MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2
- Washington University Medical School, United States
- Hope Center for Neurological Disorders, United States
Figures
Figure 1

MKK4/7 are necessary for NAD+ and ATP loss after axotomy but not in response to constitutively active SARM1.
MKK4/7 are necessary for NAD+ and ATP depletion in axons following axotomy but not following direct SARM1 activation. Axonal NAD+ (A) and ATP (B) levels were assayed at indicated timepoints after axotomy with lentiviral knockdown of MKK4 and MKK7 (sh4/7, grey bars) or Luciferase (ctrl, black bars) control. There is no change in the rate of NAD+ (C) or ATP (D) depletion after direct activation of SARM1 via dimerization of the SARM1-TIR domains in the absence of injury when MKK4/7 are depleted compared to controls. The compound AP20187 was used to induce SARM1-TIR dimerization. Values are presented as mean ± SEM. All measurements were normalized to time zero. N = 6; p values: *≤ 0.05, **≤ 0.01, ***≤ 0.001, ****≤ 0.0001 by ANOVA.
Figure 2 with 1 supplement
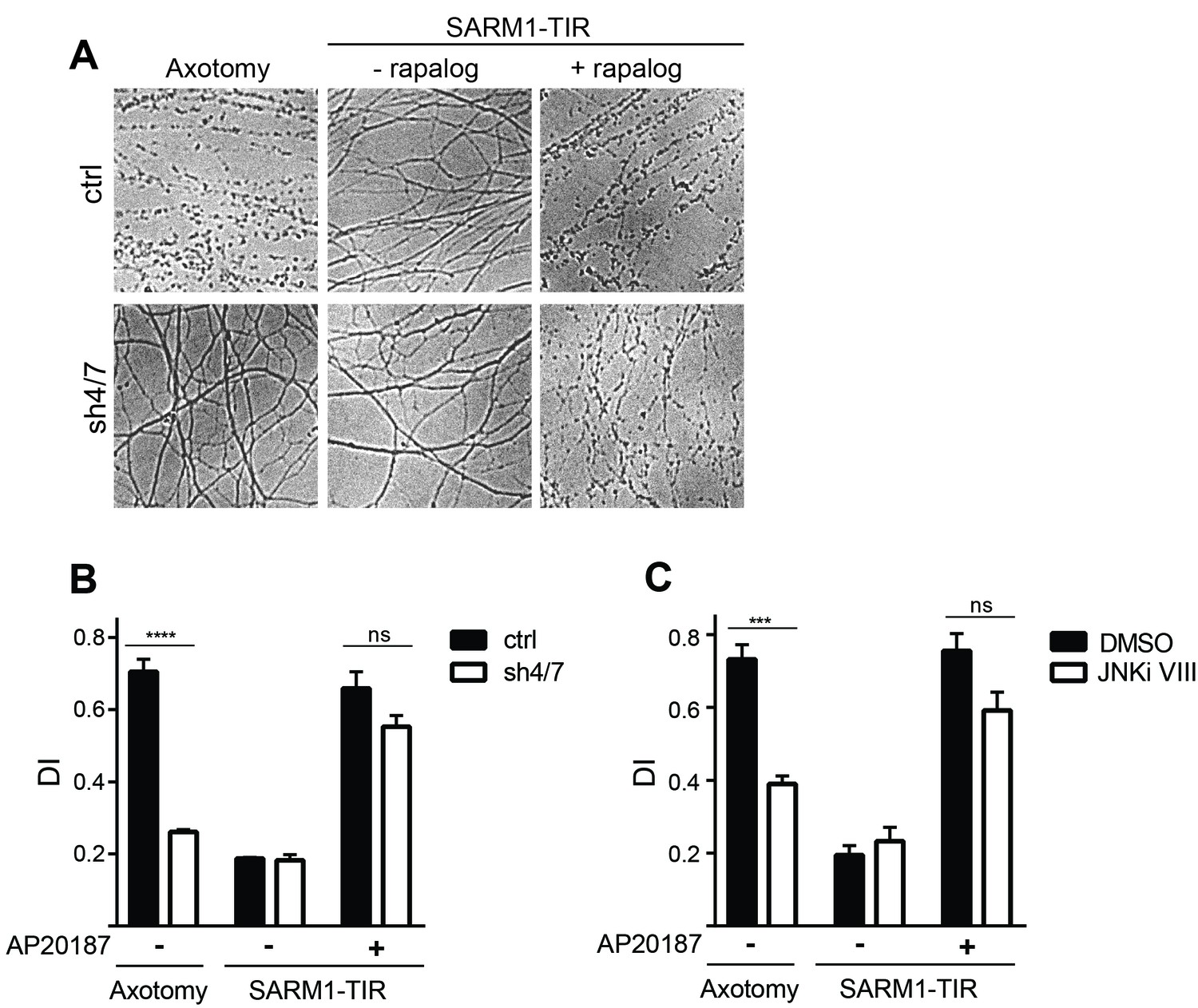
MAPKs are required for injury-induced but not SARM1-TIR-induced axon degeneration.
(A) Depletion of MKK4/7 (sh4/7) protects axons for 24 hours after axotomy but not when axon degeneration is induced by dimerization of the SARM1-TIR domains with addition of the ligand AP20187 (n = 4). Knockdown of MKK4/7 is compared to controls (ctrl) expressing an shRNA targeting luciferase (B) Quantification of degeneration index (DI). (C) Treatment with JNK inhibitor VIII delays axon degeneration 24 hours after axotomy but not when axon degeneration is induced by dimerization of the SARM1-TIR domains with addition of the ligand AP20187 as compared to DMSO vehicle control (n = 3). The data for each histogram are derived from neurons that were cultured in the same dish with treatments (i.e. axotomy or TIR-dimerization) performed in parallel. p value: ***≤ 0.001 and ****≤ 0.0001 by ANOVA; non-significant (ns). Values are presented as mean ± SEM. See also Figure 2—figure supplement 1.
Figure 2—figure supplement 1
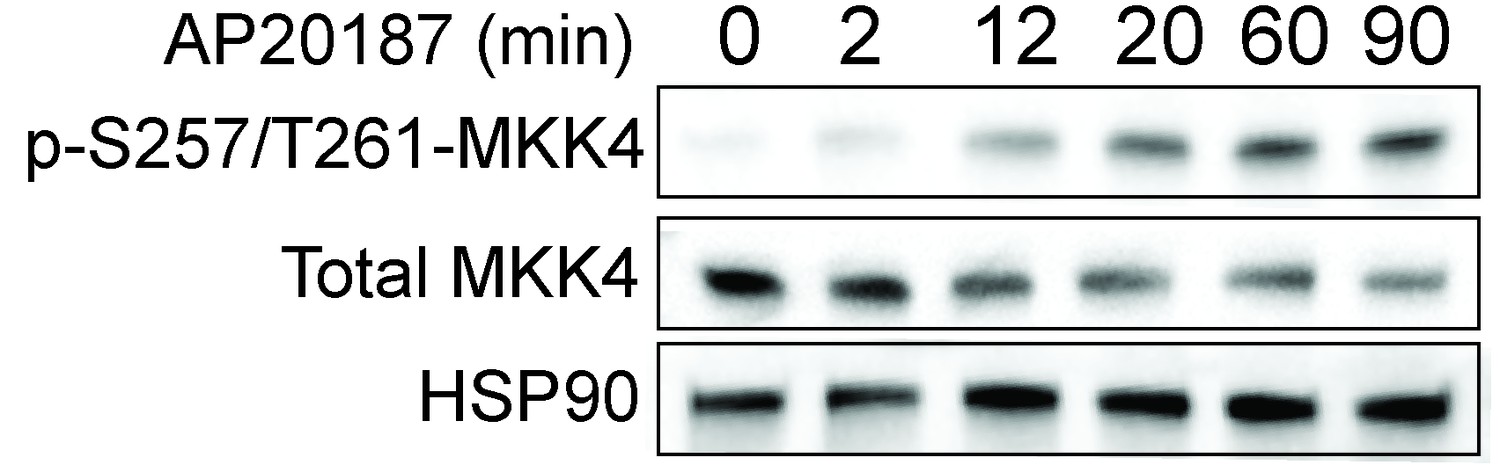
Dimerization of the SARM1-TIR domains with the ligand AP20187 results in MKK4 phosphorylation on Ser257/Thr261.
