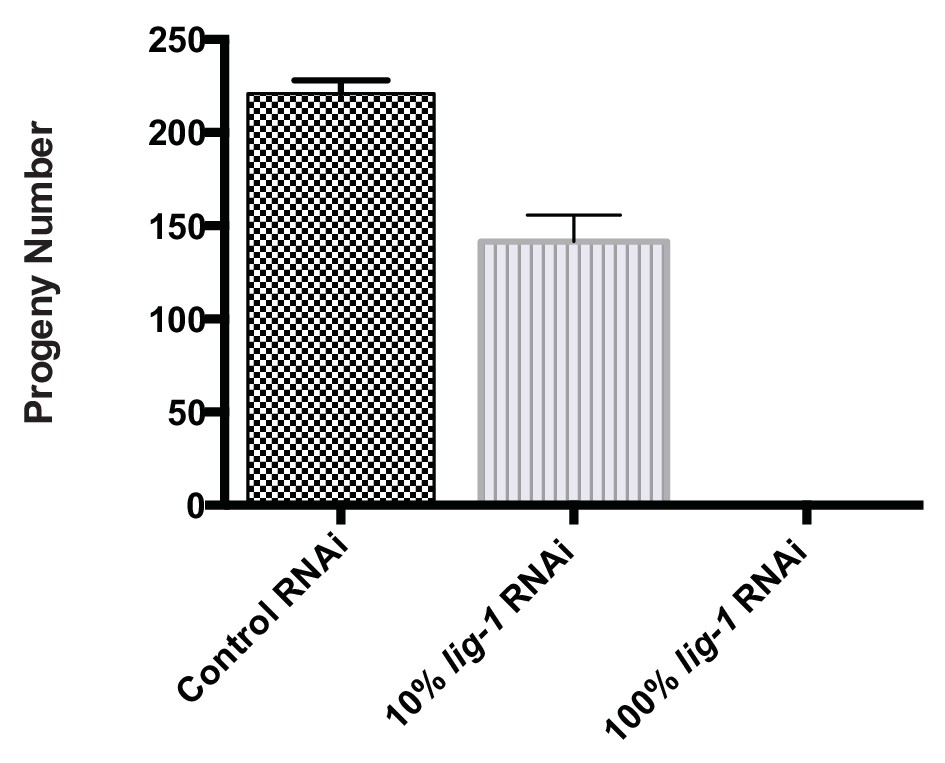
Average progeny number in C. elegans treated with 100% and 10% lig-1 RNAi.
Mean ± SD is presented, n=10.
Figure 1—figure supplement 2

Global measures of origin efficiency and spacing.
(A and B) Scatter dot-plots of replication origin efficiencies and inter-origin distances from mixed early embryos (ME), median values are shown as a black horizontal bars.
