Stella modulates transcriptional and endogenous retrovirus programs during maternal-to-zygotic transition
- University of Cambridge, United Kingdom
- European Bioinformatics Institute, United Kingdom
- Daegu Gyeongbuk Institute of Science and Technology, Republic of Korea
- Wellcome Trust Sanger Institute, United Kingdom
- European Molecular Biology Laboratory - Monterotondo, Italy
Figures
Figure 1 with 2 supplements
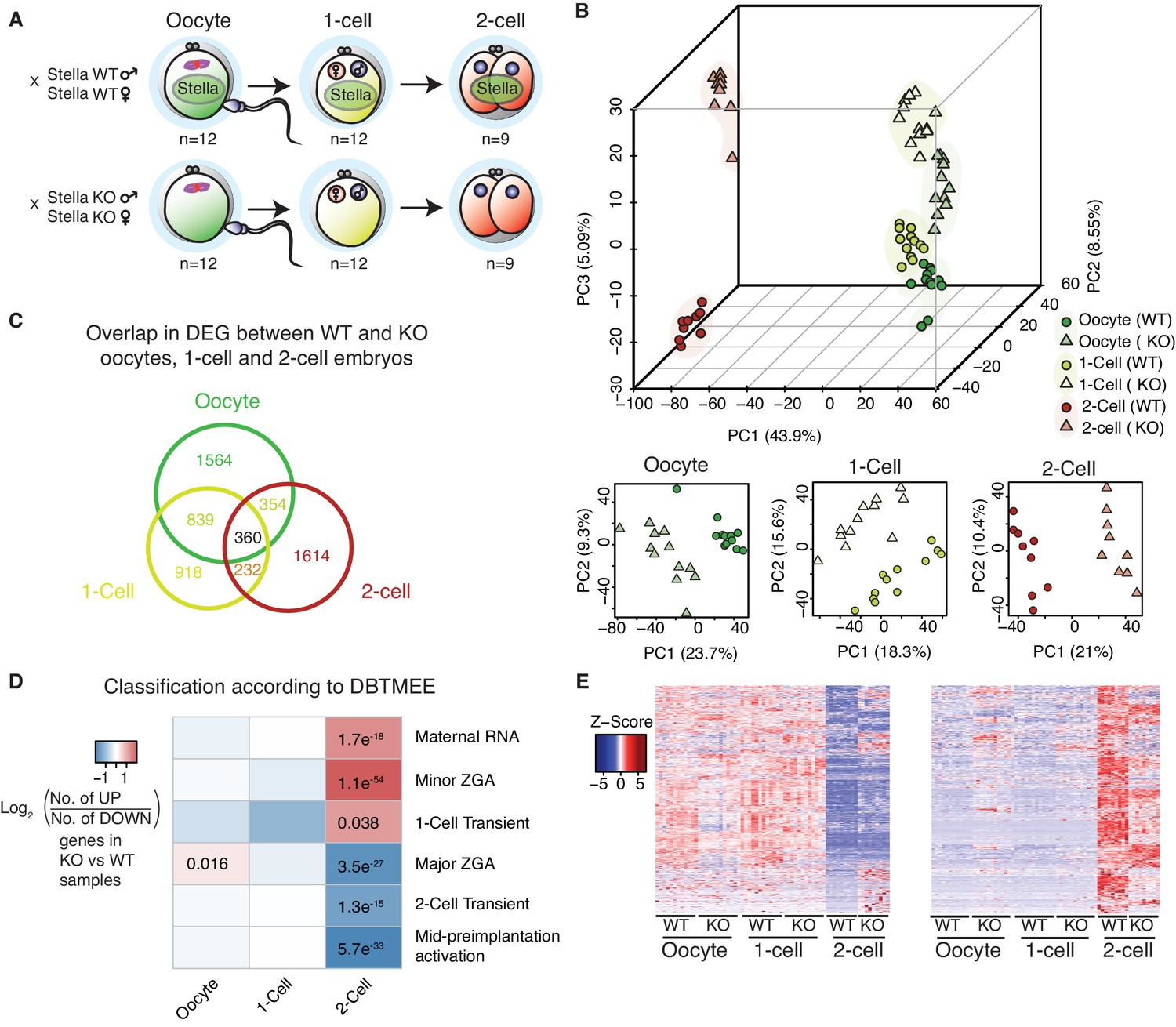
Stella M/Z KO embryos are impaired in maternal-to-zygotic transition.
(A) A schematic illustration of the single-cell / embryo RNA-seq experimental setup. The total number of oocytes and embryos collected are indicated. The colour scheme represents the transition from maternal (green) to zygotic (red) transcripts. Maternal Stella is represented in green circle. (B) A score plot of the first three principal components for 66 cells using gene counts. The lower panels represent the score plots of the first two principal components using cells belonging to a specific developmental time point. The developmental time points are indicated by colour and the genotypes for Stella are indicated by shape. (C) A Venn diagram illustrating the overlap of differentially expressed genes (DEG) between WT and KO oocytes, 1-cell and 2-cell embryos (adjusted p-value<0.05) (Figure 1—source data 2). (D) The heatmap represents the log2 ratio of the number of upregulated to downregulated genes in KO compared to WT at oocytes, 1-cell and 2-cell stage, belonging to a given cluster of DBTMEE (Park et al., 2015). Fisher’s exact test performed and statistically significant p-values are stated. (E) A time-series clustering of gene expression dynamics across oocyte to 2-cell embryo (see Materials and methods). (Left) Heatmap shows a cluster of WT maternal transcripts (ED), which are differentially expressed in KO samples (adjusted p-value<0.05, Figure 1—source data 3). (Right) Heatmap shows a cluster of WT zygotically activated genes (ZAG) (EU), which are differentially expressed in KO samples (adjusted p-value<0.05, Figure 1—source data 4). Also see Figure 1—figure supplements 1–2 and Figure 1—source data 1–4.
-
Figure 1—source data 1
Gene counts for WT and KO oocyte, 1-cell and 2-cell embryos.
Gene counts for 66 samples from single cell / embryo RNA-seq experiments. Removal of Unwanted Variation analysis has been performed to control for potentially confounding technical factors.
- https://doi.org/10.7554/eLife.22345.004
-
Figure 1—source data 2
List of differentially expressed genes between WT and KO samples at oocyte, 1-cell and 2-cell stage, from single cell/embryo RNA-seq analysis.
ENSEMBL ID and names of genes identified as significantly differentially expressed with adjusted P-value (padj) <0.05. Log2FoldChange=Log2(WT/KO). Stage indicates the developmental stage that is analysed.
- https://doi.org/10.7554/eLife.22345.005
-
Figure 1—source data 3
List of maternal transcripts, WT class = ED, which are differentially expressed in KO samples.
‘E’ denotes equally expressed, ‘D’ denotes downregulated or ‘U’ denotes upregulated at a later time point. Adjusted p-value (padj) shows significant differential expression between WT and KO class.
- https://doi.org/10.7554/eLife.22345.006
-
Figure 1—source data 4
List of zygotically activated genes (ZAG), WT class = EU, which are differentially expressed in KO samples.
‘E’ denotes equally expressed, ‘D’ denotes downregulated or ‘U’ denotes upregulated at a later time point. Adjusted p-value (padj) shows significant differential expression between WT and KO class.