Lystate from DRGs was collected at the indicated timepoints after addition of AP20187.
Figure 3 with 1 supplement
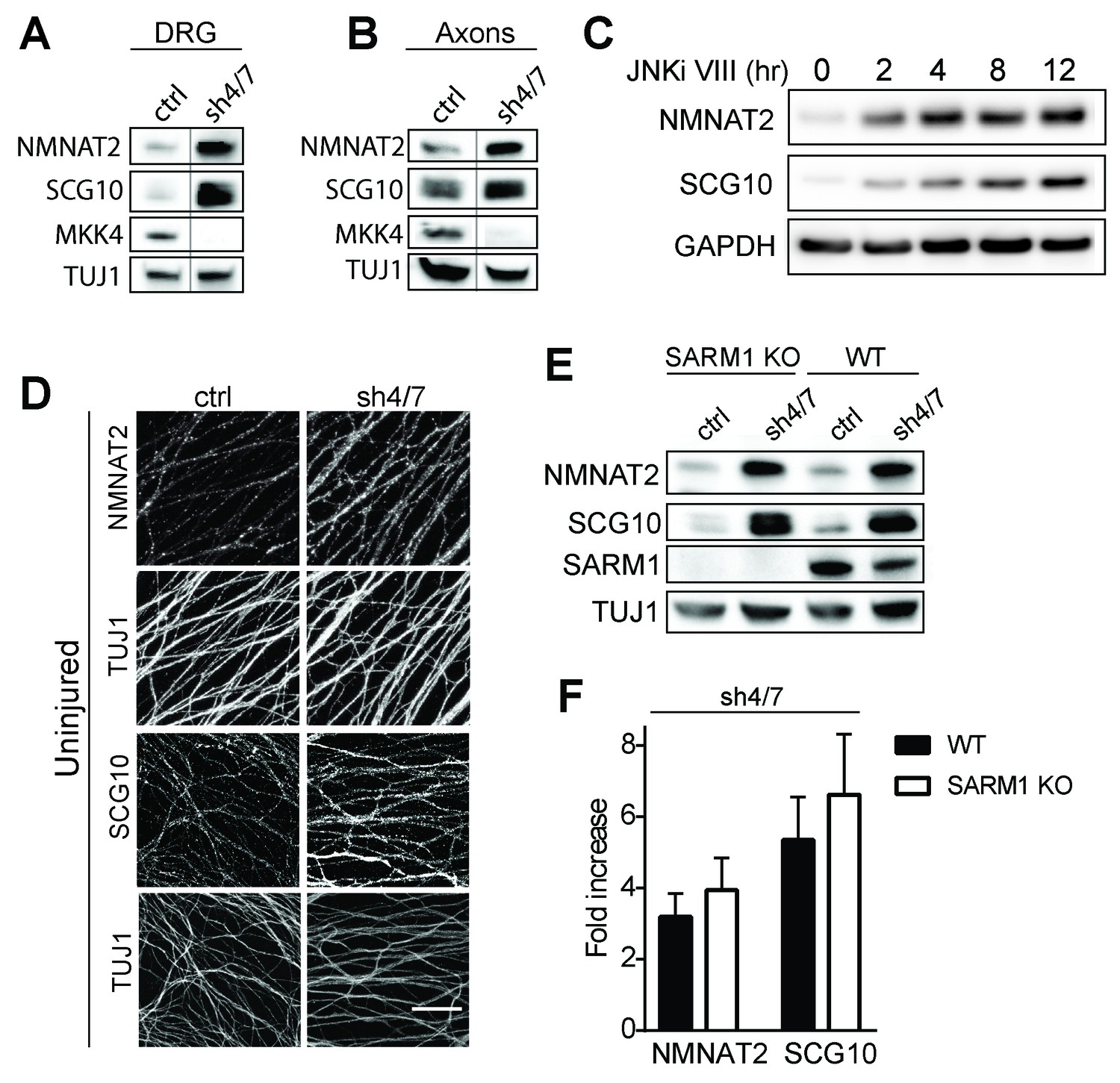
MKK4/7 regulates the levels of axon survival factors NMNAT2 and SCG10.
Loss of MKK4/7 (sh4/7) increases levels of endogenous NMNAT2 and SCG10 within cultured DRG neurons (A) and axons (B) as observed via western blot as compared to shLuciferase (ctrl) controls (n = 3). Vertical line in westerns blots denotes where lanes were removed; however, samples were run on the same gel and images are from the same exposure. We confirm that MKK4/7 functions through the canonical JNK pathway, as timecourse treatment with JNK inhibitor (JNKi) VIII (10 uM, 0–12 hr treatment; n = 4) increases endogenous NMNAT2 and SCG10 within neurons within two hours. (C) MKK4/7 regulate levels of exogenously-expressed NMNAT2-myc and endogenous SCG10 (D) as observed by immunostaining within uninjured DRG axons, indicating the modulation is post-transcriptional. The anti-Tuj1 antibody stains tubulin and labels all axons. Scale bar: 25 µm, n = 4 (E) Western blot from embryonic DRG neurons reveals that knockdown of MKK4/7 leads to an increase in the levels of endogenous NMNAT2 and SCG10 in both wildtype (WT) and SARM1 knockout (SARM1 KO) neurons. (F) Quantification of endogenous NMNAT2 and SCG10 in WT and SARM1 knockout neurons with depletion of MKK4/7 by shRNA. Data were normalized to protein levels with shLuciferase controls (not significant, n = 3). Also refer to Figure 3—figure supplement 1.
