CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin
- Max Planck Institute of Biophysics, Germany
Figures
Figure 1 with 3 supplements
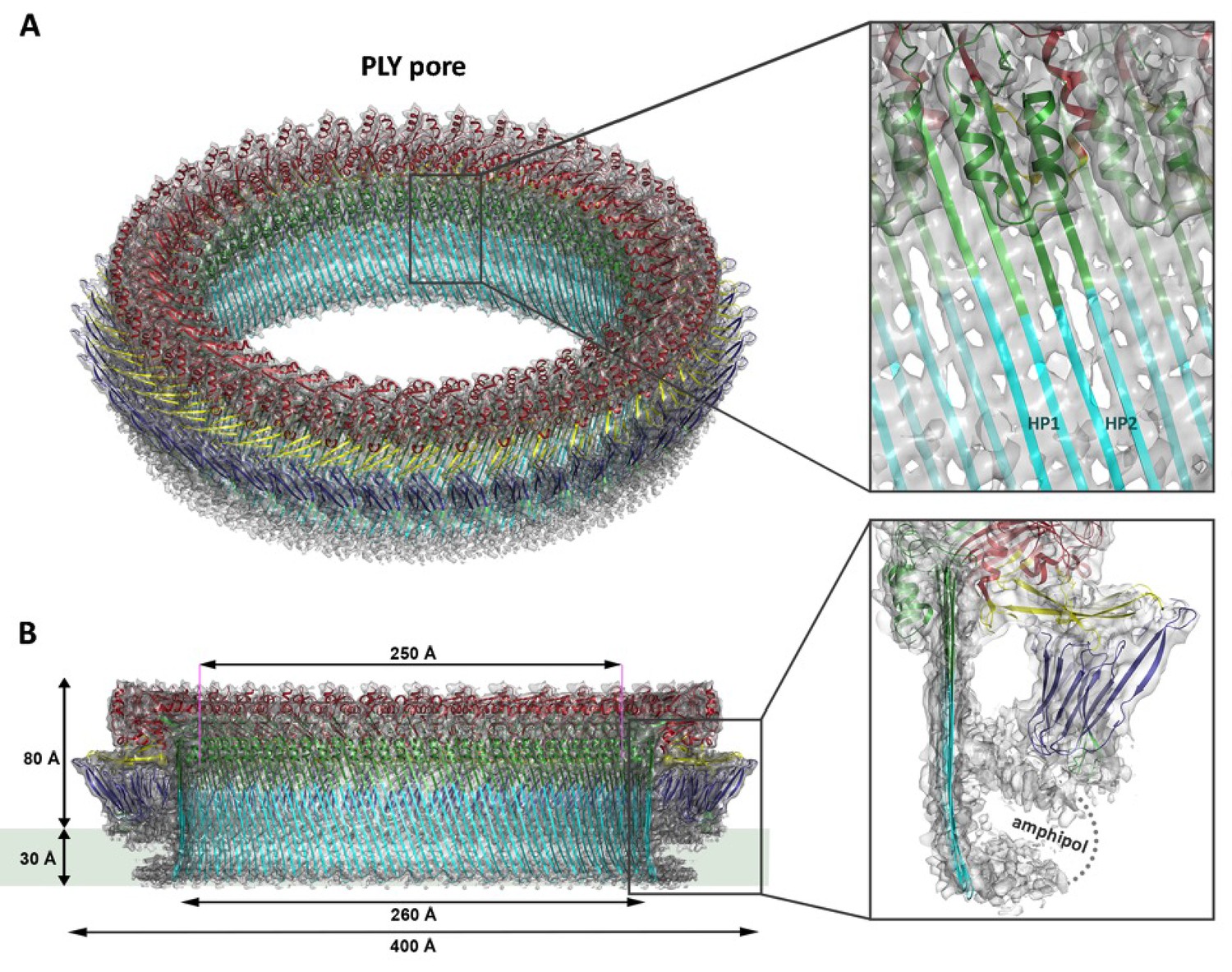
Overall structure of the PLY pore complex.
(A) Single-particle cryoEM map of PLY at 4.5 Å resolution with fitted model. The four PLY domains (D1–D4) are red (D1), yellow (D2), green/cyan (D3) and blue (D4). Inset: refolded β-hairpins (HP1 and HP2) fitted to the map. Cyan β-strands have refolded from helix bundles in D3 of the soluble form. (B) Cross-section with overall dimensions of the pore complex. The grey bar indicates the position of the lipid bilayer. Inset: side view of membrane-inserted monomer with toroid density of disordered amphipol (broken line).
Figure 1—figure supplement 1

Negative stain and cryoEM of PLY solubilized in DDM, Cymal-6 and Amphipol.
PLY rings solubilized with DDM vary in size and tend to orient on the specimen support or the air/water interface. Rings solubilized with Cymal-6 are more homogenous in size but tend to stack in pairs. Exchange of detergent against Amphipol A8-35 results in uniform, randomly oriented, single PLY rings. Scale bar: 50 nm.
Figure 1—figure supplement 2
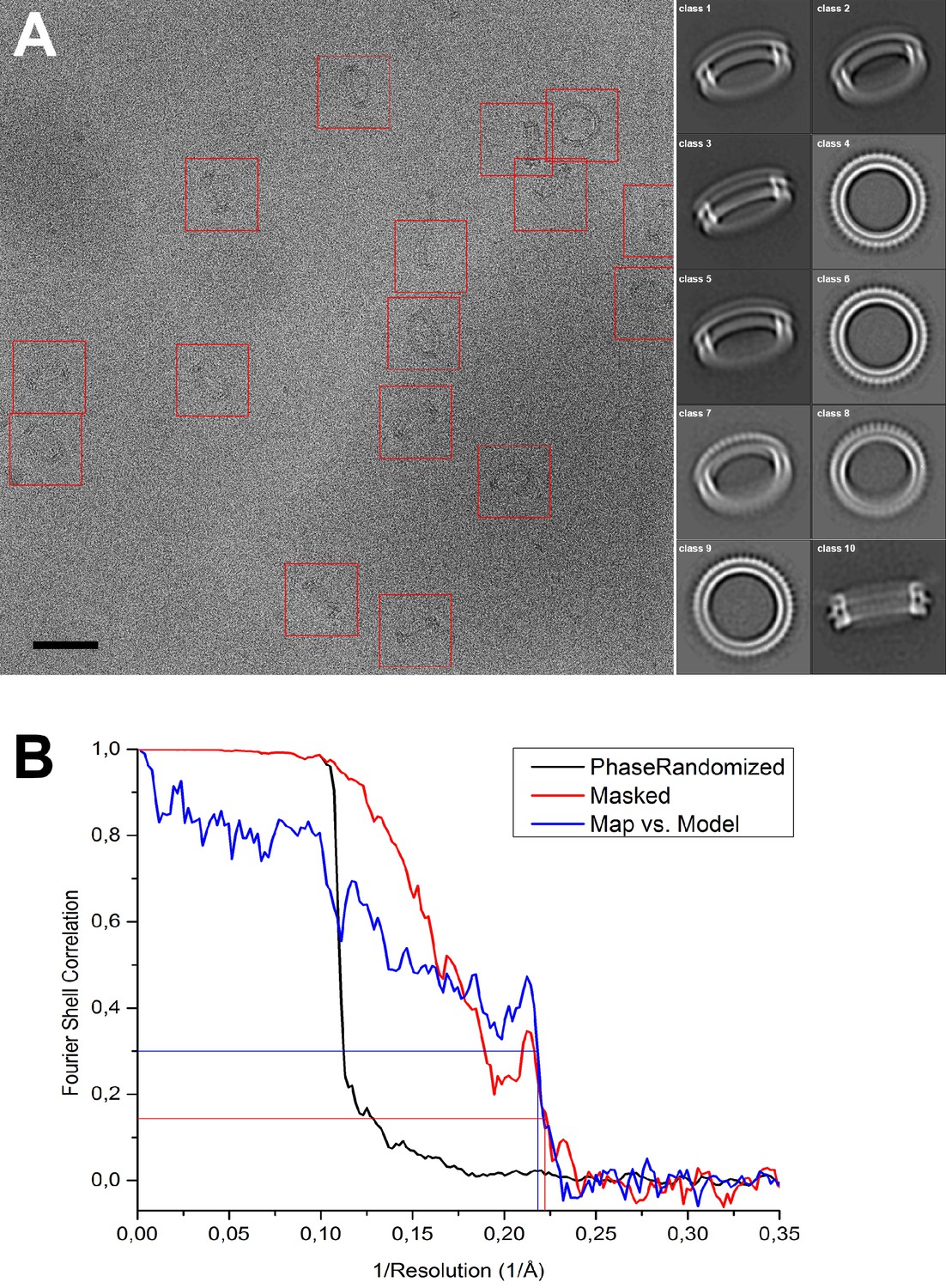
Image processing of PLY rings.