Figure 1—figure supplement 3
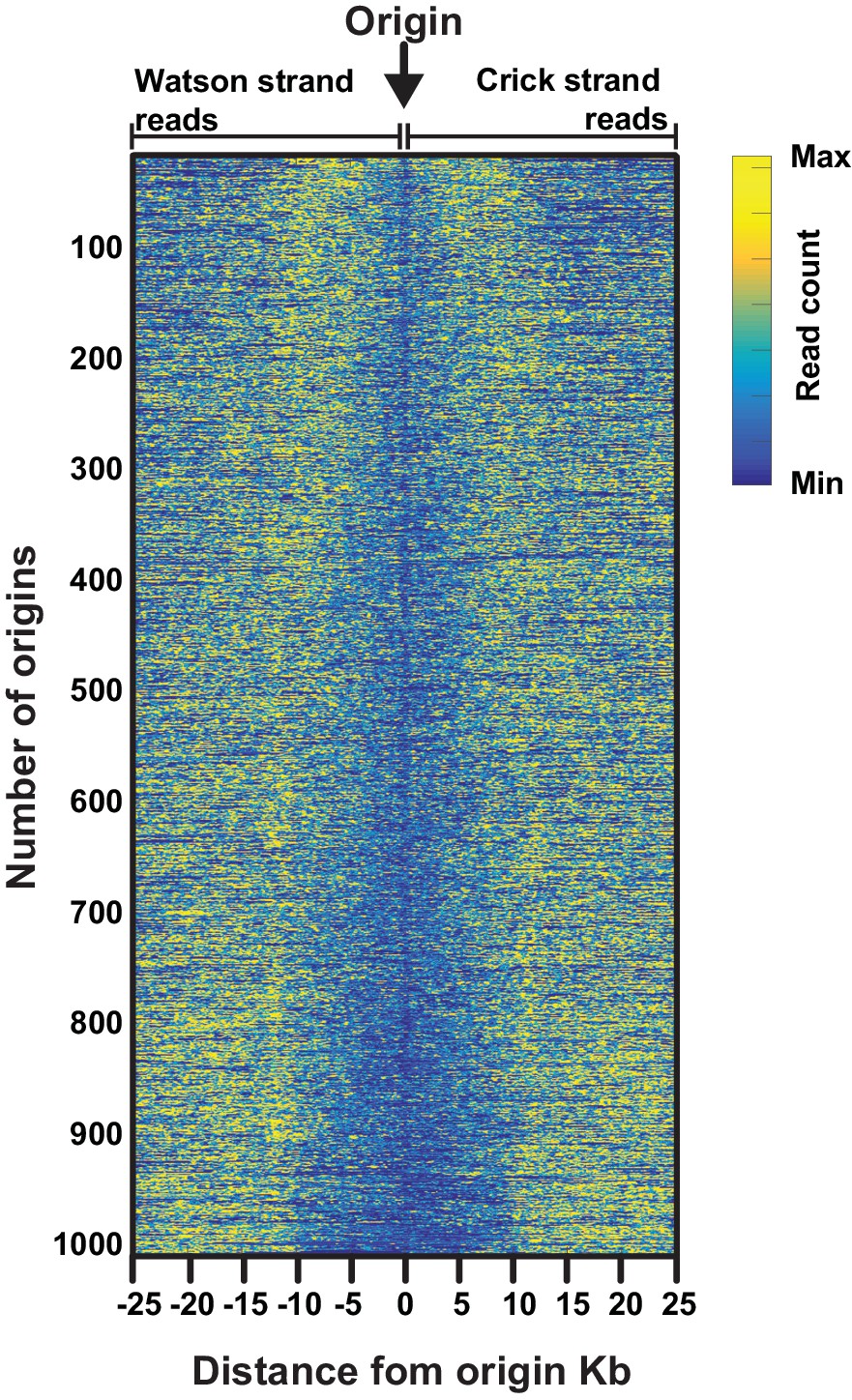
Heatmap illustrating the varying size of the 'initiation zone' at replication origins.
Data are from the most efficient replication origins (n=1000, efficiency >0.5). Mapped sequencing reads for either the Watson strand (leftward moving forks, left of the origin) or Crick strand (rightward moving forks, right of the origin) are displayed for each replication origin. To allow comparisons across origins with different efficiencies, the mapped sequencing data for Watson or Crick reads ±25 kb from each origin was mean centered and normalized such that sum of the squares of the values = 1.0. All origins are then ranked according the size of transition zone, with the smallest at the top.