Figure 3—figure supplement 1
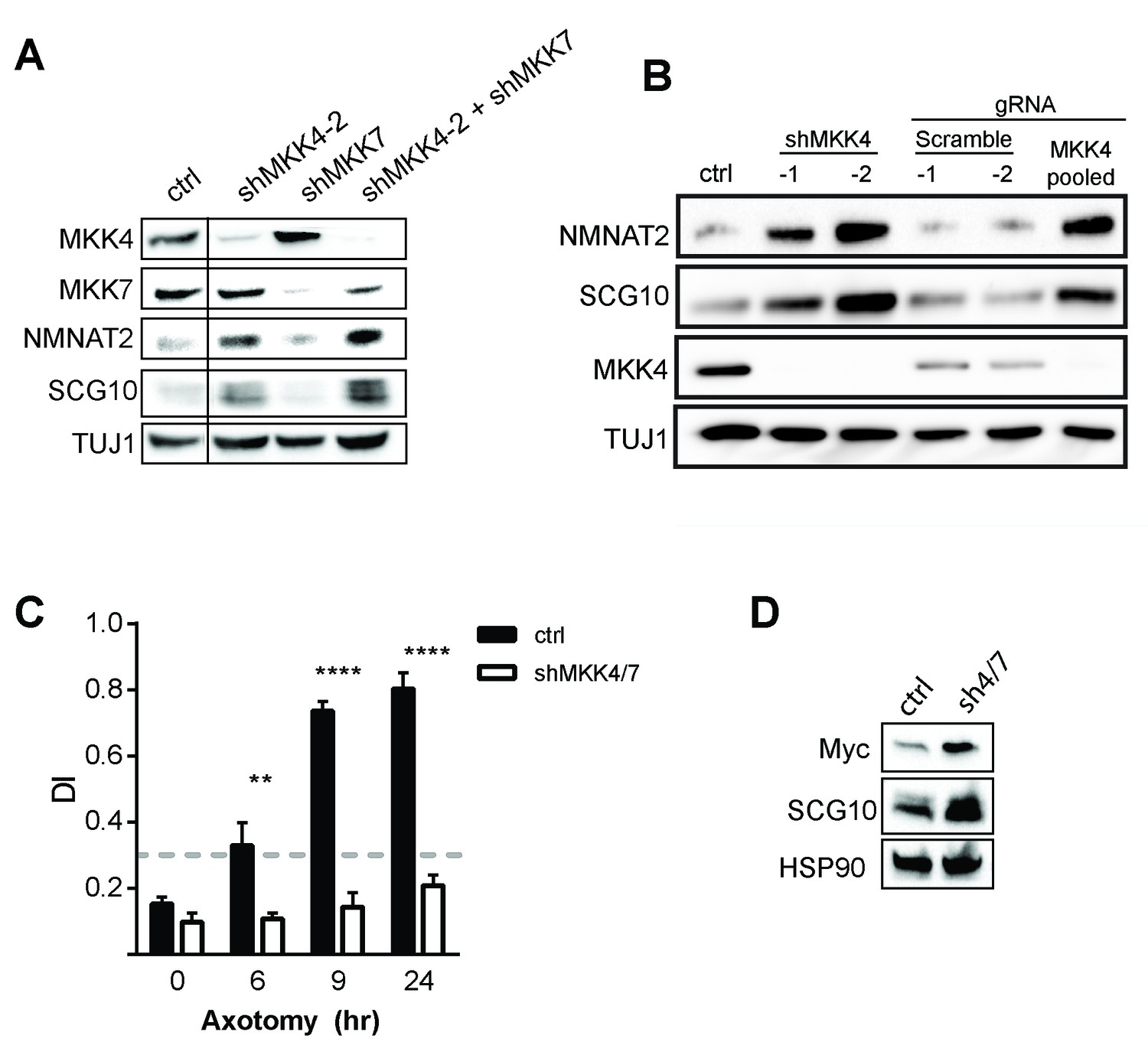
Depletion of MAPKs elevates survival factors and protects axons.
(A) Validation of specificity of shMKK4-2 and shMKK7 used in this study. All samples were run on the same gel. Vertical line indicates lanes cropped out of blot. (B) Western blot from embryonic DRGs derived from a Cas9 knock-in mouse demonstrating that depletion of MKK4 using two independent shRNAs or pooled guide RNAs (gRNA) can deplete MKK4 and modulate expression of NMNAT2 and SCG10. The shRNA used in this study to knockdown MKK4 was shMKK4-2. (C) Depletion of MKK4/7 protects axons 24 hr after injury compared to shLuciferase (ctrl) controls. Values represent the Degeneration Index, with higher DI values representing axon degeneration. Values are presented as mean ± SEM. ****p≤0.0001, **p≤0.01, n = 3–6. (D) Western blot from axon-only lysate demonstrating MKK4/7 regulate exogenously-expressed NMNAT2-myc. The NMNAT2-myc construct is expressed from the ubiquitin C promoter instead of the endogenous NMNAT2 promoter, suggesting that MAPK-dependent regulation of NMNAT2 is post-transcriptional. Samples were blotted with an anti-myc antibody (n = 3).
Figure 4 with 1 supplement
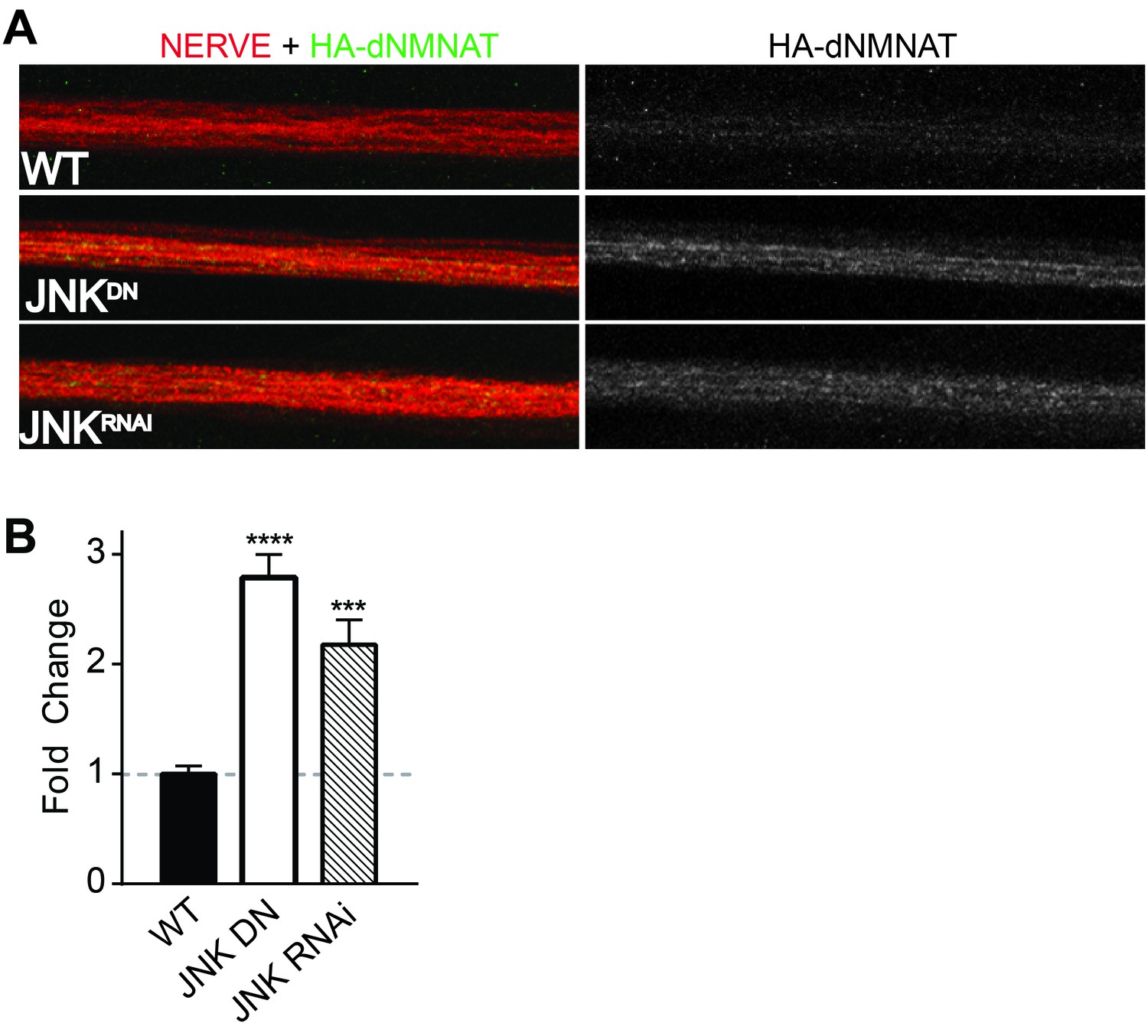
MAPKs modulate dNMNAT in vivo in Drosophila larvae.