(A) Sample micrograph of amphipol-solubilized PLY rings picked for single-particle processing (red boxes). Scale bar: 50 nm. Class averages after 2D classification are shown on the right. (B) FSC curve for phase-randomized, masked map (red curve) indicates 4.5 Å resolution by the FSC0.143 criterion (red line). The sharp peak at 4.8 Å, reflecting the repeat distance between the 168 β-strands in the β-barrel, and a steep drop beyond that, indicating a resolution of 4.5 Å at the 0.143 theshold for the masked FSC, and of 4.6 Å at the 0.3 threshold for the map-vs-model FSC. The two values are in excellent agreement.
Figure 1—figure supplement 3

Local resolution estimate of the PLY monomers and of the complete pore complex.
(A) Local resolution map of one PLY monomer from the final density map of the pore complex. The monomer is rotated in 90° steps around the y-axis. (B) Local resolution map of the PLY pore complex.
Figure 2 with 5 supplements
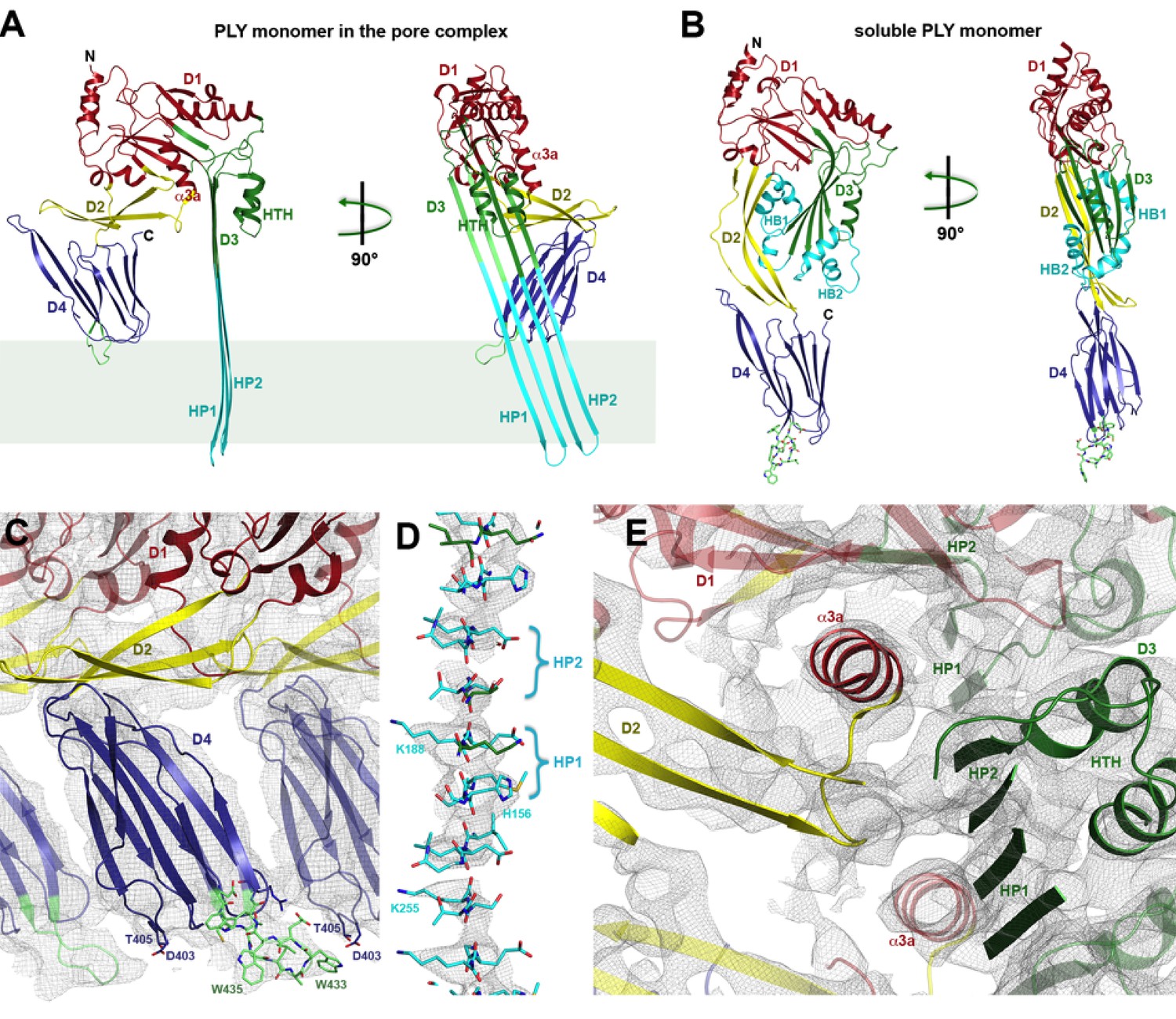
Soluble and membrane-inserted PLY monomer.
One subunit of membrane-inserted form of PLY in the cryoEM structure (A) and x-ray structure of soluble PLY (van Pee et al., 2016) (B) seen from the side (left) and from the pore center (right). Both helix bundles (HB1 and HB2, cyan) in the PLY monomer refold to form two long, membrane-spanning β-hairpins (HP1 and HP2). The upper end of the β-hairpins is sandwiched between a helix-turn-helix motif (HTH, green) on the inside and helix α3a (red) of the next monomer on the outside of the pore. One of the helices is a refolded β-strand of D3 (green). Helix α3a is the refolded linker that connects D2 to D1 in the soluble monomer. (C) D4 contains the conserved undecapetide (green sticks) that confers cholesterol specificity to PLY. The peptide includes three resolved Trp sidechains, one of which interacts with Thr405 in the adjacent monomer. (D) Cross section through a segment of the 168-strand β−barrel with resolved bulky sidechains. The two β-hairpins of one monomer are labelled. (E) Helix α3a interacts with HP1 and D1 of the neighbouring monomer, stabilizing the pore complex.