Figure 1—figure supplement 4
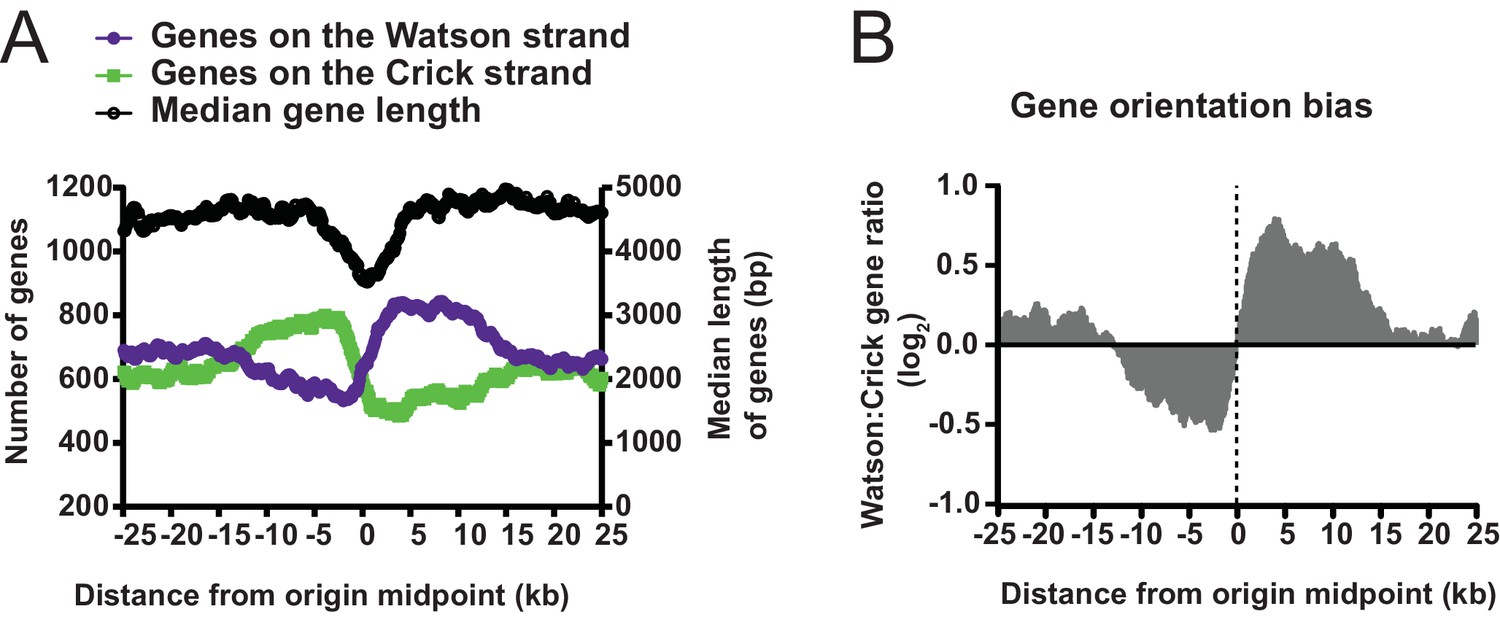
Gene orientation around replication origins.
(A) Graph displaying the length and strand bias of genes within ±25 kb of replication origins. Median gene length, defined as from translation start to end, is shown as black line on right y-axis. Strand bias of gene orientation is shown on the left y-axis; Watson strand genes (purple) are defined as genes whose transcribed RNA is complementary to the Crick strand; Crick strand genes (green) have RNA product complementary to the Watson strand. (B) Gene orientation bias from A is displayed as a log2 ratio of Watson: Crick genes.