Immunostaining in third instar Drosophila larvae of HA-dNMNAT (green) expressed in nerves from the motorneuron driver OK6-Gal4, with HRP labeling (red) to counterstain the nerve. (A) Expression of JNK dominant negative (DN) or depletion of JNK with RNAi (BL57035) increases levels of HA-dNMNAT in the nerve in vivo. (B) Quantification of fluorescence intensity of HA-dNMNAT normalized to wildtype (WT) controls. N = 8–10 animals. p values: ***≤ 0.001, ****≤ 0.0001 by ANOVA. Also refer to Figure 4—figure supplement 1.
Figure 4—figure supplement 1
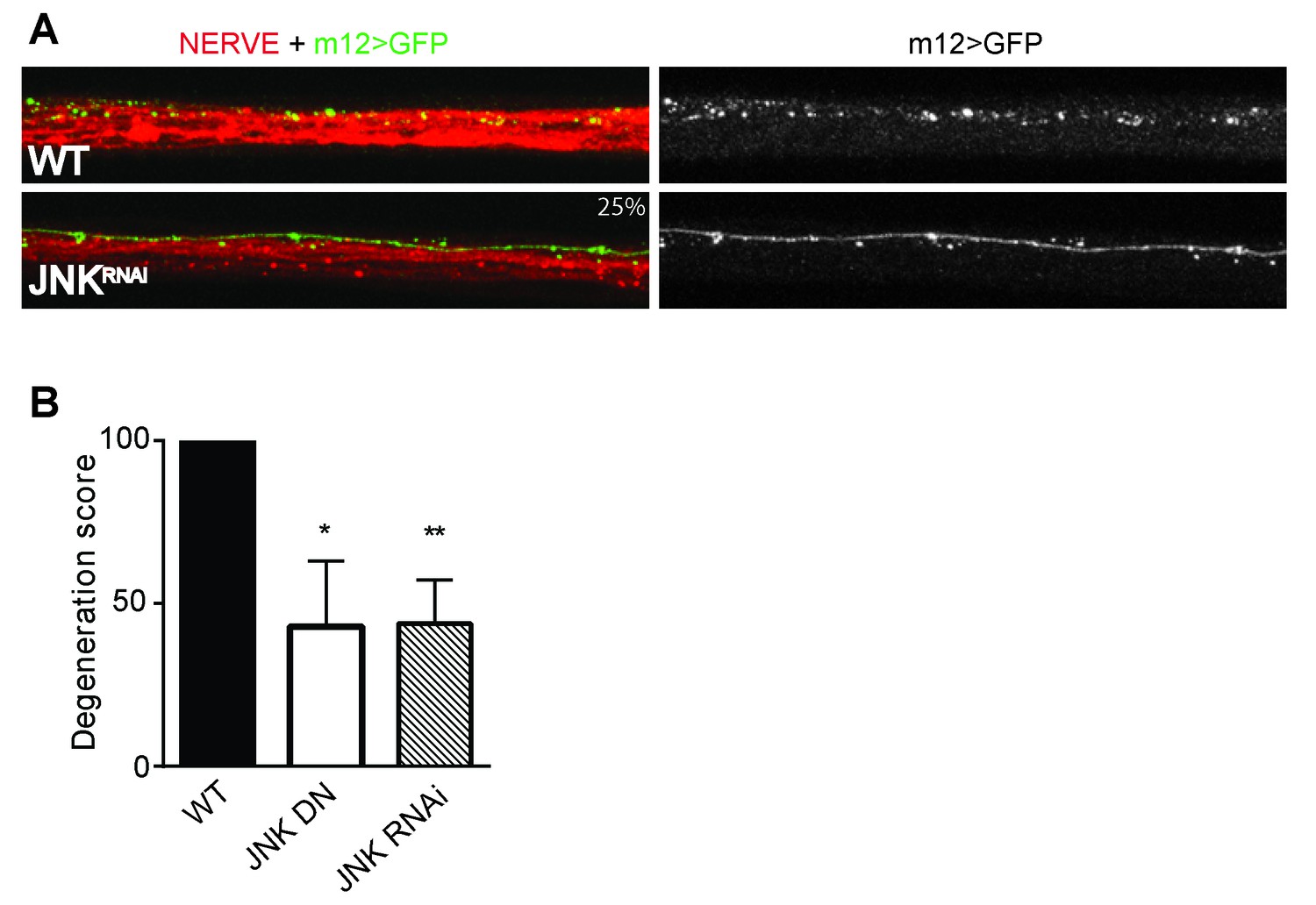
Depletion of JNK with RNAi or expression of JNK dominant negative (DN) protects Drosophila larval axons 24 hr after pinch injury.
(A) Representative images of injured axons. Usually two single axons within the nerve are labeled with GFP driven by m12-GAL4. Total nerve is labeled with HRP. (B) Quantification of axon degeneration, with 100 being complete fragmentation and 0 being an intact axon. Values are presented as mean ± SEM. N = 7–12 nerves. **p≤0.01, *p≤0.05.