Figure 2—figure supplement 1
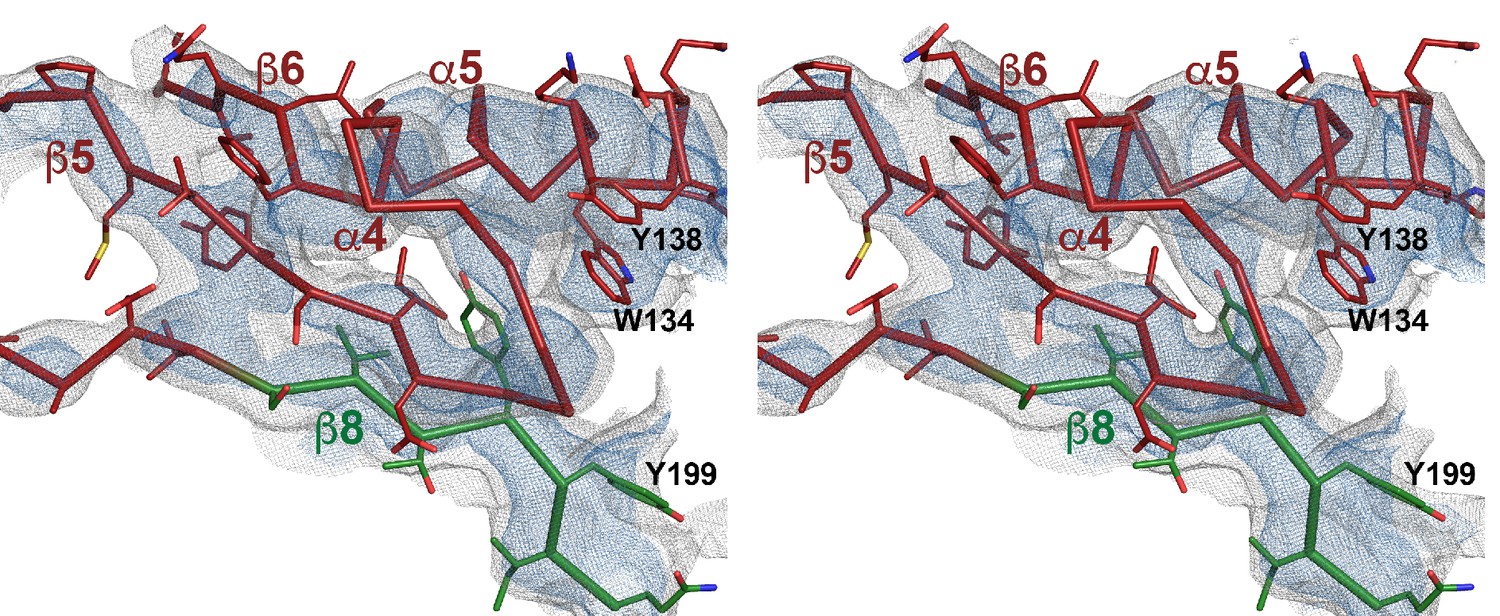
Stereo view of domain 1 presented as ribbon-and-stick model.
Selected large sidechains are shown as sticks. The map is contoured at 7.0 and 9.0 σ.
Figure 2—figure supplement 2
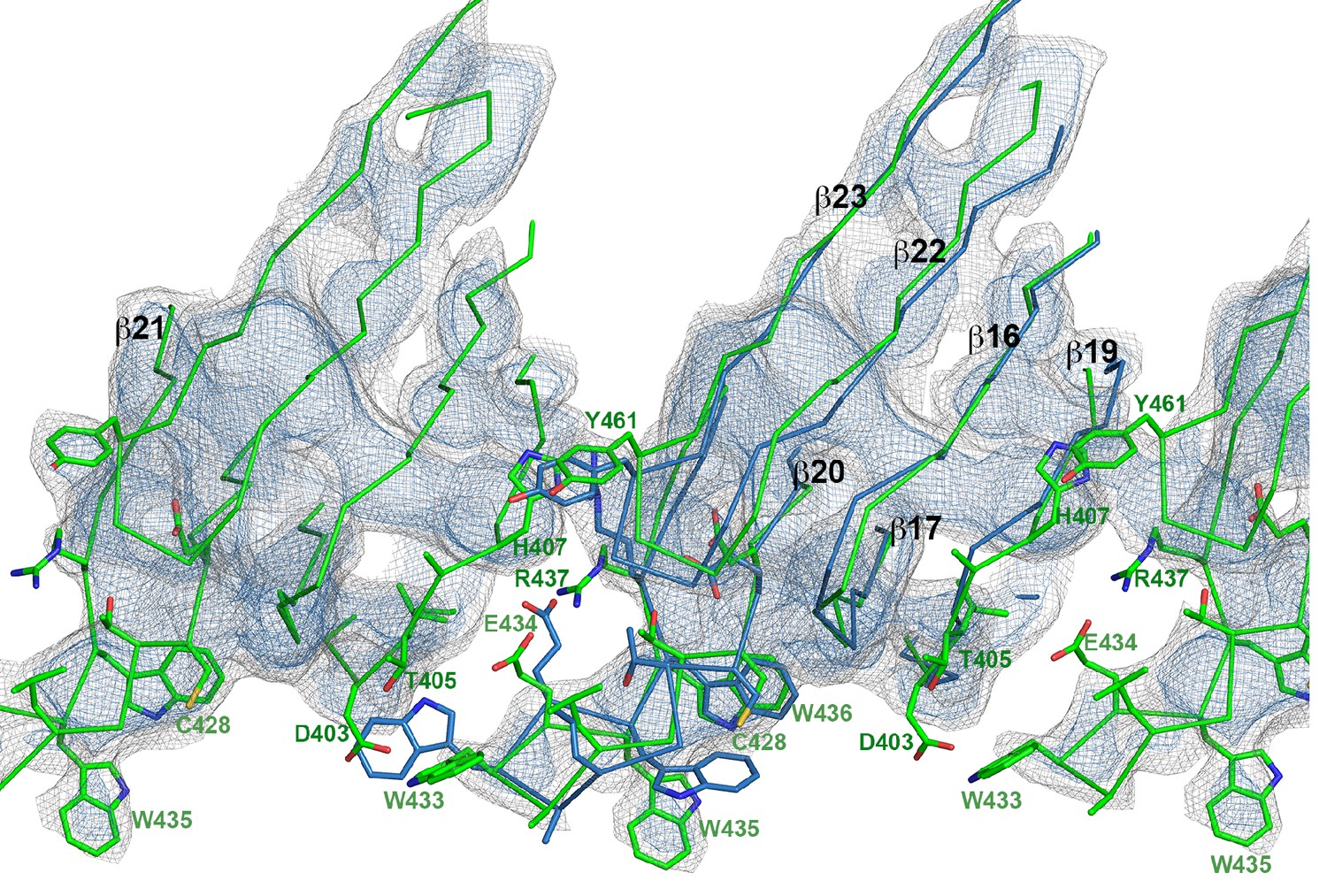
Superposition of domain 4 of the PLY monomer in the crystal structure (blue) and in the pore complex (green), viewed from the pore center.
Residues of the undecapeptide and selected residues in other loops are shown as sticks. The map is contoured at 6.5 and 7.5 σ.