Figure 2 with 1 supplement
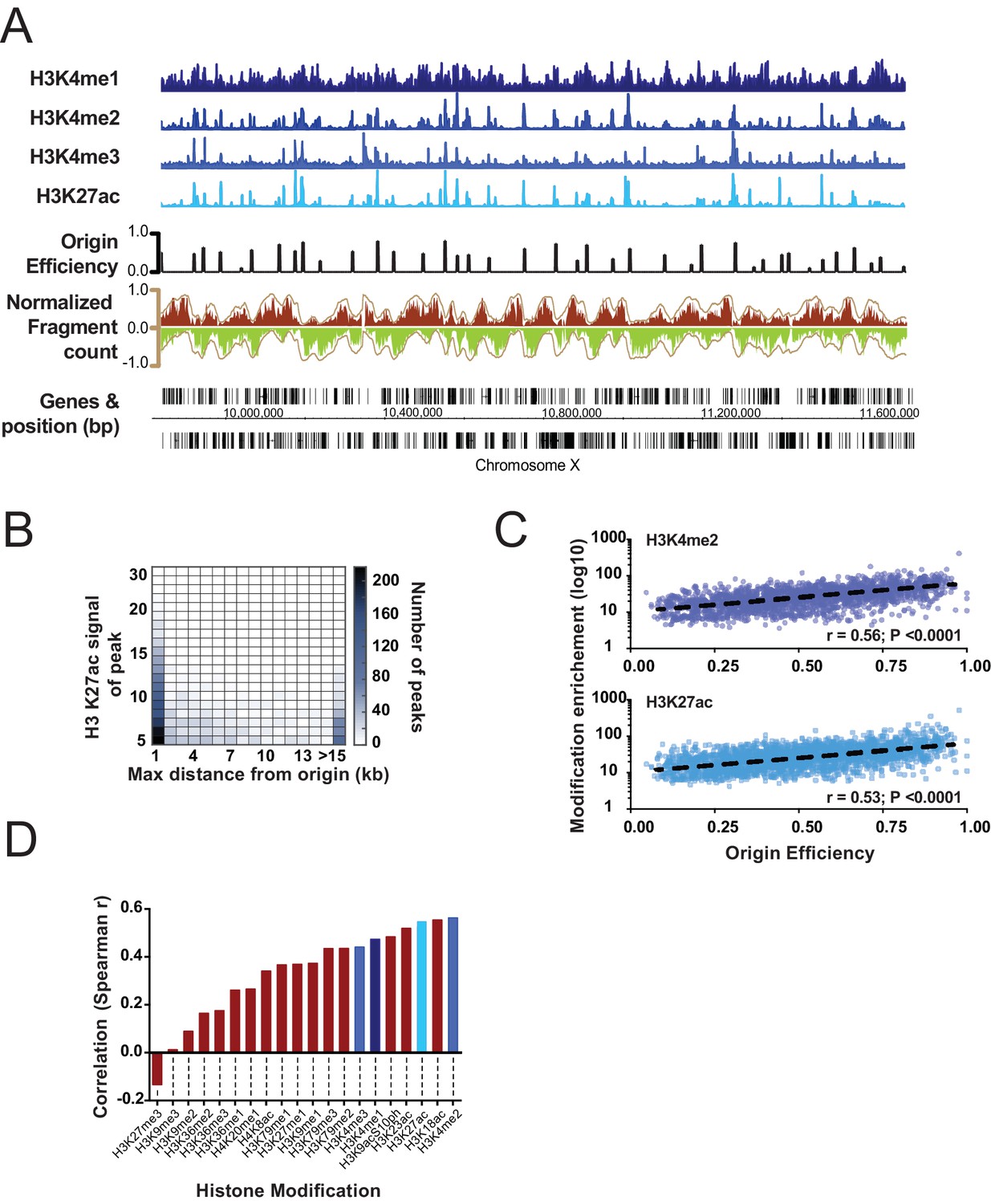
Correlation of select histone modifications with replication origins.
(A) Representative ChIP signal for select histone modifications are shown for a ~1 Mb region of Chromosome X. Mapped replication origins (black) are broadly coincident with peaks in the ChIP signal. Okazaki fragment reads are shown below and are colored according to Figure 1B. (B) Correlation between H3K27ac and replication origins is displayed as a heatmap. Replication origins and H3K27ac peaks were defined computationally and the distance between peaks measured using Intervalstats (Materials and ethods); the number of H3K27ac peaks that lie within a maximum distance, defined on the x-axis, is plotted as heatmap; the relative intensity (fold enrichment over background) of the ChIP signal at H3K27ac peaks is plotted on the y-axis. (C) ChIP signal scales with increasing origin efficiency; level of histone modifications at replication origins (±2.5 Kb) is plotted for H3K27ac and H3K4me2 relative to the efficiency of the origin. (D) Data were analyzed as C to compute the correlation of several histone modifications with replication origin efficiency; blue shades correspond to histone marks shown in A.