Figure 5 with 2 supplements
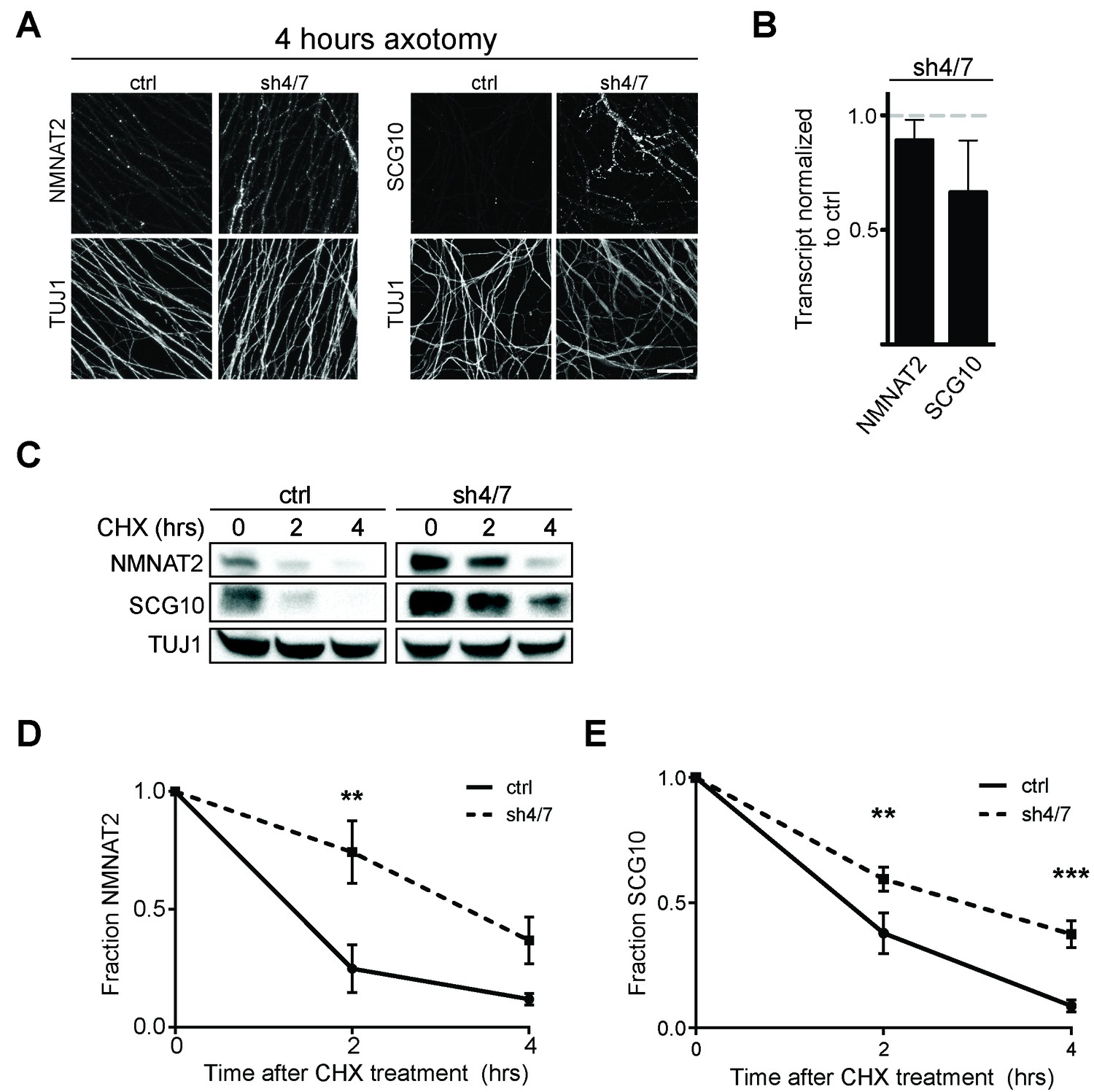
MKK4/7 promote turnover of axon survival factors.
(A) Immunostaining for NMNAT2-myc and SCG10 in distal axons four hours after axotomy. Fluorescence signal is diminished four hours after cut in control axons; however, with depletion of MKK4/7 (sh4/7) levels of NMNAT2-myc and SCG10 are maintained after injury compared to shLuciferase (ctrl) control (p≤0.01 for NMNAT2-myc and p≤0.001 for SCG10, comparing fluorescence between conditions at four hours, n = 4). Scale bar: 25 µm (B) rtPCR reveals no significant change in transcript levels for NMNAT2 or SCG10 after depletion of MKK4/7 when normalized to shLuciferase controls. N = 3, p=0.29. (C) Western blot analysis from axon-only lysate from DRG cultures treated with cycloheximide (CHX) for the indicated timepoints to block protein synthesis. Quantification from CHX timecourse experiments for fraction of NMNAT2 (D) and SCG10 (E) as compared to time zero. The turnover rate of both NMNAT2 and SCG10 is slowed upon MKK4/7 knockdown. N = 4; p values: **≤ 0.01, ***≤ 0.001. Also refer to Figure 5—figure supplements 1 and 2.
Figure 5—figure supplement 1
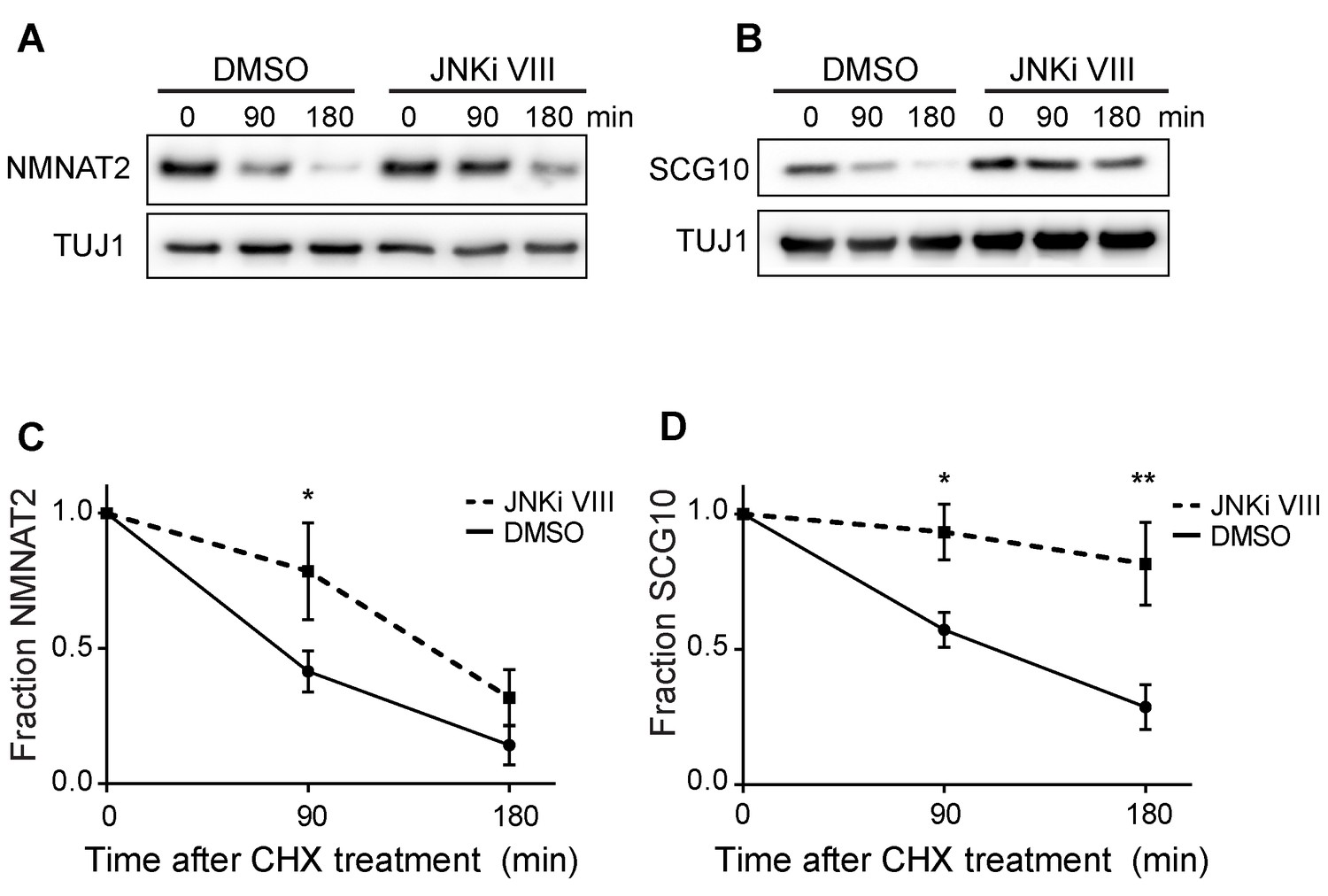
Pre-treatment with JNKi VIII (30 min) delays degradation of NMNAT2 and SCG10 after cycloheximide (CHX).