- https://doi.org/10.7554/eLife.22345.007
Figure 1—figure supplement 1
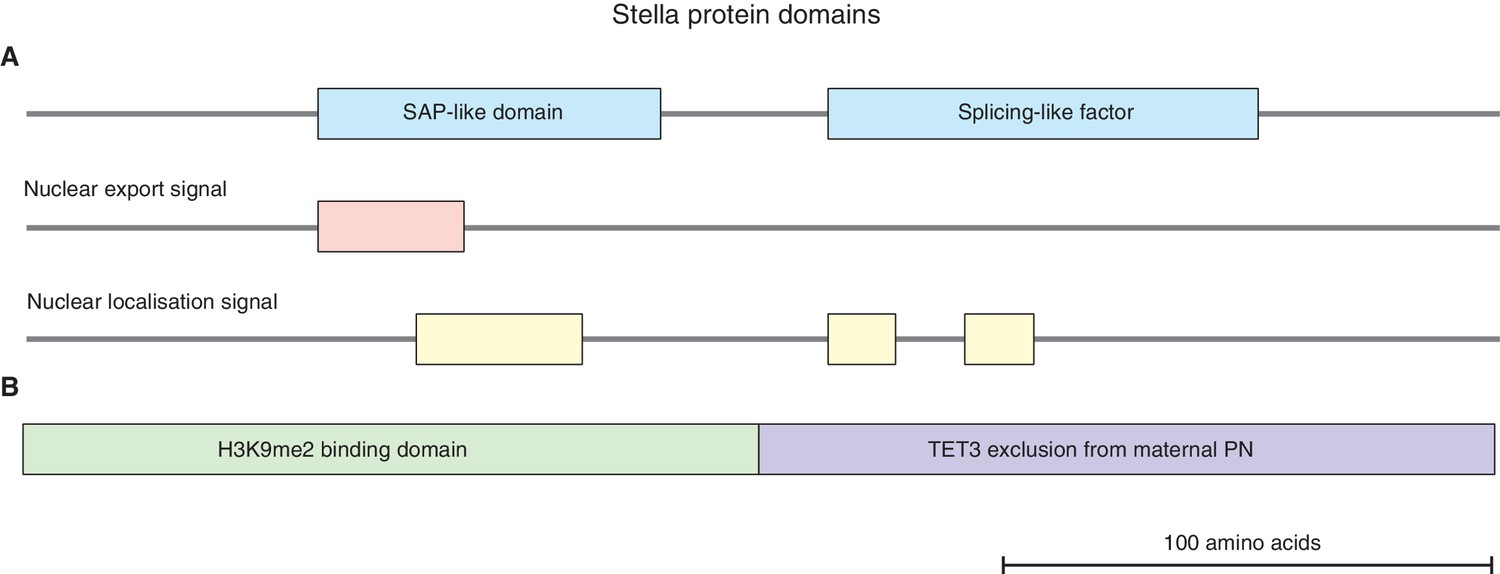
Stella protein domains.
(A) A schematic diagram illustrating the putative Stella protein domains: SAP-like domain, splicing-like factor, nuclear export and nuclear localization signals (Aravind and Koonin, 2000; Payer et al., 2003). (B) A schematic diagram illustrating the N-terminus of Stella (amino acids 1–75) is required for H3K9me2 binding in maternal pronuclei (PN), while the C-terminus of Stella (amino acids 76–150) is required to exclude TET3 from the maternal PN (Nakamura et al., 2012).
Figure 1—figure supplement 2
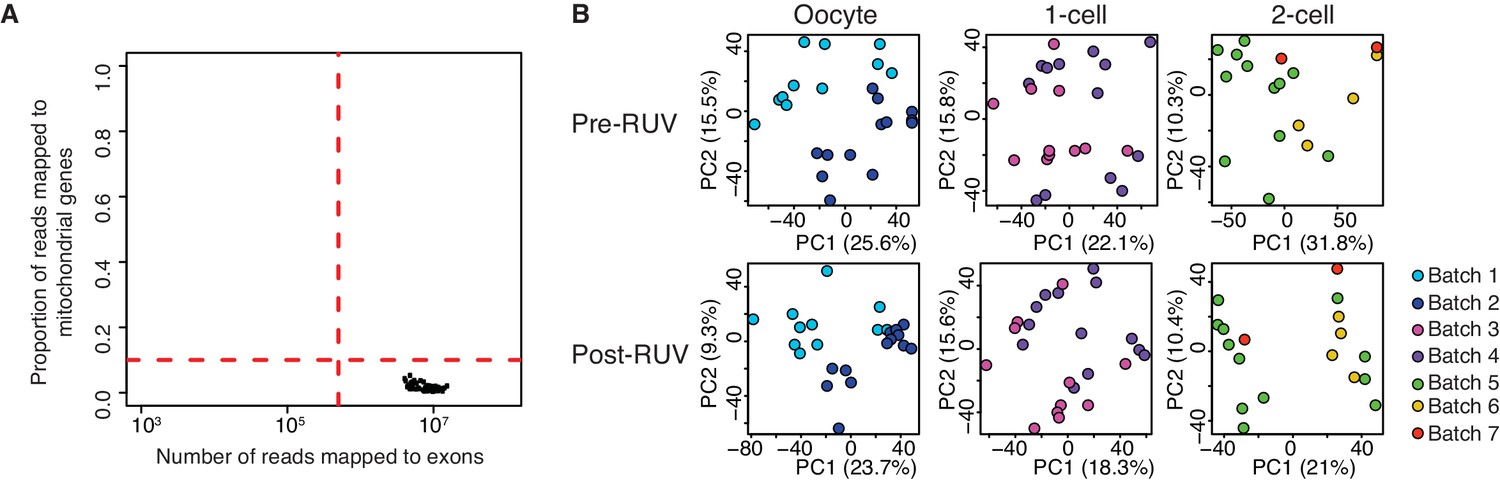
Quality control of single-cell / embryo RNA-seq.
(A) A scatter plot of the number of reads mapped to exons against the proportion of reads mapped to mitochondrial genes. The dashed red line indicates threshold of a good quality sample if: (1) cells have a total number of reads mapped to exons greater than 500,000; (2) cells have less than 10% of the proportion of reads mapped to 37 genes on the mitochondria chromosome. All 66 samples are of good quality. (B) A score plot of the first two principle components for cells of each developmental stage. RUV = removal of unwanted variation (see Materials and methods, Figure 1—source data 1). Top panel shows samples clustering pre-RUV, and bottom panel shows samples clustering post-RUV. Each batch represents independent experiments in which samples are collected, and this is indicated by a different colour (Supplementary file 1).
Figure 2 with 1 supplement

Stella is associated with the activation of essential zygotic genes.
(A) Revigo plots (Supek et al., 2011) of a selection of gene ontology (GO) terms enriched in 2-cell DEG (Figure 2—source data 1). The colour of the circle represents the log10 p-value. Semantic space clusters GO terms of similar functions together. (B) A boxplot of normalised counts of genes belong to the Ribosome KEGG pathway between WT and KO across the developmental stages. Two-sided Wilcoxon rank sum test performed between WT and KO and statistically significant p-value is stated. (C and D) shows boxplots of single-embryo qRT-PCR validation of RNA-seq in WT (white) and KO (light blue) 2-cell embryos. All genes are normalised to housekeeping genes, relative to one WT embryo and log2 transformed. (C) shows genes significantly upregulated and (D) downregulated in KO relative to WT 2-cell embryos (p<0.05 = *; p<0.01 = **, p<0.001 = *** and p<0.0001 = ****). (E) A table of the top 10 GO terms enriched in 710 genes whose expression levels are positively correlated with Dppa3 (Pearson’s correlation coefficient >0.9), identified from the genome-wide co-expression network analysis (Figure 2—source data 2). (F) Scatter plots of the expression of Snrpd1 and Snrpb2 against Dppa3. Gene expression was log2 transformed (counts per million + 1) and Pearson’s correlation analysis was performed. Also see Figure 2—figure supplement 1 and Figure 2—source data 1–2.
-
Figure 2—source data 1
List of gene ontology terms enriched in differentially expressed genes between WT and KO 2-cell embryos.
Gene ontology terms enriched for biological processes were calculated using DAVID software (version 6.7).
- https://doi.org/10.7554/eLife.22345.011
-
Figure 2—source data 2
Genome-wide Dppa3 co-expression network analysis.
Table shows ensemble ID and official names of genes exhibiting significant expression correlation with Dppa3 (Pearson Correlation |r| > 0.9).