Figure 2—figure supplement 3
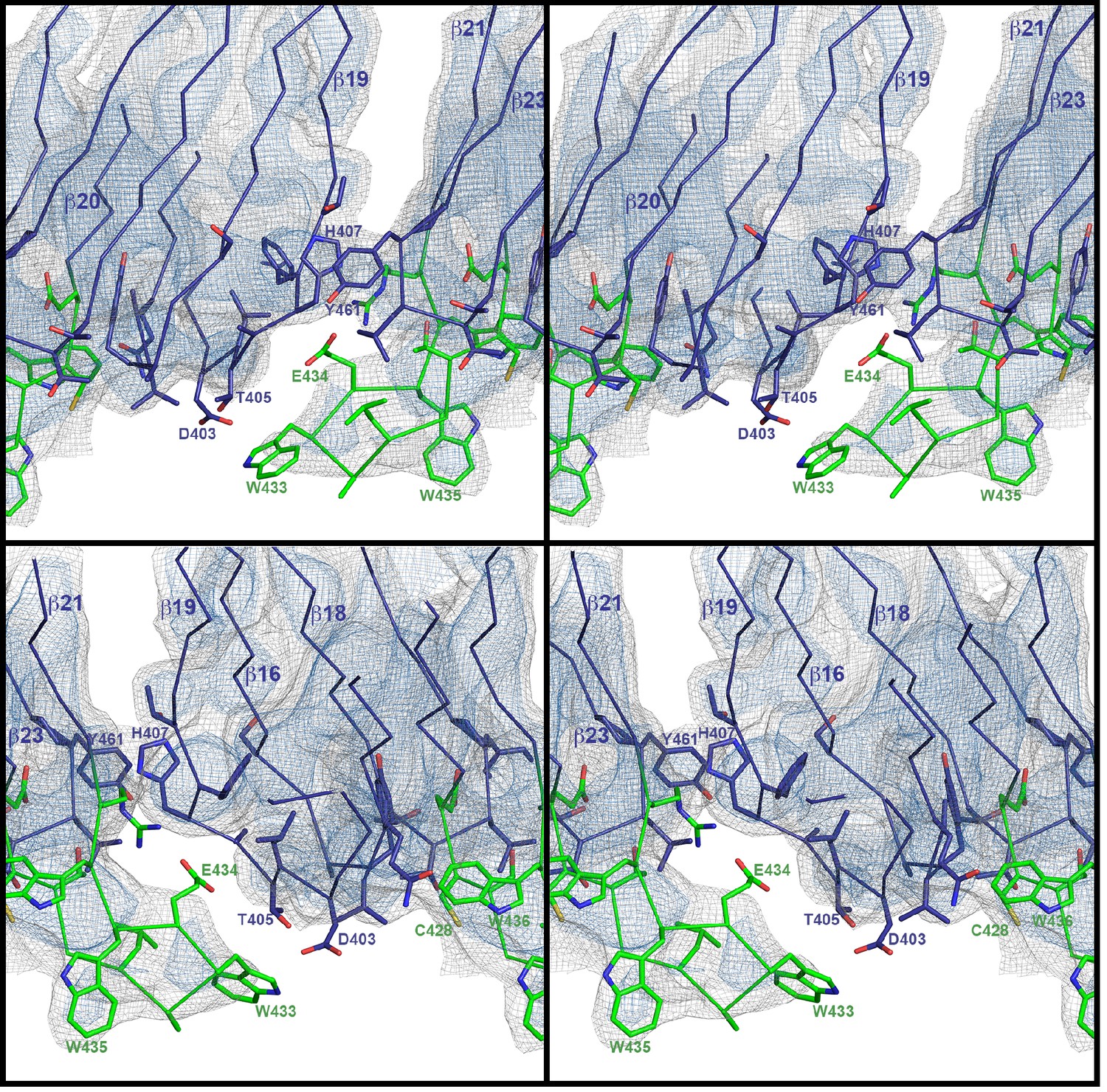
Stereo view of domain 4 in the pore complex with potential inter- and intramolecular interactions of loops viewed from two sides.
The undecapeptide is green and selected residues are shown as sticks. The closest distance between the loops from different monomers is 4–5 Å. The map is contoured at 6.0 and 7.5 σ.
Figure 2—figure supplement 4
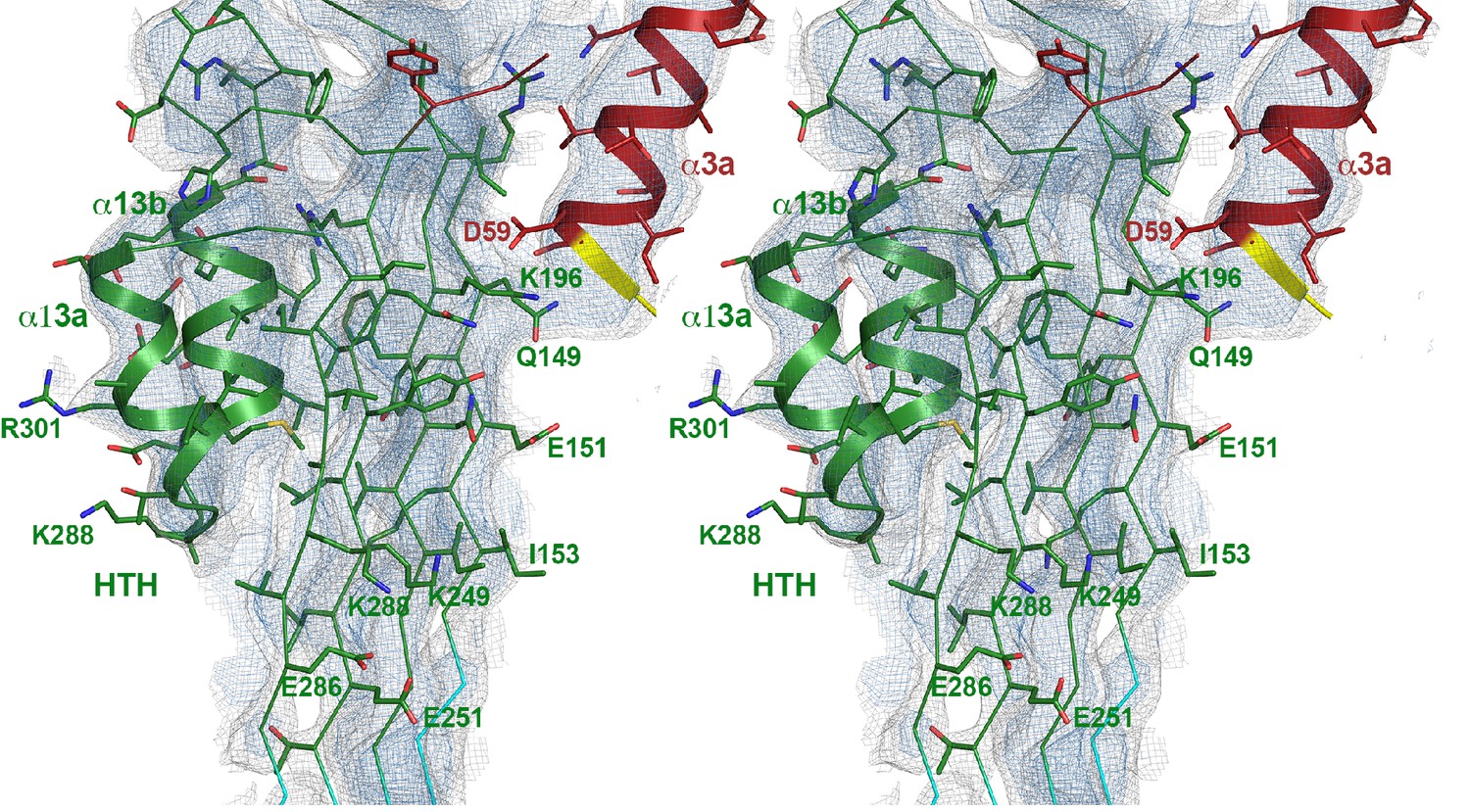
Stereo view of domain 3 with the upper part of the β-barrel and the new helix-turn-helix motif (HTH) that forms the α-barrel inside the pore.