Figure 2—figure supplement 1
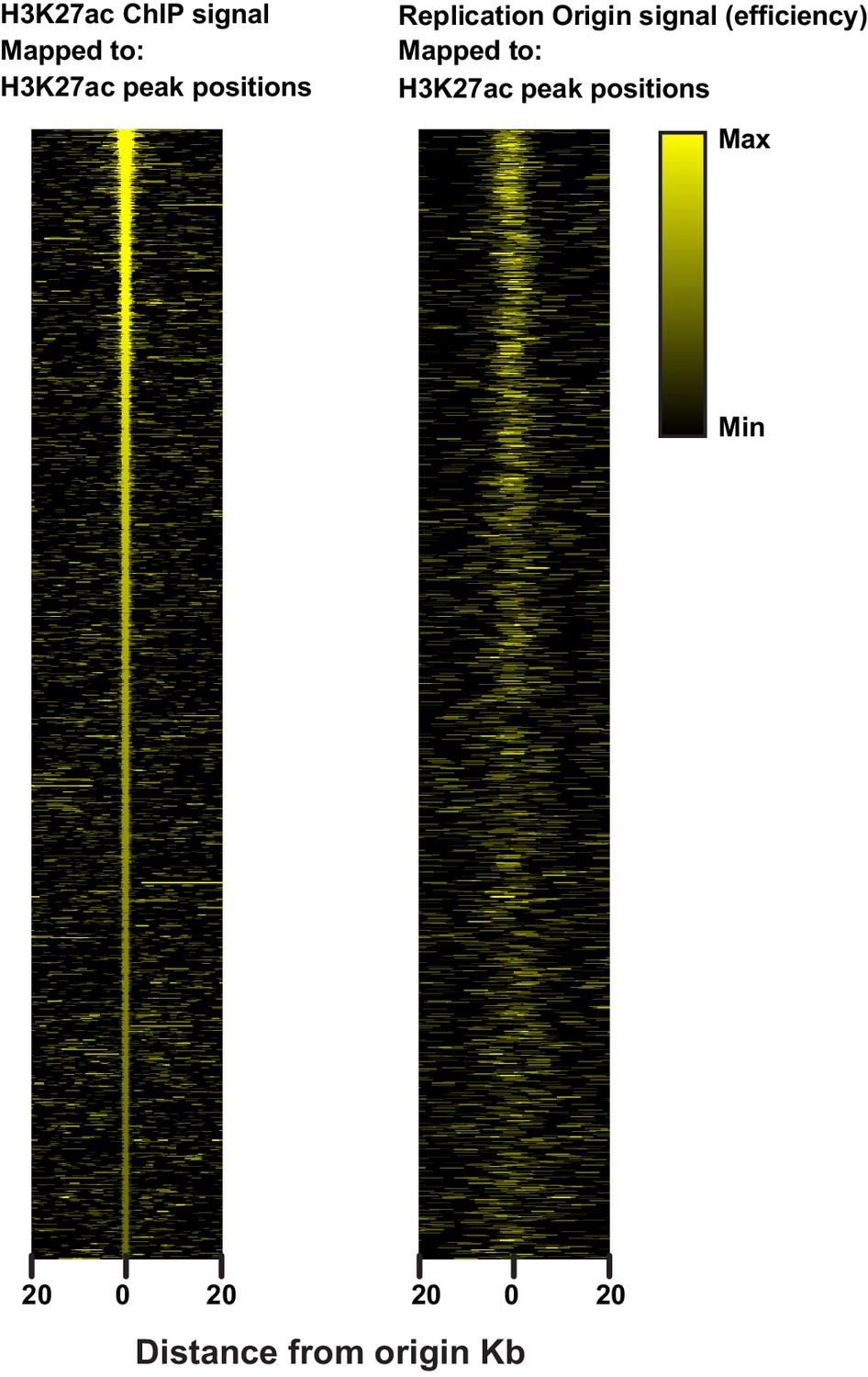
Heatmap illustrating the relationship between H3K27ac and replication origins.