- https://doi.org/10.7554/eLife.22345.012
Figure 2—figure supplement 1
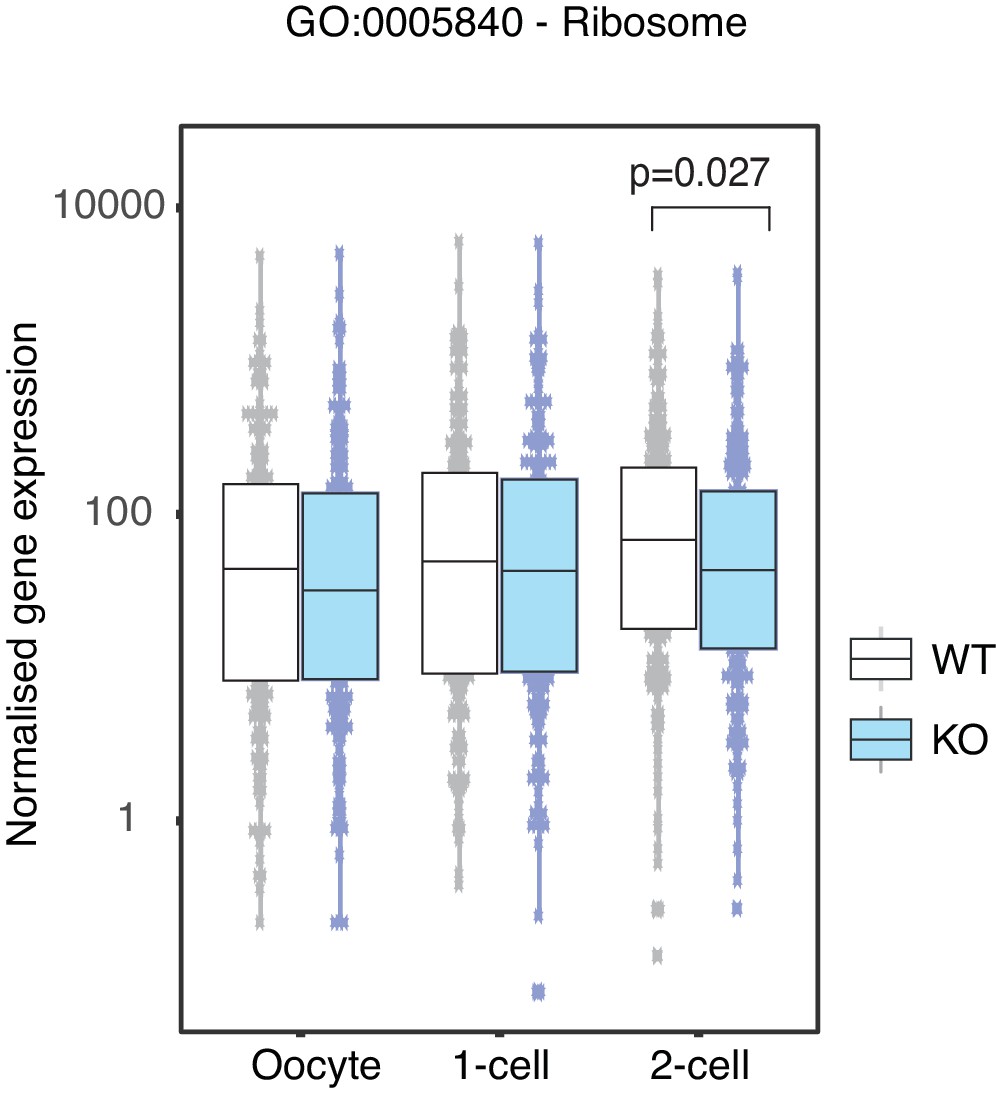
Ribosome associated genes are depleted in Stella M/Z KO 2-cell embryos.
Comparison of normalised counts of genes belonging to the Ribosome associated gene ontology pathway between WT (white) and KO (blue) across the developmental stages. Two-sided Wilcoxon rank sum test was performed between WT and KO samples and statistically significant p-value stated.
Figure 3 with 3 supplements
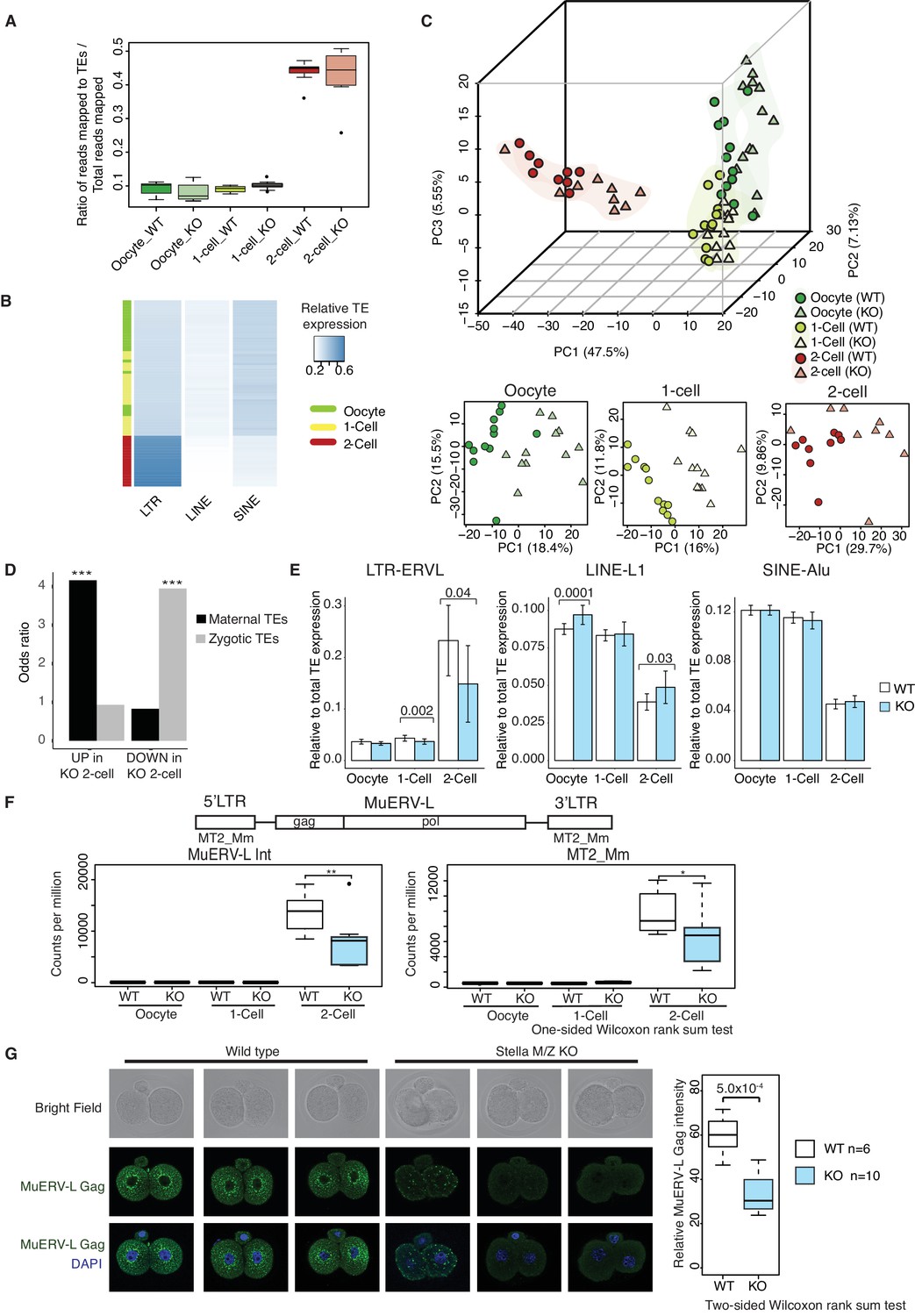
TEs are mis-expressed in the absence of Stella.
(A) Top panel shows boxplots of the ratio of reads mapped to TEs to total reads mapped in WT and KO oocytes, 1-cell and 2-cell embryos. The number of reads mapped to TEs are based on uniquely and multi-mapped counts (Figure 3—source data 2). (B) A heatmap of the relative expression of LTR, LINE and SINE in oocyte (green), 1-cell (yellow) and 2-cell embryos (red). Blue indicates higher expression and white indicates lower expression. Samples are clustered by row. (C) A score plot of the first three principal components for 66 cells using uniquely mapped TE counts (Figure 3—source data 1). The lower panels represent the score plots of the first two principal components using cells belonging to a specific developmental time point. The developmental time points are indicated by colour and the genotypes of Stella are indicated by shape. (D) A bar chart of the odds ratio of TEs up and downregulated in KO 2-cell embryos compared to WT, intersected with ‘maternal TEs’ and ‘zygotic TEs’. For maternal TEs enriched in TEs upregulated in KO 2-cell, ***p=5.35 × 10−6; for zygotic TEs enriched in TEs downregulated in KO 2-cell, ***p=8.13 × 10−7. (E) Bar plots showing the relative expression of TE families (LTR-ERVL, LINE1-L1 and SINE-Alu) in WT (white) and KO (light blue) oocytes, 1-cell and 2-cell embryos, data analysed from single-cell / embryo RNA sequencing. Two-sided Wilcoxon rank sum test performed between WT and KO samples and statistically significant p-values stated. (F) Top is an illustration of the structure of the full-length MuERV-L element flanked by 5’ and 3’ LTRs. Bottom is boxplots of the relative expression (counts per million) of MuERV-L Int and MT2_Mm transcript in WT (white) and KO (light-blue) oocytes, 1-cell and 2-cell embryos (p<0.05 corresponds to * and p<0.01 corresponds to **). (G) Immunofluorescence staining against MuERV-L Gag antibody in 2-cell embryos. (Left) The top panel shows bright-field, middle panel is MuERV-L Gag (green) and bottom panel are merged images of MuERV-L and DAPI, which counterstains DNA. Representative projections of z-stacks are shown for WT and KO embryos. (Right) A boxplot of the z-stack quantifications of the relative fluorescence intensity of MuERV-L Gag protein between WT and KO 2-cell embryos. Also see Figure 3—figure supplements 1–3 and Figure 3—source data 1–2.