The new helix α3a (red) from the neighboring monomer is on the right. Hydrophobic residues at the interface between the β-sheet and HTH and intermolecular interactions to the next helix α3a are shown as stick models. The map is contoured at 6.5 and 8 σ.
Figure 2—figure supplement 5

Stereo view of domain 2 seen along the ring axis.
The main chain is presented as cartoon and the side chains as sticks. The map is contoured at 7.0 and 9.0 σ.
Figure 3
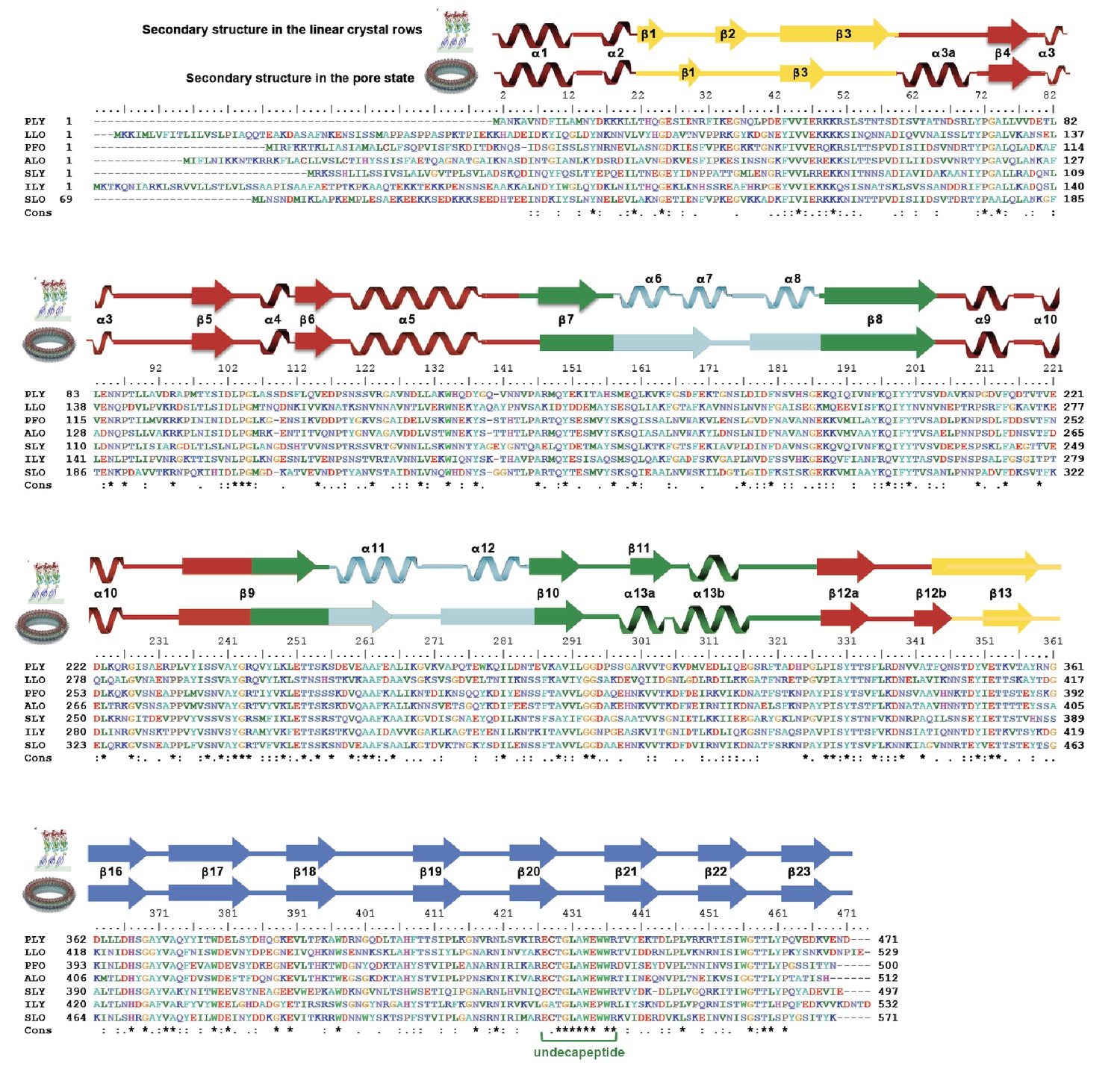
Sequence alignment of selected cholesterol-dependent cytolysins.
Elements of secondary structure are shown for the crystal structure of soluble PLY (above) and for the cyroEM structure of the pore complex (below). Colors of the secondary structure elements and residue numbers correspond to the PLY crystal structure (Figure 2B; pdb code 5aod). Asterisks indicate conserved residues.
Figure 4

Intermolecular interactions in the 4.5 Å cryoEM map of the PLY pore complex.
Slices parallel (A–C) and perpendicular (D–F) to the membrane reveal interactions of secondary structure elements between adjacent monomers in the pore complex. Section planes are indicated on top. Note that the scale of sections D to F is 50% larger to show molecular detail more clearly. The β5-α4-β6 region in domain D1 and the long membrane-parallel helix α5 (red box in A and D) alternate around the top of the pore complex. On the outside, the 168-strand β-barrel is flanked by helix α3a (orange box in B and D) and by the α-barrel of the helix-turn-helix motifs (green box in B and F) on the inside. The 4-strand β-sheets of domain D4 (blue box in C and E) are offset against D4 of the next monomer, forming a ring of 8-strand β-sheets. The 85 Å-long β-strands forming the 168-strand β-barrel are clearly resolved (cyan box in C and E).
Figure 5
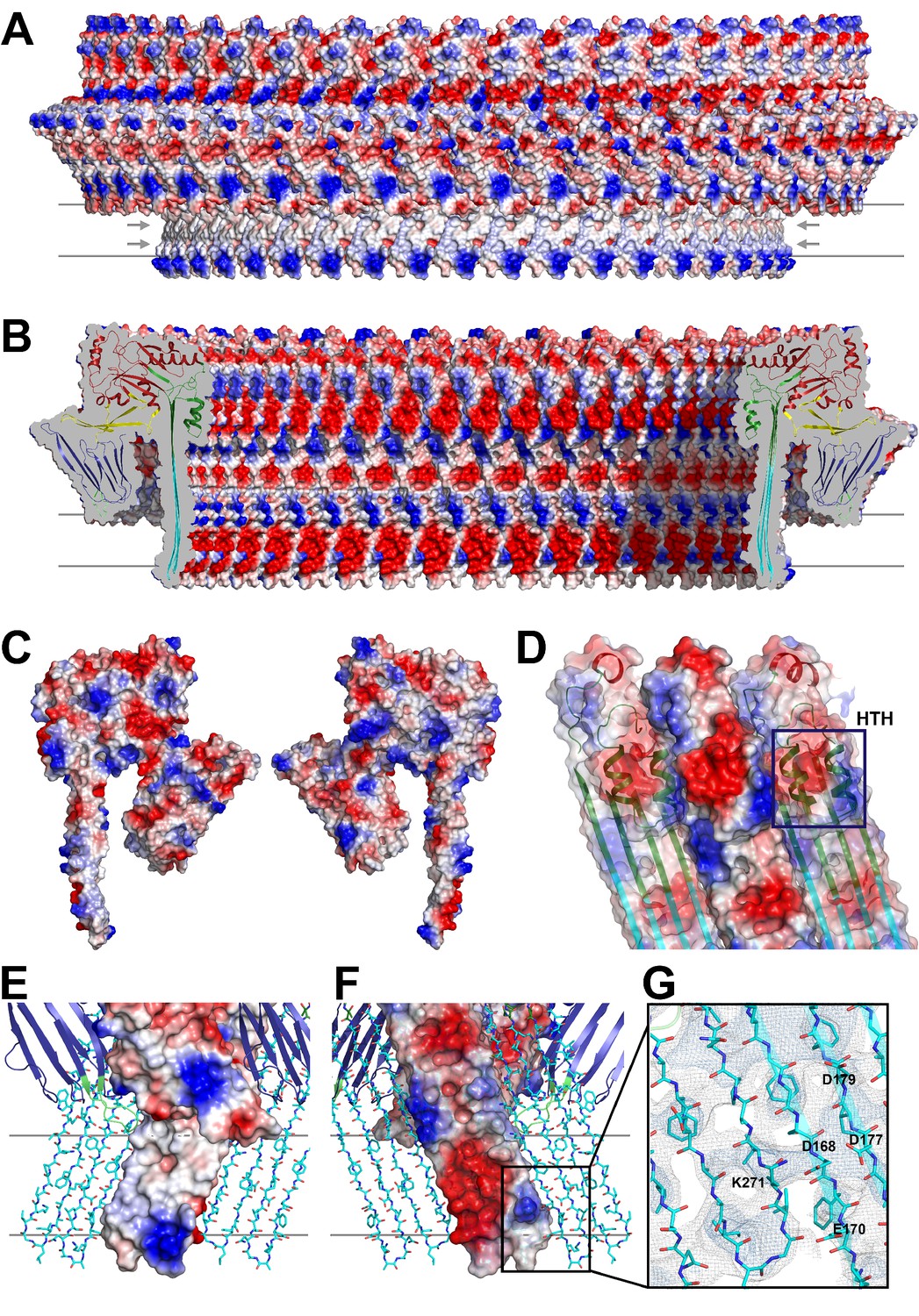
Charge distribution in the PLY pore complex.