Western blot analysis from axon-only lysate from DRG cultures treated with CHX for the indicated timepoints to block protein synthesis to show turnover of endogenous NMNAT2 (A) and SCG10 (B). TUJ1 is a loading control. Quantification from CHX timecourse experiments for fraction of NMNAT2 (C) and SCG10 (D) as compared to time zero. The turnover rate of both NMNAT2 and SCG10 is slowed upon treatment with JNKi VIII compared to vehicle (DMSO) controls. N = 4; P values: * ≤ 0.05, ** ≤ 0.01.
Figure 5—figure supplement 2
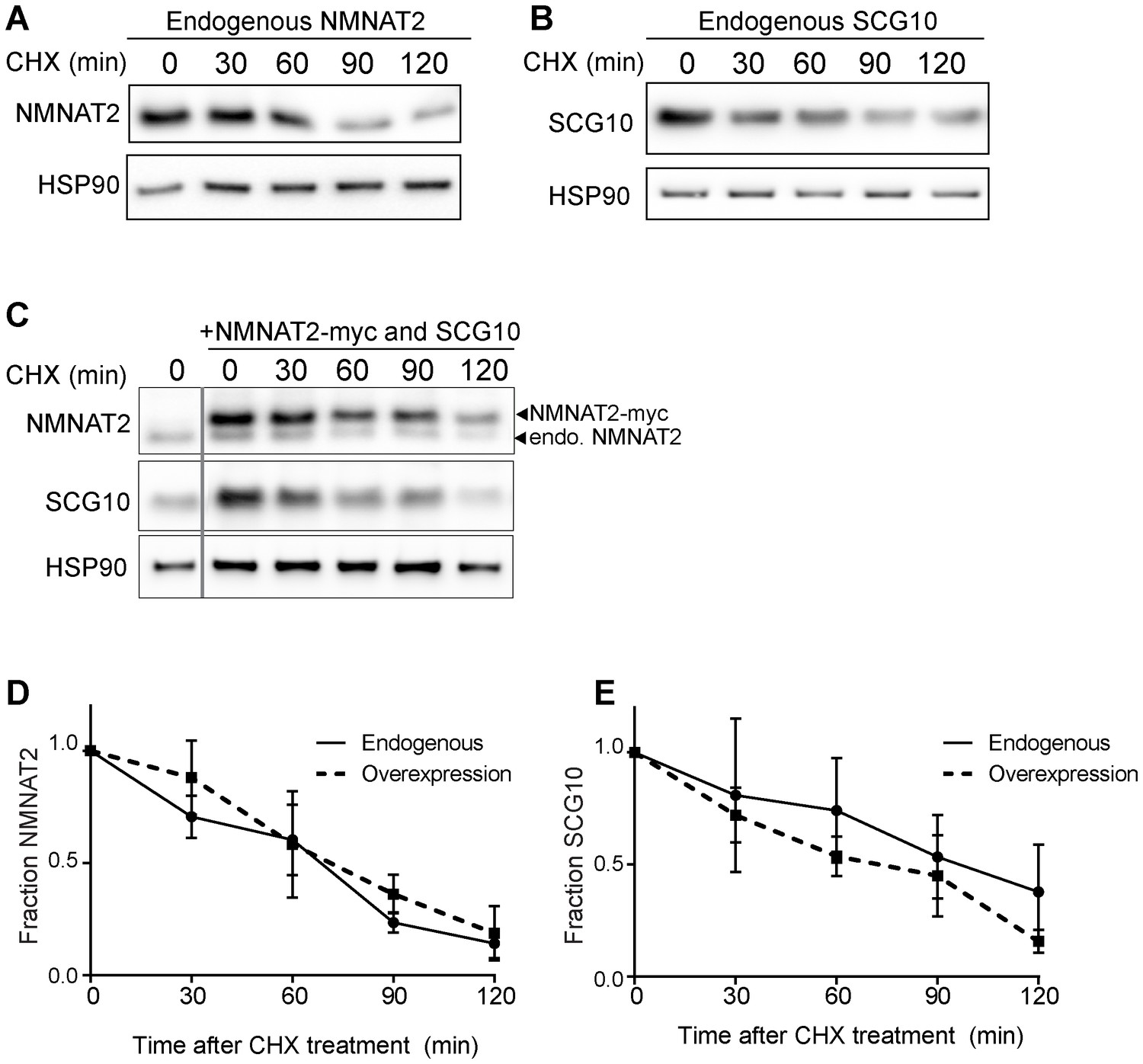
Elevation of NMNAT2 and SCG10 does not delay turnover of survival factors within axons.
Representative western blots from axon-only lysate from DRG cultures treated with cycloheximide (CHX) for the indicated timepoints, showing turnover of endogenous NMNAT2 (A) and endogenous SCG10 (B). (C) Representative western blot from axon-only lysate from DRG cultures that were uninfected (left lane; endogenous) or overexpressing both NMNAT2-myc and wildtype SCG10 and treated with CHX. NMNAT2-myc and SCG10 were expressed at levels 5- to 8-fold higher than wildtype protein. The vertical line denotes where bands were cropped out of the blot, but all samples within (C) were run on the same gel. The bands for NMNAT2-myc and endogenous NMNAT2 are labeled. Quantification from CHX timecourse experiments for fraction of NMNAT2 (D) and SCG10 (E) as compared to time zero. The turnover rate of both NMNAT2 and SCG10 is not statistically different when comparing endogenous survival factors in non-infected cultures to the turnover rate in cultures overexpressing high amounts of NMNAT2-myc and SCG10.