-
Figure 3—source data 1
Uniquely mapped transposable element (TE) counts.
TE counts for 66 samples from single cell / embryo RNA-seq experiments. Uniquely mapped reads only.
- https://doi.org/10.7554/eLife.22345.015
-
Figure 3—source data 2
Uniquely and multi-mapped transposable element (TE) counts.
TE counts for 66 samples from single cell/embryo RNA-seq experiments. Uniquely and multi-mapped reads.
- https://doi.org/10.7554/eLife.22345.016
Figure 3—figure supplement 1
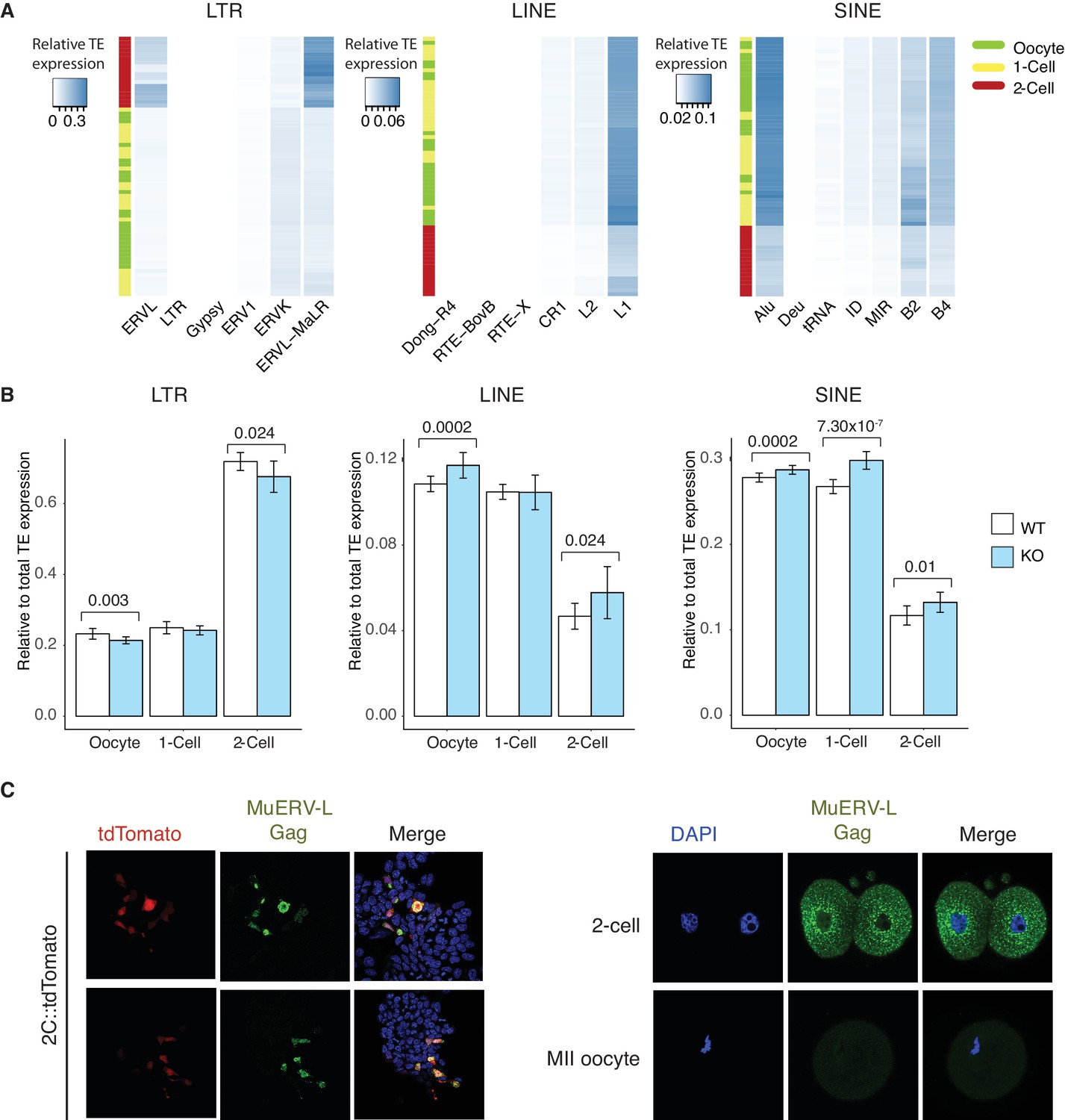
TEs are mis-expressed in the absence of Stella.
(A) Heatmaps of the relative expression of individual families of LTR, LINE and SINE elements in oocyte (green), 1-cell (yellow) and 2-cell (red) embryos. Samples are clustered by row. Blue indicated relatively high expression while white indicates relatively low expression of TEs. (B) Bar charts of relative expression of LTR elements, LINE and SINE in WT (white) and KO (light blue) oocytes, 1-cell and 2-cell embryos. Two-sided Wilcoxon rank sum test performed between WT and KO samples and statistically significant p-values are stated. (C) Validation of MuERV-L Gag antibody. (Left) IF staining of an ESC reporter line for MuERV-L – 2C::tdTomato (Macfarlan et al., 2012), illustrating the MuERV-L Gag (GFP) staining overlaps well with tdTomato+ ESC. (Right) IF staining showing robust detection of MuERV-L Gag (GFP) in 2-cell embryos, while it is virtually undetectable in metaphase II (MII) oocytes. DNA is counterstained with DAPI.
Figure 3—figure supplement 2
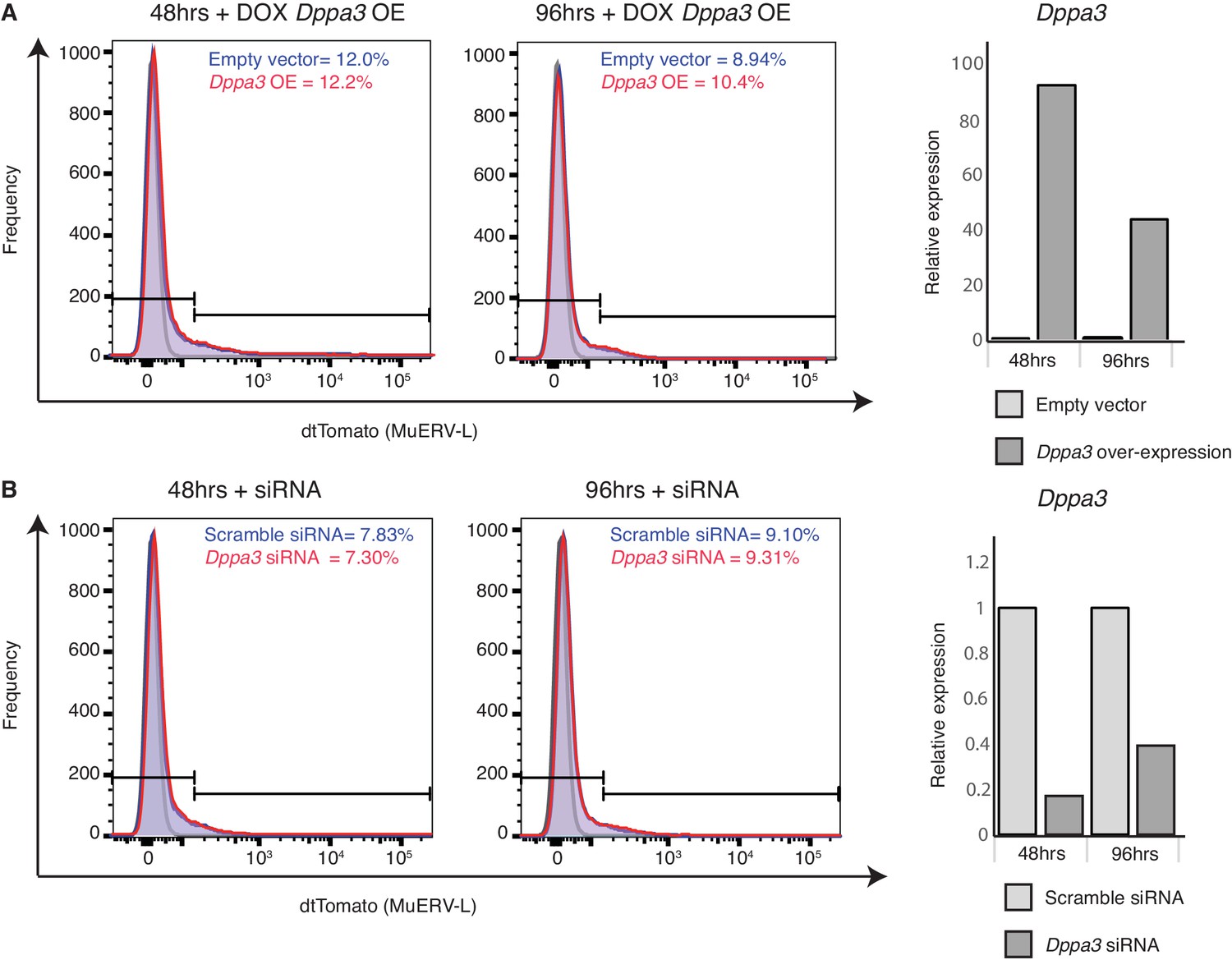
Dppa3 does not affect MuERV-L expression in mESCs.