(A) Positive (blue) and negative charges (red) are evenly distributed on the polar outer surface of the pore complex. The membrane-inserted region is marked by a band of neutral hydrophobic residues (arrows). (B) The inner surface of the pore complex is highly charged. (C) Contact surfaces of adjacent PLY monomers in the pore complex. (D) Charge complementarity of the two helices in the helix-turn-helix motif forming the internal α-barrel. Positive charges are shown in blue and negative charges in red. (E) The membrane-inserted region of the β-barrel is hydrophobic on the outside and negatively charged on the inside (F). The inset (G) shows map density for the salt bridge between Asp168 of one PLY monomer with Lys271 in the next monomer along the ring. Glutamates and aspartates forming the negatively charged patches on the inner surface of the β-barrel are drawn as sticks. The map is contoured at 5.0 and 6.0 σ.
Figure 6 with 1 supplement
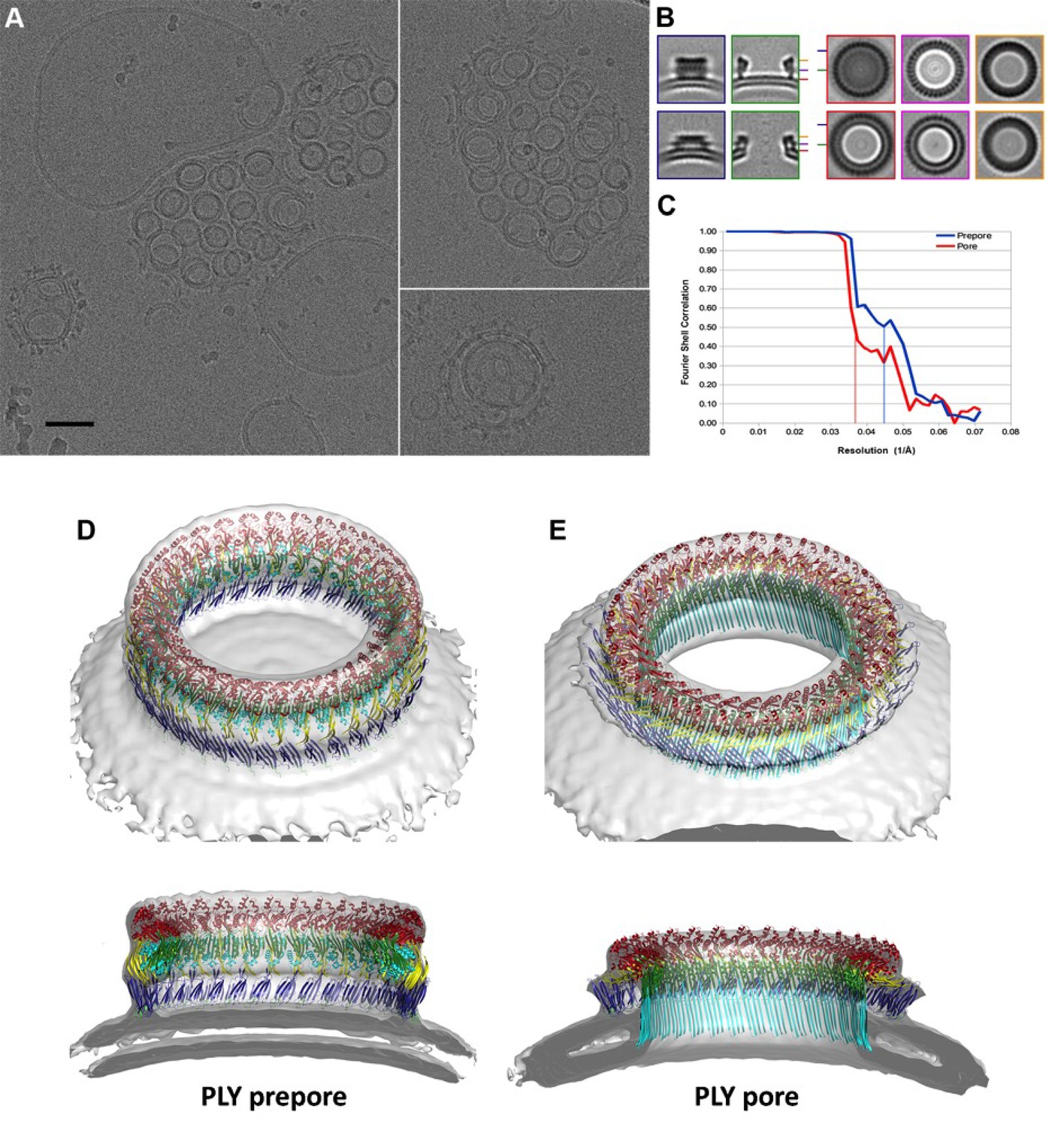
CryoET of PLY prepores and pores.
(A) PLY assembles into prepores and pores upon incubation with cholesterol-containing liposomes. Scale bar, 50 nm. (B) Sections through subtomogram average volumes of the prepore (top) and pore complex (below). Left panel: sections perpendicular to the membrane; right panel: sections parallel to the membrane. Colors indicate section planes. (C) Fourier shell correlation for subtomogram averages indicate 22 Å resolution for the prepore and 27 Å for the pore at FSC0.5 or 20 Å and 21 Å resolution at at FSC0.3. Oblique view (D) and cross section (E) of PLY prepore (left) and pore (right). Both maps accommodate 34 PLY monomers. The prepore map was fitted with the crystal structure of the soluble PLY monomer (Marshall et al., 2015), and the pore map with the cryoEM structure of the pore monomer (Figure 2A). PLY domains are red (D1), yellow (D2), green/cyan (D3) and blue (D4). The lipid bilayer is continuous in the prepore complex, but absent in the pore complex.