Figure 6 with 1 supplement
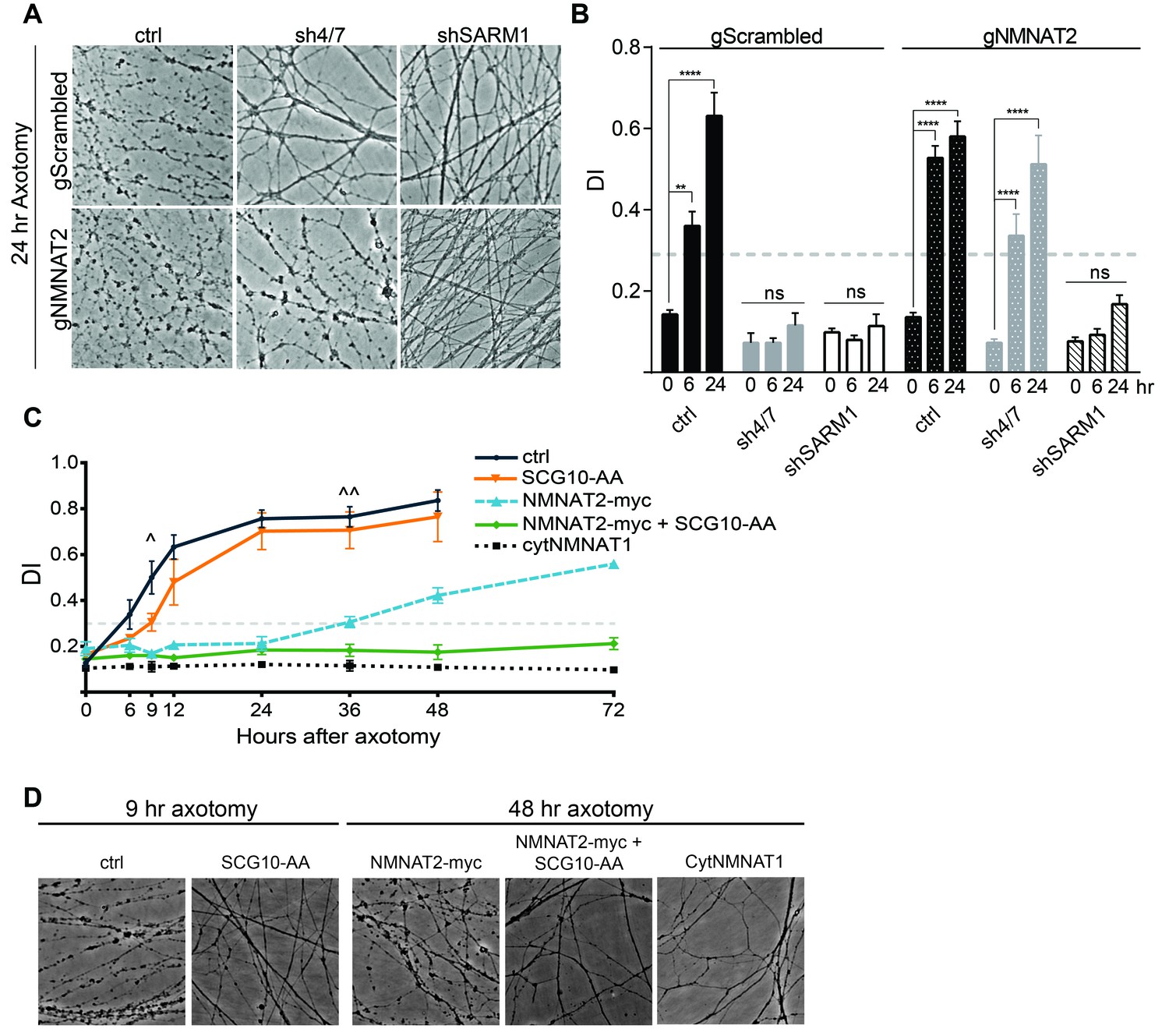
MAPK signaling promotes axonal degeneration via regulation of survival factors.
MKK4/7 requires NMNAT2 to protect axons. Cas9 knock-in DRGs were cultured and treated with scrambled (gScrambled) or NMNAT2 (gNMNAT2) guide RNAs. (A) Knockdown of MKK4/7 (sh4/7) protects axons at 24 hr after axotomy; however, knocking out NMNAT2 using guide RNAs suppresses this protection (p≤0.0001). In contrast, axons from SARM1 -/- DRG neurons do not require NMNAT2 to maintain integrity. (B) Quantification of degeneration index (DI) from images shown in A and in Figure 6—figure supplement 1 plus additional replicates. NMNAT2 guide RNAs revert protection from shMKK4/7 at 6 hr. (C) The protection conferred by overexpression of Scg10-AA (stable, with JNK sites mutated) and NMNAT2-myc is synergistic. Expression of SCG10-AA protects axons for 9 hr (∧), while overexpression of NMNAT2-myc delays degeneration for 36 hr (∧∧). When expressed together, SCG10-AA and NMNAT2-myc protect axons for 72 hr, comparable to protection due to expression of cytoplasmically-localized NMNAT1 (cytNMNAT1). At 9 hr, all conditions are statistically different from non-recombinant vector controls (ctrl; p≤0.01) and axons are protected. After 48 hr, NMNAT2-myc is statistically different from cytNMNAT1 and NMNAT2-myc + SCG10-AA (p≤0.01); however, cytNMNAT1 and NMNAT2-myc + SCG10-AA are essentially equivalent. The horizontal dotted lines indicate the DI value above which axons have begun to degenerate. (D) Representative images from figure C at 9 hr or 48 hr after axotomy, as noted. N = 3–4. Also refer to Figure 6—figure supplement 1.
Figure 6—figure supplement 1
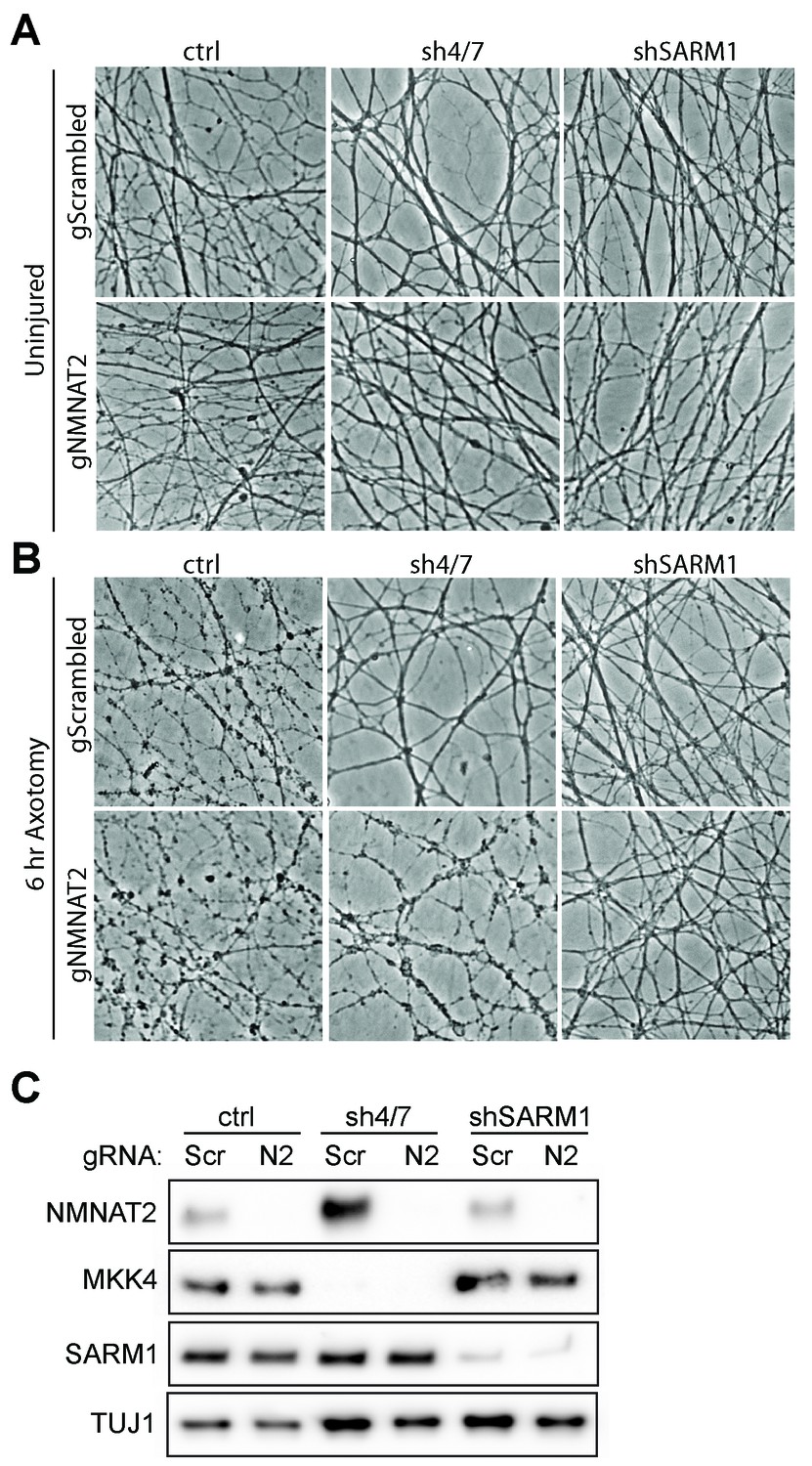
MKK4/7 requires NMNAT2 to protect axons.