H3K27ac peaks as defined by Macs2 (see Materials and methods) were used as reference positions to map either H3K27ac ChIP signal (left) or origin efficiency (right).
Figure 3
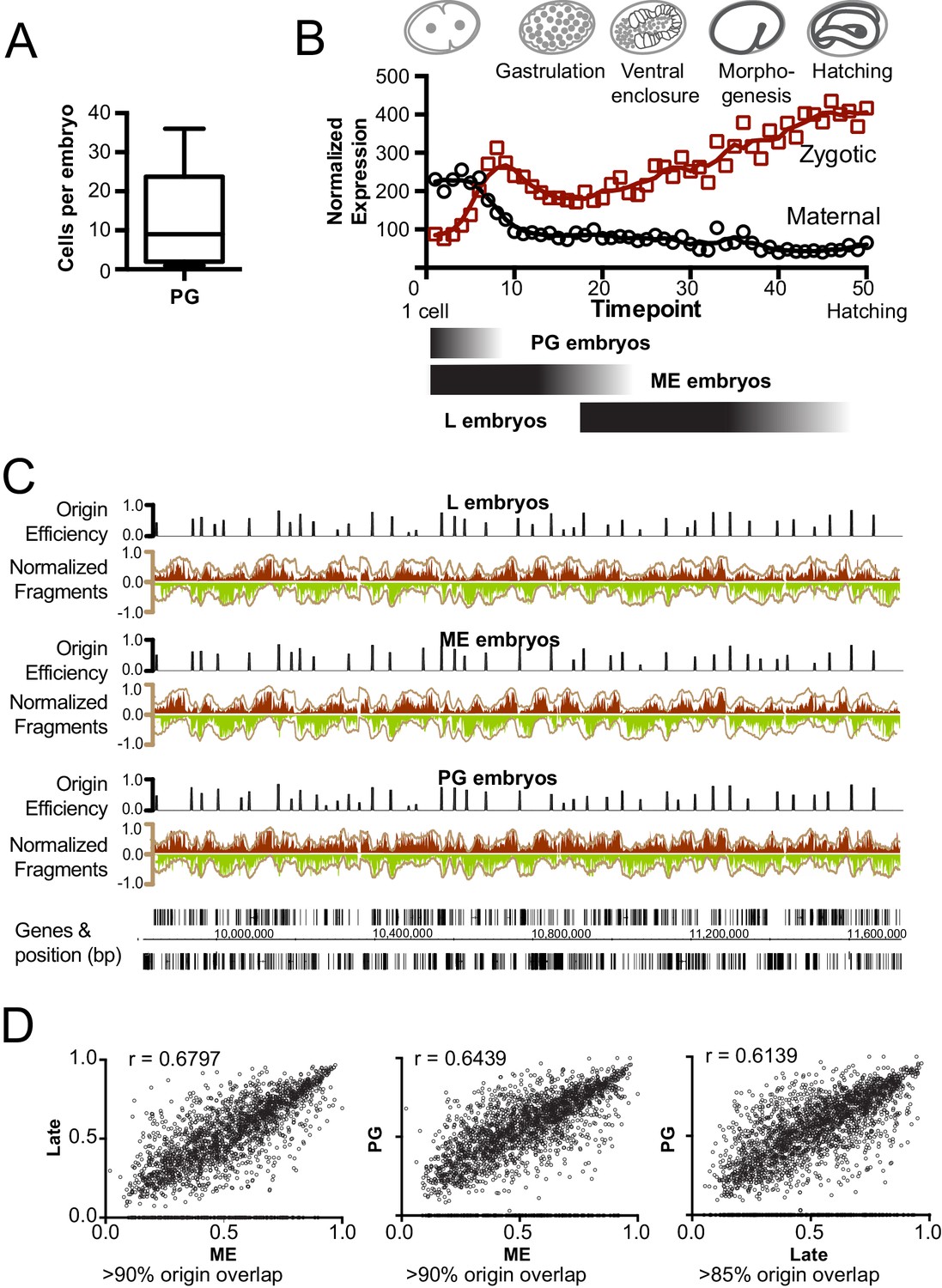
Mapping replication origins through early embryogenesis.