Figure 6—figure supplement 1
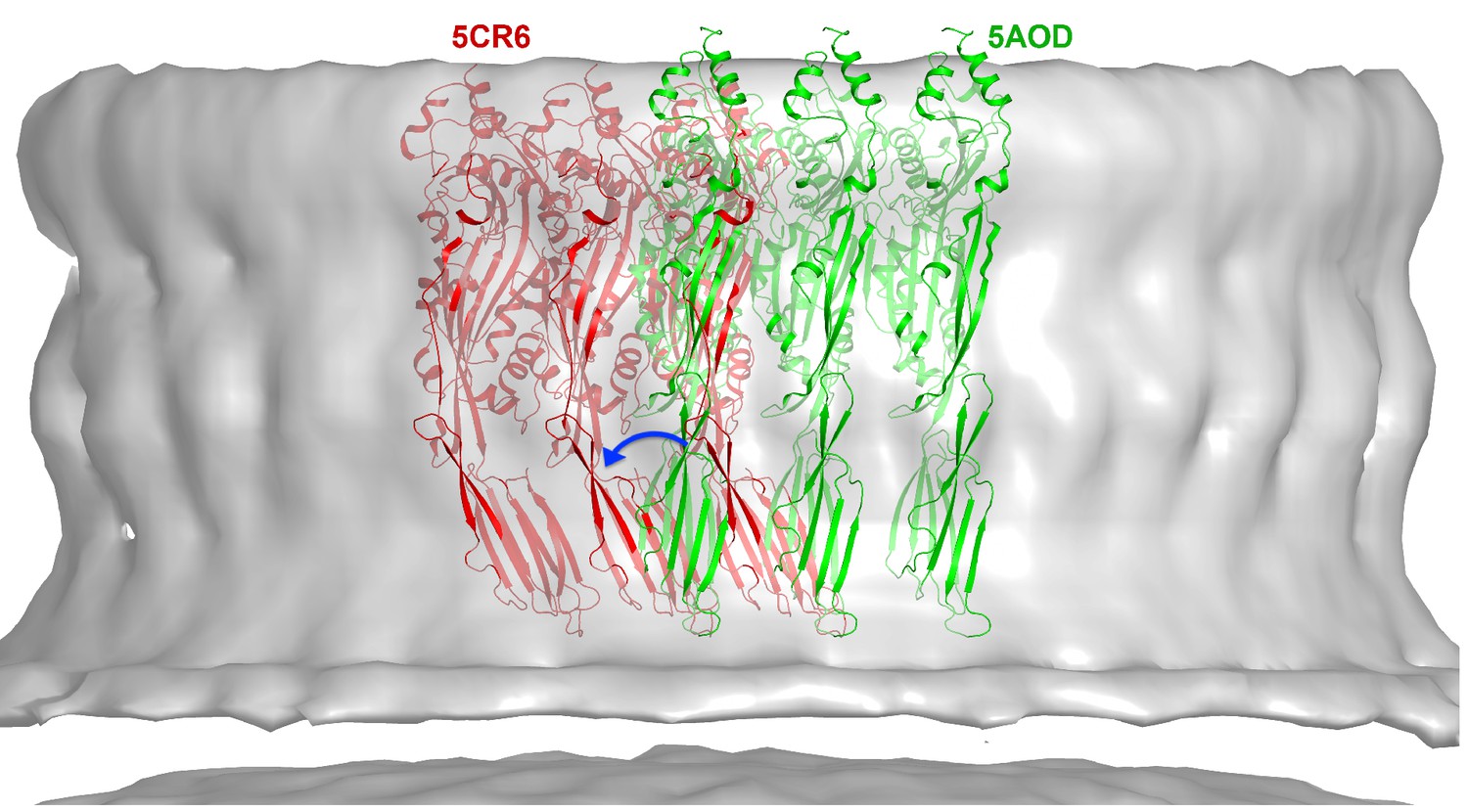
CryoET map of the PLY prepore with rigid-body fitted x-ray structures of the water-soluble toxin that forms rows in the 3D crystals.
PLY monomers of 5AOD (green) are straight and protrude by 10 Å at the top of the map. Monomers of 5CR6 (red) are bent at the hinge between domain 2 and 4 (blue arrow) and match the map volume closely, indicating that this structure represents the pre-pore form of PLY.
Figure 7
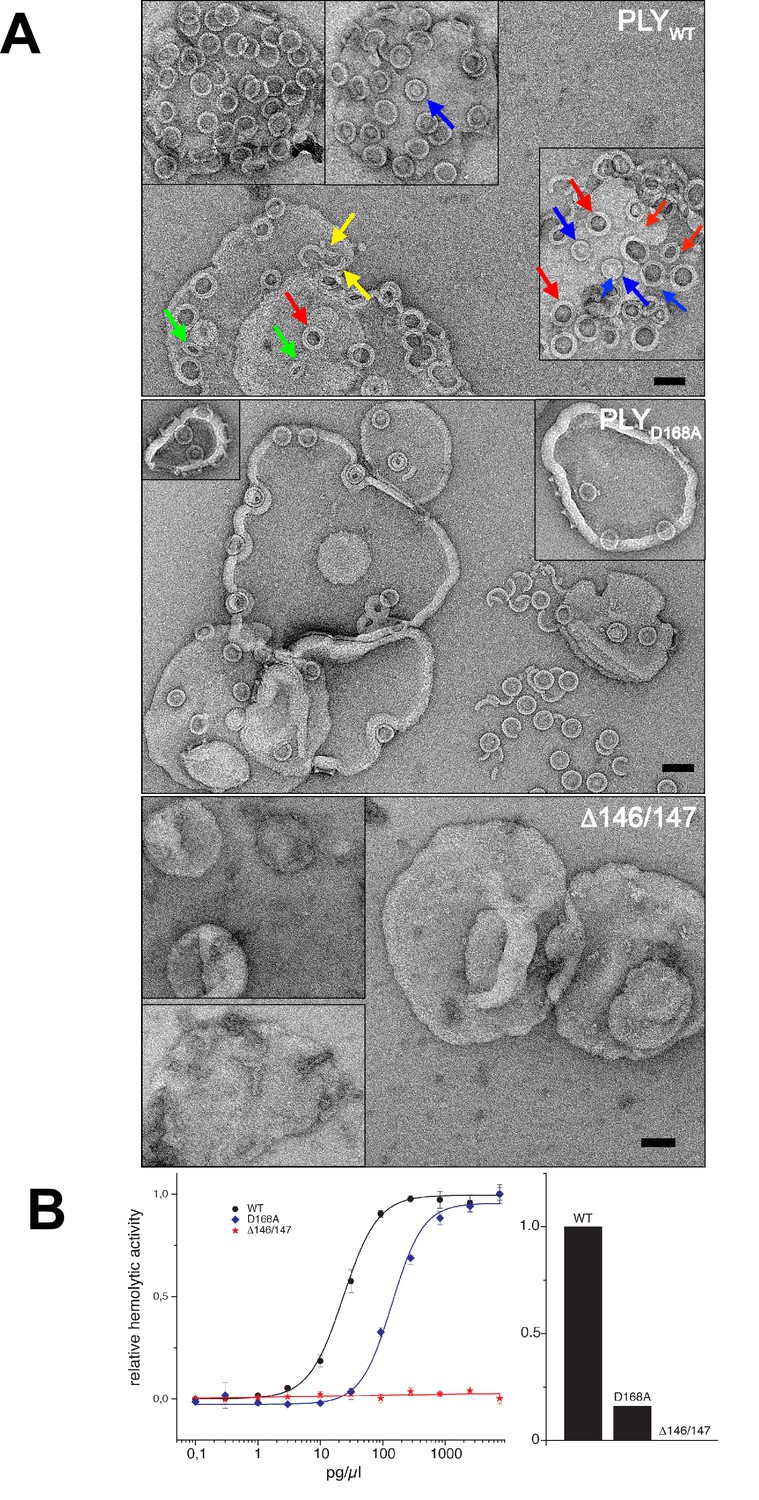
Membrane binding and hemolytic activity of PLY .