Cas9 knock-in DRGs were cultured and treated with scrambled (gScrambled) or Nmnat2 (gNMNAT2) guide RNAs (gRNA). (A) Uninjured Cas9 knock-in DRGs are intact when NMNAT2 is knocked out using guide RNA (gNMNAT2) and when MKK4/7 (sh4/7) or SARM1 (shSARM1) are depleted with shRNAs. (B) Knockdown of MKK4/7 protects axons at 6 hr after axotomy compared to shLuciferase (ctrl) controls; however, knocking out NMNAT2 using guide RNAs suppresses this protection (p≤0.0001). Scrambled gRNAs (gScrambled) have no effect on sh4/7 axon protection. In contrast, depletion of SARM1 with shRNAs (shSARM1) in DRG neurons does not require NMNAT2 to maintain integrity. Quantification of degeneration is shown in Figure 6 . (C) Representative western blot demonstrating efficacy of shRNAs and guide RNAs (Scr is scrambled gRNA, N2 is Nmnat2 gRNA). TUJ1 is a loading control.
Figure 7
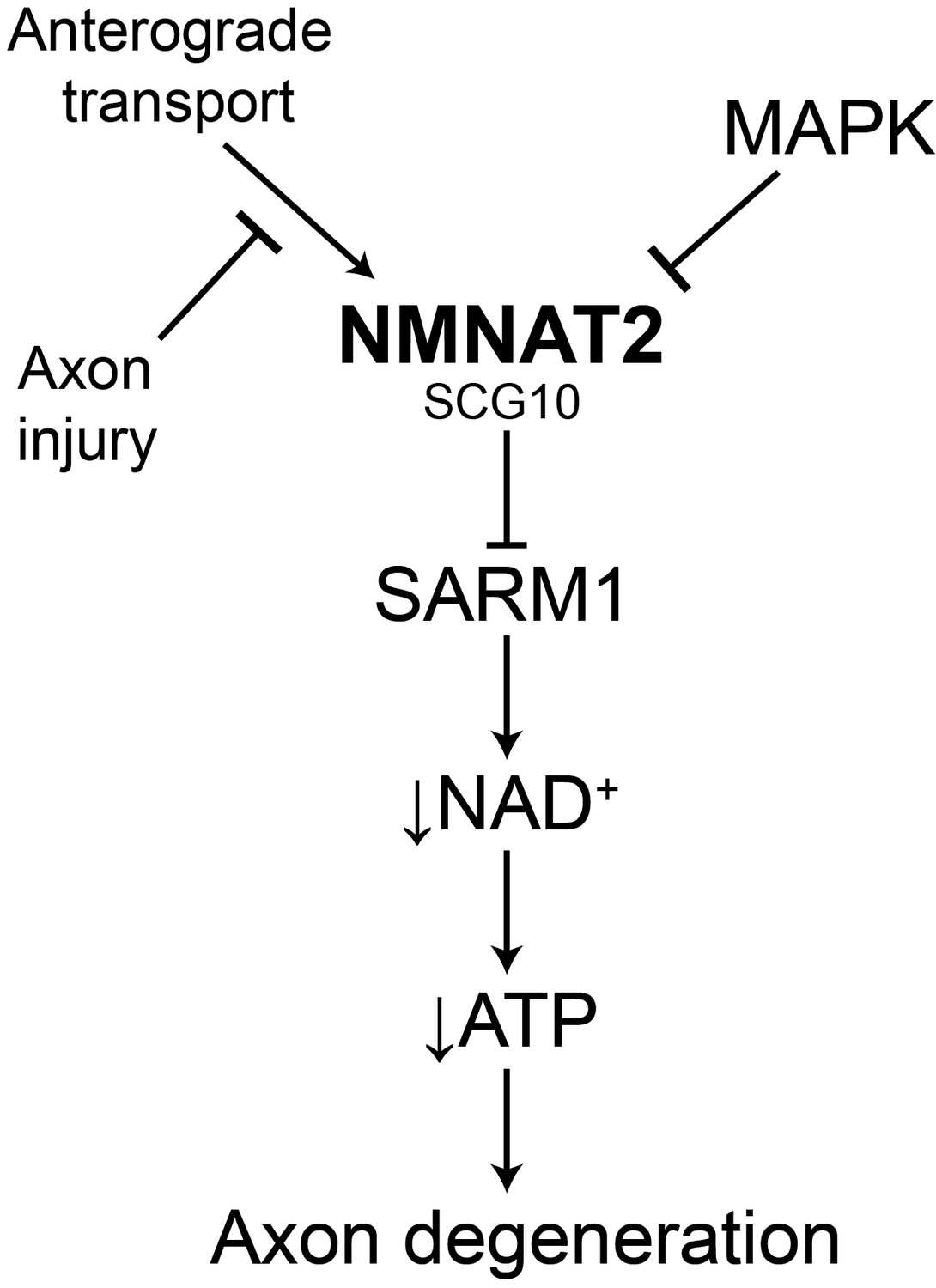
Model of the axonal degeneration program.
In this model, MAPK signaling limits the levels of NMNAT2 and SCG10. NMNAT2 blocks activation of SARM1, and SCG10 may facilitate NMNAT2 function. Upon axon injury, delivery of the labile survival factors via anterograde transport is blocked. The continued degradation of survival factors allows levels of survival factors to drop below a critical threshold, thereby activating SARM1. Activated SARM1 rapidly depletes NAD+, leading to depletion of ATP and induction of a metabolic crisis that drives axon fragmentation. Our studies identify a critical function for MAPK signaling upstream of endogenous SARM1, however forced dimerization of SARM1-TIR can activate JNK, and so there could be additional roles for MAPK signaling downstream of SARM1.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2
eLife 6:e22540.
https://doi.org/10.7554/eLife.22540




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}