(A) Left shows histogram plots of tdTomato expression in 2C::tdTomato mESCs 48 hr and 96 hr after addition of DOX to induce Dppa3 expression. Red represents tdTomato frequency with Dppa3 over-expression (OE), while blue represents empty vector transfection control. The horizontal bars depict the expression threshold set for dtTomato and the proportion of tdTomato+ cells in each condition is stated. Right shows a bar plot of Dppa3 expression 48 hr and 96 hr after addition of DOX. Dppa3 expression is normalized to a housekeeping gene (GAPDH) and relative to empty vector sample. (B) Left shows histogram plots of tdTomato expression in 2C::tdTomato mESCs 48 hr and 96 hr after siRNA transfection. Red represents tdTomato frequency with Dppa3 siRNA, while blue represents scramble siRNA control. The bar depicts the expression threshold set for dtTomato and the proportion of tdTomato+ cells in each condition is stated. Right shows a bar plot of Dppa3 expression 48 hr and 96 hr after siRNA transfection. Dppa3 expression is normalized to a housekeeping gene (GAPDH) and relative to scramble siRNA sample.
Figure 3—figure supplement 3
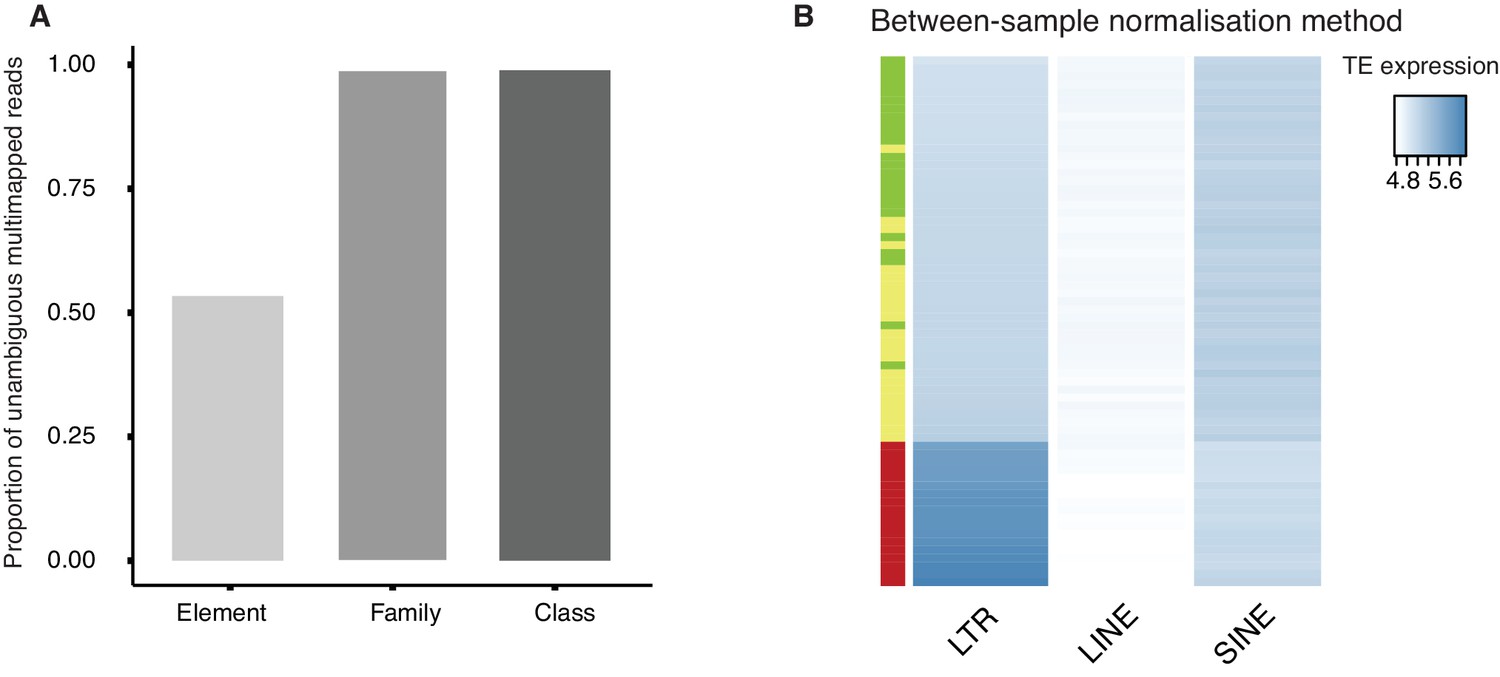
Quality control of TE reads mapping and normalization.
(A) A bar plot of the proportion of multi-mapped TE reads (Figure 3—source data 2) which can be unambiguously assigned at the level of class (repClass), family (repFamily) and element (repName) based on the RepeatMasker annotation provided by UCSC table browser. (B) A heatmap of the expression of LTR, LINE and SINE in oocyte (green), 1-cell (yellow) and 2-cell (red) embryos based on between-sample normalization method (See Materials and methods). Samples are clustered by row.
Figure 4 with 1 supplement
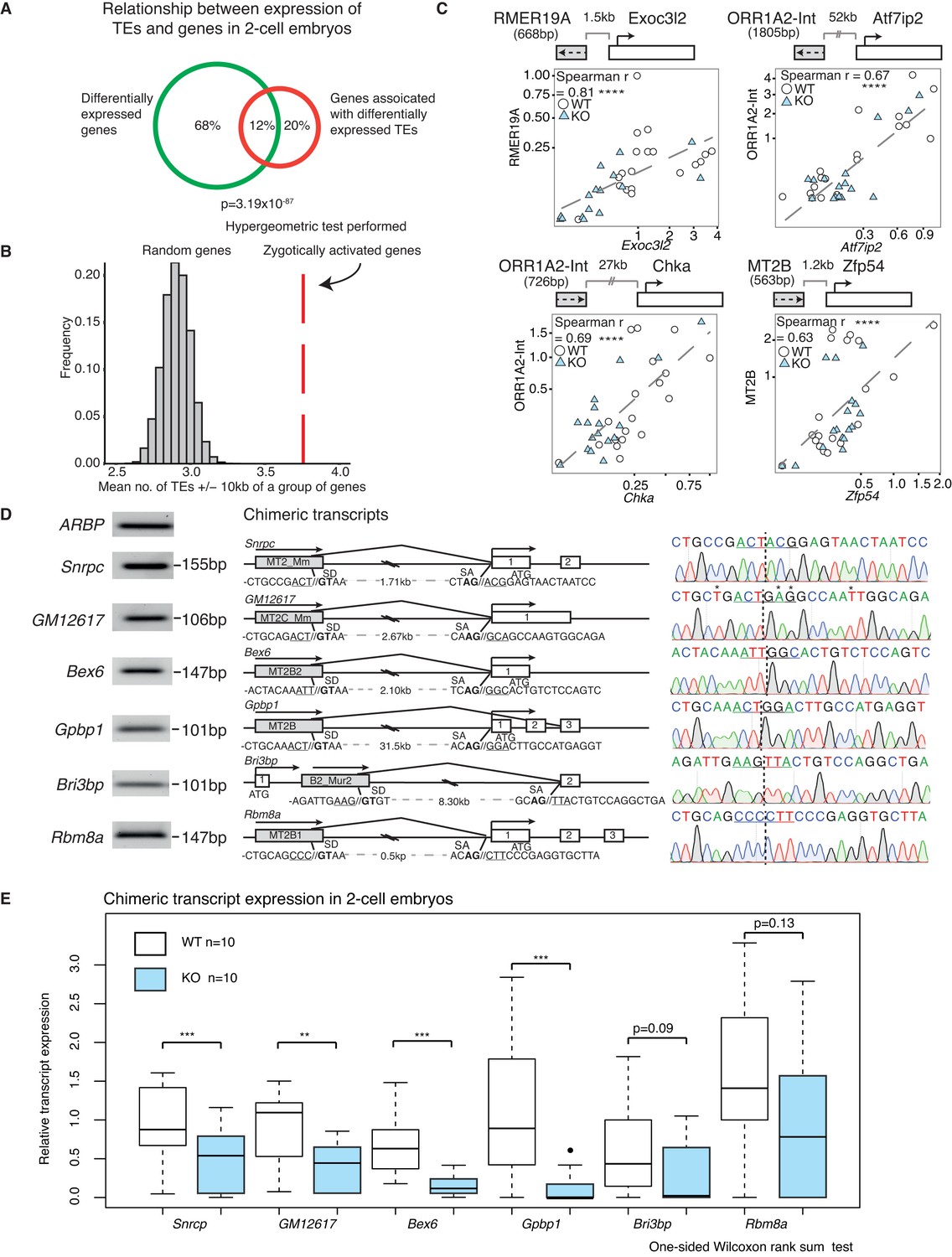
A positive correlation between the expression of a subset of TEs and their nearest gene.