(A) Box plot showing the number of cells/embryo for a representative sample of pre-gastrula (PG) embryos, whiskers indicate the range of values. (B) Graph showing maternal and zygotic transcripts through embryogenesis. Data from whole embryo transcriptome from Hashimshony et al (Hashimshony et al., 2015) were normalized so that the sum expression for each gene through the time course = 1; total transcript levels for all genes at time point is shown. Below, black shaded bars illustrate the approximate age range for the purified embryo populations; late (L), mixed early (ME) and pre-gastrula (PG). (C) Okazaki fragment sequencing of DNA from L, ME and PG embryos revels broadly similar replication patterns. Graphs colored according Figure 1B. (D) Scatterplot comparing origin the efficiencies of overlapping origins within the L, ME and PG datasets. Spearman correlation (r) is shown on graph. The % of origins that overlap spatially is shown below.
Figure 4 with 2 supplements

Gene transcription is coupled with replication origin use.
(A) Gene ontology analysis for genes at varying distances from the midpoint of a replication origin. Gene ontologies were calculated using the Gene Ontology Consortium (http://geneontology.org/). Select GO terms with greatest significance are shown that lie within ‘Biological Process’ annotation datasets. Left, observed/expected log2 ratio is shown at varying distances from replication origins for each ontology term as a heatmap: color key is below – yellow indicates enrichment, blue is depletion. Right, heatmap showing the calculated p-values (hypergeometric distribution) for the data on left; color key is below. (B) Normalized transcript abundance within ±25 kb of the replication origin was summed for the top 1000 origins for each of the 50 time points through embryogenesis. Data are displayed as a heatmap and arranged according to sample number shown on left, corresponding time in embryogenesis is also indicated. Timing of gastrulation and mid development transition (MDT) calculated by (Levin et al., 2016) are also shown. (C) Live cell nuclei per embryo as calculated by (Sulston et al., 1983) is plotted as a function of time through embryogenesis and aligned with B.
Figure 4—figure supplement 1
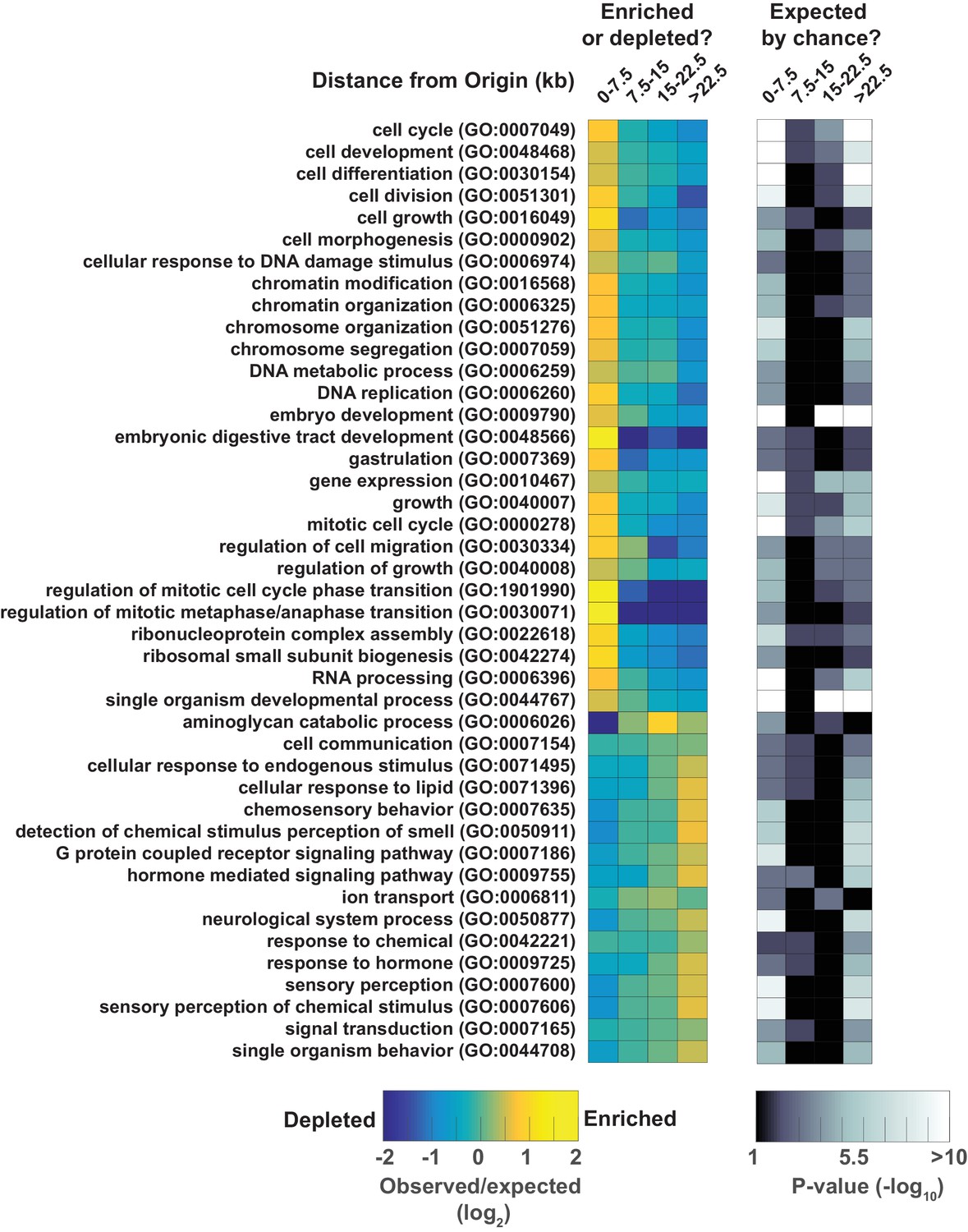
Gene ontology analysis for genes at varying distances from the midpoint of a replication origin.
Gene ontologies were calculated using the Gene Ontology Consortium (http://geneontology.org/). Select GO terms with greatest significance are shown that lie within 'Biological Process' annotation datasets. Left, observed/expected log2 ratio is shown at varying distances from origins for each ontology term as a heatmap: color key is below – yellow indicates enrichment, blue is depletion. Right, heatmap showing the calculated p-values (hypergeometric distribution) for the data on left; color key is below.
Figure 4—figure supplement 2

Heatmap showing the relationship between transcribed genes and replication origins through embryogenesis.
Normalized data from each of the 50 time points from the whole embryo transcriptome (16) were plotted relative to the positions of the most efficient ~2000 replication origins. Origins are ranked from most efficient (0, top) to least efficient (2000, bottom). Genes within ±25kb from the origin are shaded according to their relative level of expression; origin midpoints are indicated by arrows. Sample numbers and relative time through embryogenesis are shown above.
Additional files
-
Supplementary file 1
All origin efficiencies: Origin efficiencies and locations are included in supplementary file 1.
- https://doi.org/10.7554/eLife.21728.013
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans
eLife 5:e21728.
https://doi.org/10.7554/eLife.21728




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}