(A) A Venn diagram illustrating a significant overlap between differentially expressed genes and genes within 20 kb of differentially expressed TEs in WT v KO 2-cell embryos. (B) A histogram of the mean number of depleted TEs in Stella M/Z KO 2-cell embryos within 10 kb of the TSS of a group of genes. The red line represents zygotically activated genes (ZAG, n = 698) that belong to WT class EU and differentially expressed in KO samples (adjusted p-value<0.05) (Figure 1—source data 4). The grey bars represent each of 10,000 random sets of 698 genes. (C) Top is schematic illustrations of the relationship between the TE and downstream gene, where grey box represents the TE and its orientation; white box represents the gene and its orientation. Bottom is scatter plots of qRT-PCR expression in 2-cell embryos. WT (white circle) n = 19 and KO (light blue triangle) n = 17. Spearman’s correlation analysis was performed and p<0.0001 corresponds to ****. (D) Characterisation of chimeric transcripts. (Middle) A schematic illustration of the alternative splicing event that occurred between the TE and the downstream gene. SD = splice donor site and SA = splice acceptor site. The endogenous starting codon (ATG) is indicated. (Left) The PCR product of the chimeric transcript where the forward primer originates from the TE and reverse primer originates from the gene. The PCR product is significantly shorter than the annotated distances between them, confirming a splicing event. (Right) Sanger sequencing chromatographs from the PCR product for each chimeric transcript, confirming the site of chimeric junction. * Indicates a different nucleotide to annotation. (E) Box plots of single-embryo qRT-PCR of the relative expression of chimeric transcripts in WT (white) and KO (light blue) 2-cell embryos. p<0.01 corresponds to ** and p<0.001 corresponds to ***. All expression data are normalised to three housekeeping genes and relative to 1 WT embryo. Also see Figure 4—figure supplement 1.
Figure 4—figure supplement 1
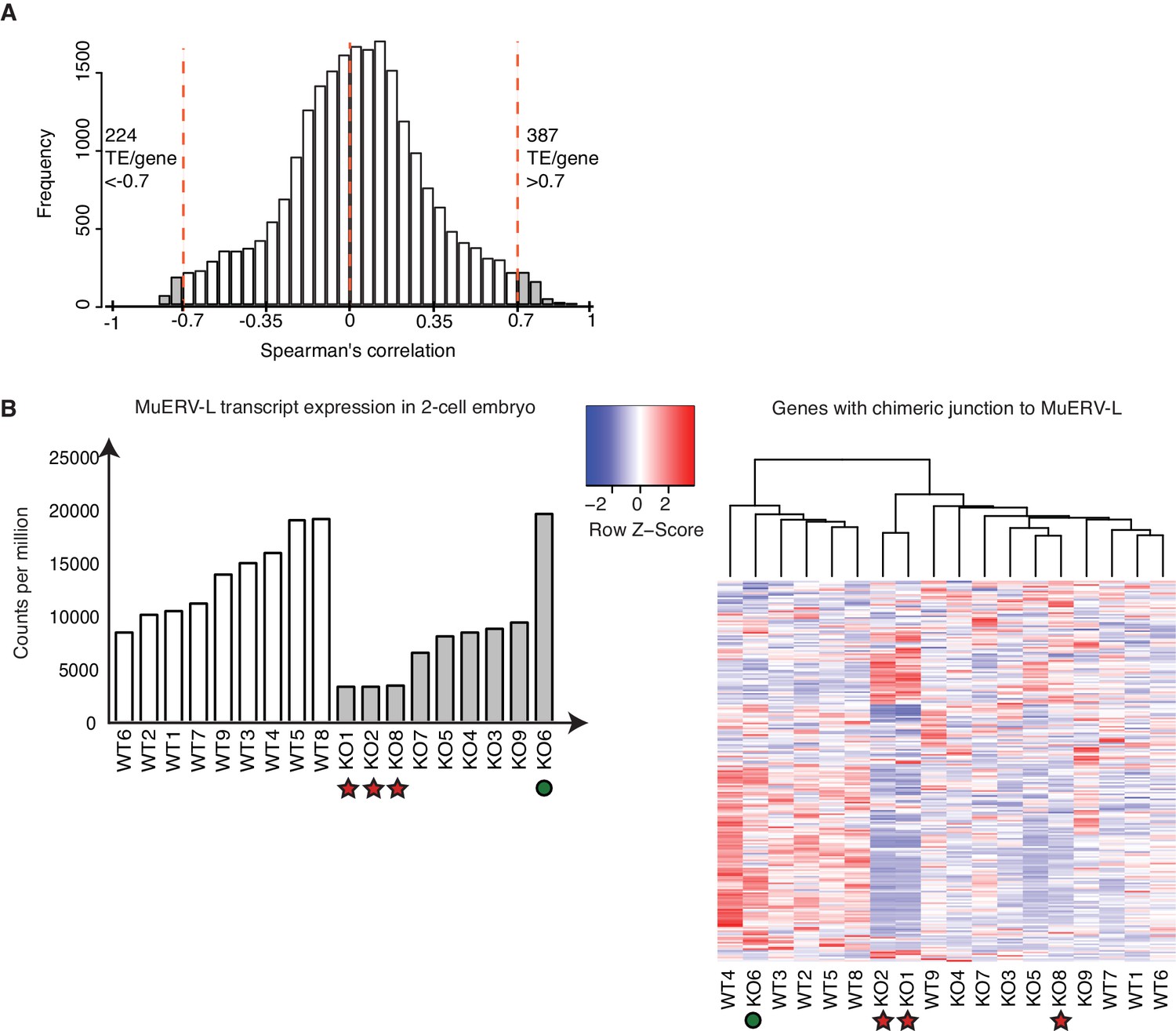
A subset of TEs is positively correlated with its nearest genes.
(A) A histogram of Spearman’s correlation of the expression between a gene and its nearest TE within a distance of 1Mbp. The red line indicates correlation >|0.7|. 387 TE / gene pair correlation >0.7 while 224 TE/ gene pair correlation <−0.7. (B) (Left) A bar chart of MuERV-L int transcript expression in individual WT (white) and KO (grey) 2-cell embryos. Red star indicates 3 KO embryos expressing the lowest MuERV-L transcript (KO2,8,1) while green circle highlights KO6, the KO embryo which expresses the highest MuERV-L levels. The right panel shows a heatmap of expression of genes identified to make a chimeric junction with MuERV-L (Macfarlan et al., 2012). Heatmap is normalised by row and clustered by column.
Figure 5 with 1 supplement
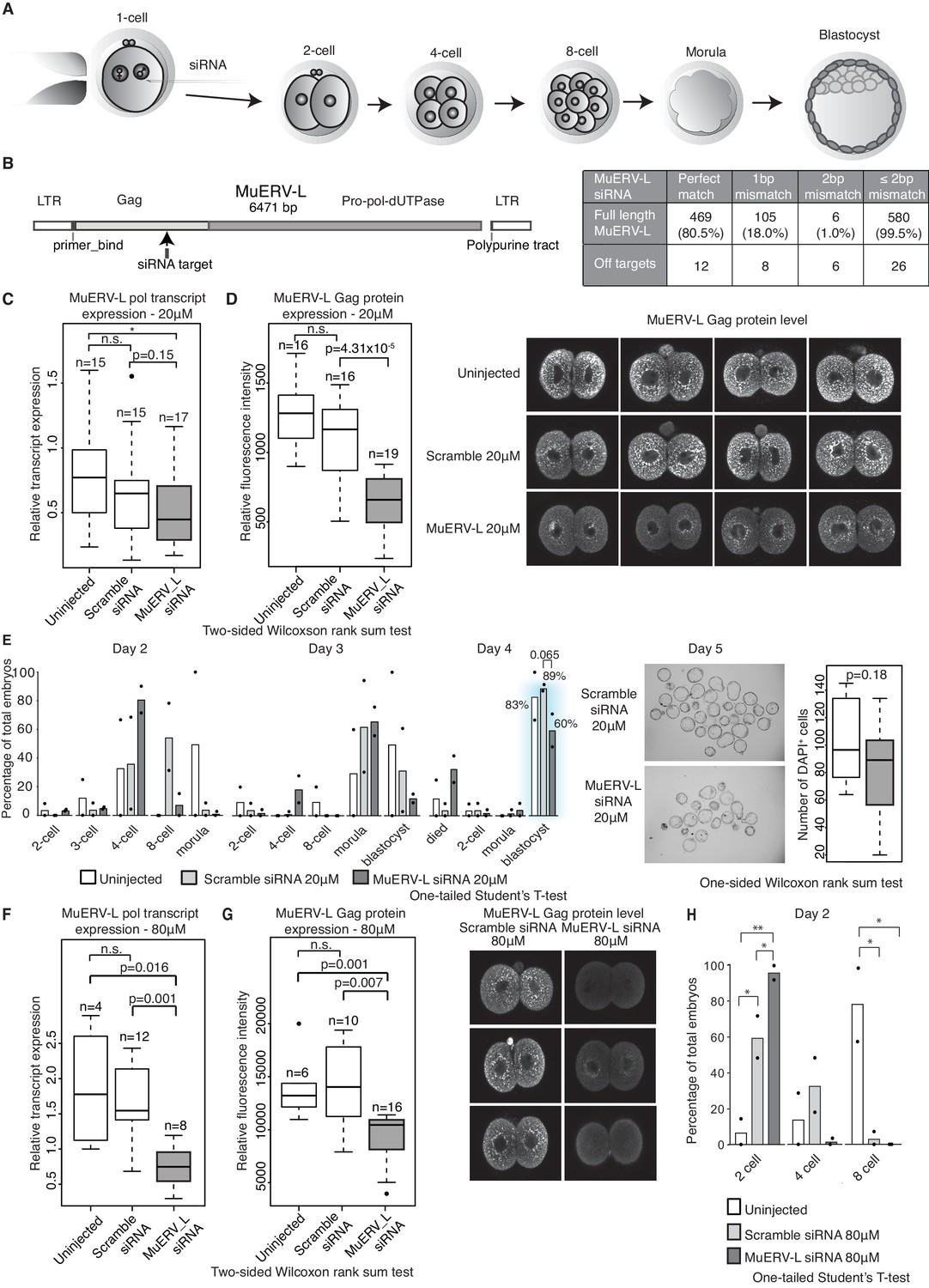
MuERV-L plays a functional role during pre-implantation development.
(A) A schematic diagram illustrating the experimental setup. (B) Left is an illustration of the structure of the MuERV-L element (GenBank accession number: Y12713), depicting the exact site of siRNA target. Right is a table showing the number of full-length MuERV-L elements targeted by MuERV-L siRNA and the number of off-targets. (C) Box plots of single-embryo qRT-PCR of MuERV-L pol transcript and D) MuERV-L Gag protein (fluorescence) intensity in uninjected controls, 20 µM scramble siRNA or 20 µM MuERV-L siRNA injection 2-cell embryos. (Right) Representative z-stack projections of immunofluorescence (IF) staining against MuERV-L Gag in 2-cell embryos. Two-tail Wilcoxon Rank Sum Test was performed for both (C) and (D) and p<0.01 corresponds to *, not significant (n.s.) or are otherwise stated. (E) The effect of MuERV-L siRNA on developmental progression of pre-implantation embryos. (Left) Bar graphs illustrating the number of embryos observed at different stages on day 2–4 of in-vitro culture. Blue background indicates area of interest. Experiments were repeated twice and the circle dots represent the data points for each replicate. A total of n = 17, n = 58 and n = 53 embryos were analysed for uninjected, scramble and MuERV-L siRNA injections respectively. One-tailed Student’s T-test performed and there were no statistical difference between uninjected, scramble or MuERV-L siRNA injected groups. (Middle) Day five bright field images of late blastocysts injected with scramble or MuERV-L siRNA. (Right) A box plot of IF quantification of number of DAPI+ cells in the late blastocyst stage on day five injected with 20 µM of scramble (white) or MuERV-L (grey) siRNA. (F and G) are box plots of single-embryo qRT-PCR of MuERV-L pol transcript and MuERV-L Gag protein (fluorescence) intensity in uninjected control, 80 µM scramble siRNA and 80 µM MuERV-L siRNA injected 2-cell embryos. (Right) Representative z-stack projections of IF staining against MuERV-L Gag antibody in 2-cell embryos. Two-tail Wilcoxon Rank Sum Test was performed for both (F) and (G) and p<0.01 corresponds to *, not significant (n.s.) or are otherwise stated. (H) A bar graph plotting the number of embryos observed at each stage in day two in-vitro culture in uninjected embryos, 80 µM scramble (white) or MuERV-L (grey) siRNA injection. Experiments were repeated twice and the circle dots represent the data points for each replicate. A total of n = 17, n = 54 and n = 50 embryos were analysed for uninjected, scramble and MuERV-L siRNA respectively. One-tailed Student’s T-test performed. p<0.05 corresponds to * and p<0.01 corresponds to **.
Figure 5—figure supplement 1

Chimeric transcript expression in MuERV-L knockdown embryos.
A scatter dot plot of qRT-PCR analysis of chimeric transcripts in uninjected (circle, n = 4), 80 µM scramble siRNA (square, n = 12) and 80 µM MuERV-L siRNA (triangle, n = 8) treated 2-cell embryos. The median and interquartile range is indicated. All transcripts are normalised to housekeeping genes, relative to one uninjected embryo. Two-sided Wilcoxon rank sum test was performed and statistically significant results are stated.
Author response image 1
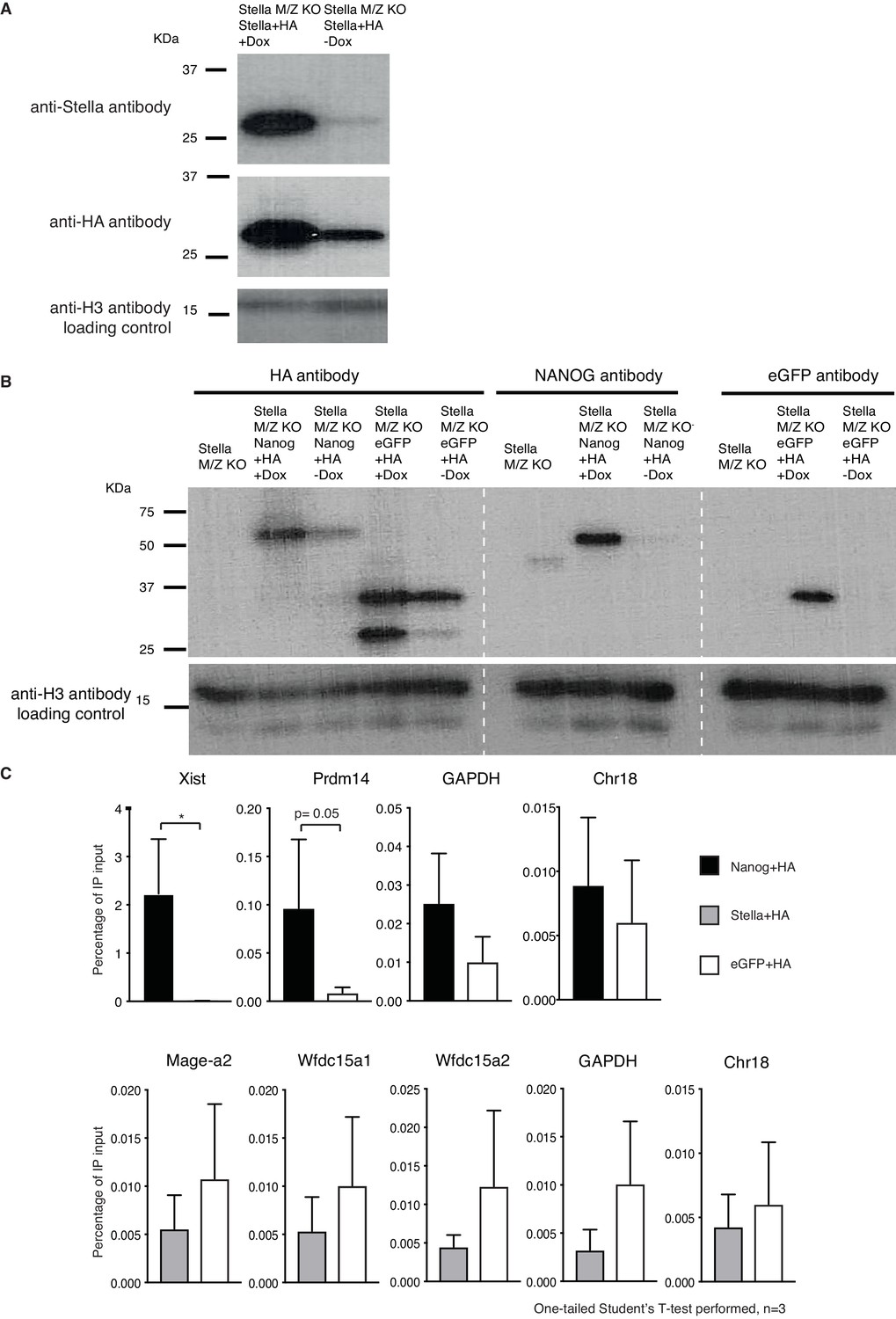
Stella-HA ChIP.
(A) Western blot of Stella and HA. Stella with 3 copies of hemagglutinin (HA) tagged to its C-terminal is over-expressed in a Dox inducible system in StellaM/Z KO ESCs. Stella’s predicted size is ~ 21kDA, and appears as ~ 27kDA due to addition of 3xHA construct. Bottom panel shows H3 loading control. (B) Western blot for NANOG+HA and eGFP+HA. Both NANOG+HA and eGFP+HA constructs were expressed in a Dox-inducible system in StellaM/Z KO ESCs. The blot show NANOG+HA construct is detected at ~55kDA while the endogenous NANOG is detected at ~45kDA. eGFP+HA construct is detected at ~37kDA, as confirmed with eGFP antibody alone. Bottom panel shows H3 loading control. (C) Top panel shows ChIP-qPCR of the relative enrichment of NANOG-HA in NANOG known binding regions (Xist, proximal enhancer of Prdm14) and gene desert regions (GAPDH and Chr18). Bottom panel shows ChIP-qPCR of the relative enrichment of Stella-HA in previously published Stella binding regions (Mage-a2, Wfdc15a) (Nakamura et al., 2012) and gene desert regions. One-tailed Student’s T-test was performed, where * corresponds to p<0.05 or otherwise stated.
Author response image 2

Effects of MT2 element knockdown on pre-implantation embryonic development.
(A) A bar graph illustrating the number of uninjected embryos, embryos injected with 20M scramble or 20M MT2_Mm siRNAs, at different stages of development in day 2 – 4 of in-vitro culture, n=1. The percentage of embryos reaching the blastocyst stage is stated. (B) Bioinformatics analysis of the number of MT2_Mm elements and off-targets that are perfect matches or 1 or 2 mismatches for MT2_Mm siRNA. A total of 2667 MT2_Mm elements are included in the analysis. (C) A bar graph illustrating the number of uninjected embryos, embryos injected with 20 M scramble or 20 M MT2 siRNAs, at different stages of development in day 2 – 4 of in-vitro culture, n=1. The percentage of embryos reaching the blastocyst stage is stated. (D) Bioinformatics analysis of the number of MT2 elements (MT2A, MT2B, MT2B1, MT2B2, MT2C_Mm) and off-targets that are perfect matches or 1 or 2 mismatches for MT2 siRNA. A total of 37002 MT2 elements are included in the analysis.
Author response image 3
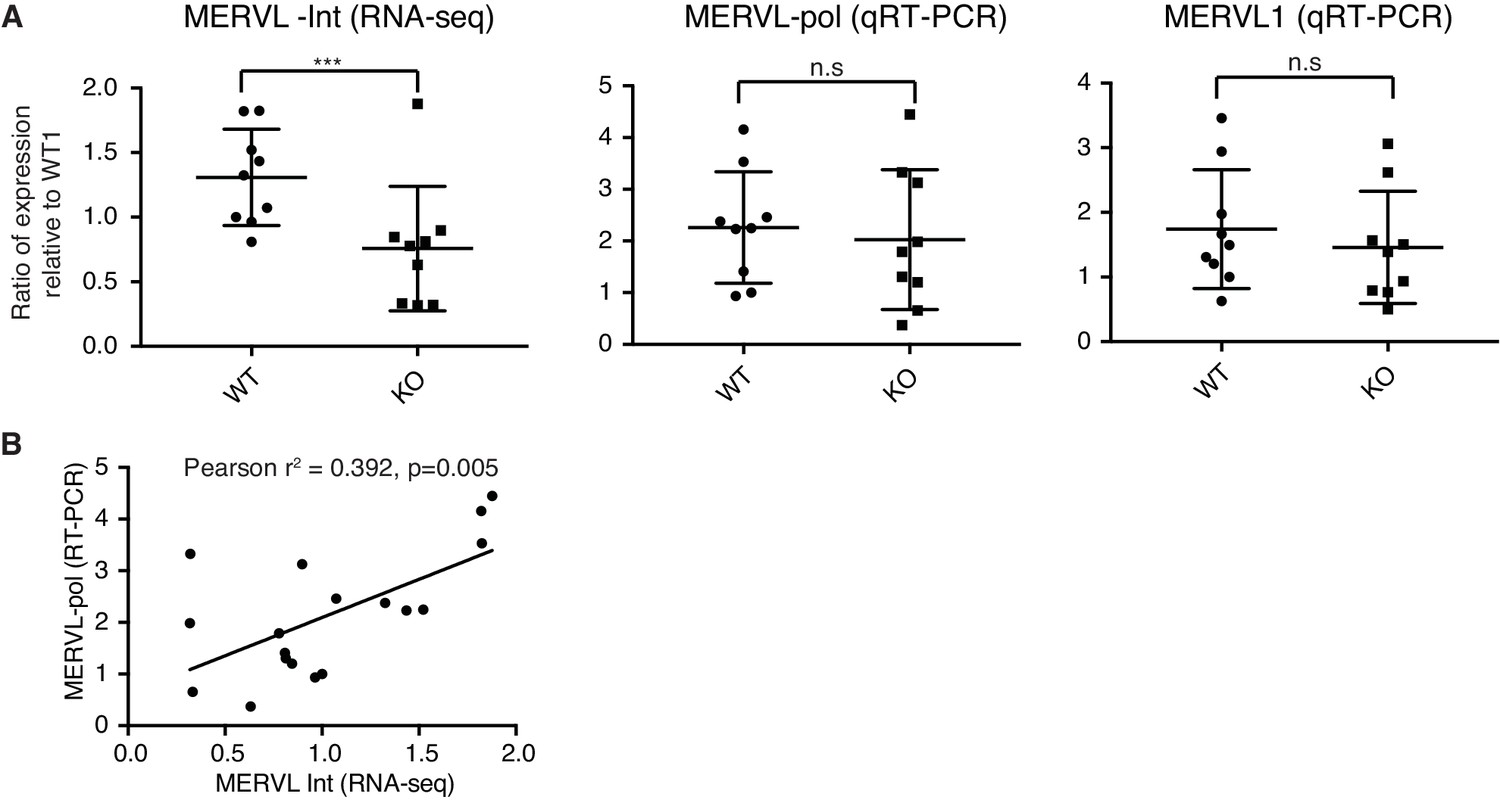
MERVL RNA-sequencing versus qRT-PCR.
(A) The first dot plot shows MERVL-Int expression in WT and KO 2-cell embryos. MERVL-Int reads were mapped from single embryo RNA-sequencing and expressed as counts per million. The latter two dot plots shows MERVL expression from 2 qRT-PCR primers (MERVL-pol and MERVL1) in the same WT and KO 2-cell embryos. Samples are expressed as a ratio relative to WT embryo 1 (WT1). The median and interquartile range is indicated. WT n=9, KO n=9. Two-sided Wilcoxon rank sum test was performed and *** corresponds to p<0.001, n.s = not significant. (B) A plot of expression correlation between MERVL-Int from RNA-sequencing and MERVL-pol from qRT-PCR, Pearson correlation analysis performed.
Additional files
-
Supplementary file 1
Sample details for single-cell / embryo RNA sequencing.
- https://doi.org/10.7554/eLife.22345.024
-
Supplementary file 2
Stella ChIP-seq enriched peaks.
- https://doi.org/10.7554/eLife.22345.025
-
Supplementary file 3
A list of primer sequences used in this study.
- https://doi.org/10.7554/eLife.22345.026
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Stella modulates transcriptional and endogenous retrovirus programs during maternal-to-zygotic transition
eLife 6:e22345.
https://doi.org/10.7554/eLife.22345




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}