(A) Wildtype PLY (PLYWT) and PLYD168A form rings on cholesterol-containing liposomes. PLYWT lyses the majority of liposomes, while PLYD168Aleaves them mostly intact. Lipid-filled rings with a narrow rim (blue arrows) are prepores, while rings with a wider rim that do not contain lipid (red arrows) are pores. Slits (green arrows) and arcs (yellow arrows) are observed occasionally, but mostly PLY forms complete rings. Mutant PLYD168A prepores detach easily from the liposomes due to reduced binding affinity, and then break into fragments. Curves indicate the hemolytic activity of PLYWT, PLYD168A, and PLYΔ146/147. PLYΔ146/147 is inactive, in line with the inability of this mutant to form oligomers on cholesterol-containing liposomes. Scale bar: 50 nm.
Figure 8

Location of functionally important PLY residues.
(A) Ala146 and Arg147 in the loop that induces refolding of the last β-strand in the central D3 β-sheet into helix α13a in the late prepore and pore complex. Deletion of both residues renders the toxin inactive. (B) In the pore complex, Asp168 near the end of one long trans-membrane β-hairpin (HP1) forms a salt bridge with Lys271 in the other trans-membrane β-hairpin (HP2) of the adjacent monomer. Replacing Asp168 by alanine inhibits membrane insertion. (C) α-carbon traces in the x-ray structures of PLYWT (pdb 5aod), PLYΔ146/147 (pdb 5aof), and PLYD168A (pdb 5aoe). Minor differences between wildtype and mutant structures are visible in the loop regions of D4 (blue arrows); D2 (yellow arrows) and HB2 (green arrows). In PLYΔ146/147, one loop connecting D1 to D3 is also slightly different (red arrows).
Figure 9
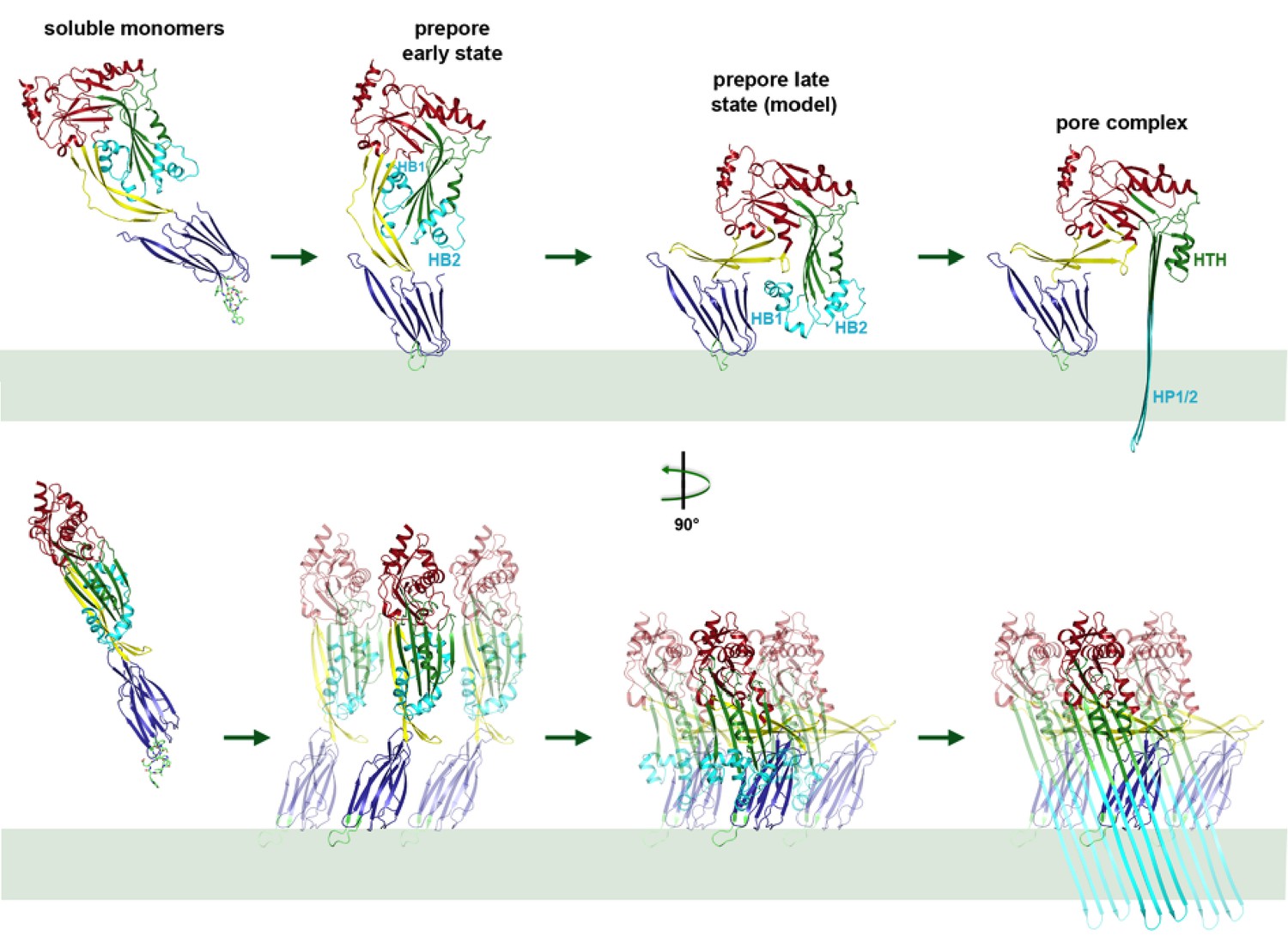
Mechanism of membrane insertion and pore formation.
Stepwise conformational changes of PLY toxin during pore formation shown for one monomer (above, side view) and for three neighbouring monomers (below, view from the pore centre). In the first step, soluble PLY monomers attach to the surface of cholesterol-containing cell membranes via the conserved D4 undecapeptide (Figure 2C) to form circular oligomers of the early prepore. A 90° rotation of D2 moves D1 and D3 towards the membrane in the late prepore. Helix bundles HB1 and HB2 are poised above the membrane surface to refold into 85 Å β-hairpins HP1 and HP2. In the final step of pore formation, both hairpins traverse the hydrophobic membrane core and assemble into a 168-strand, 260 Å β-barrel. Reorganization of the PLY monomer exposes numerous charges on the inside of the β-barrel (Figure 4) that would destabilize the lipid bilayer and repel membrane lipids, resulting in pore opening and cell lysis.
Videos
Video 1
Superposition of domain 4 of the PLY monomer from the crystal structure (blue) on domain 4 of the PLY monomer in the pore complex (green) rotated around an axis perpendicular to the membrane.
Residues of the undecapeptide and selected residues in other loops are shown as sticks.
Video 2
Mechanism of pneumolysin pore formation.
(1) Soluble PLY monomers attach to cholesterol-rich membranes by the cholesterol-binding undecapeptide (light green) of domain D4 (blue) and oligomerize into rings. For simplicity, only three ring subunits are shown. (2) Domain D2 (yellow) rotates by 90°, bringing domain D3 (green) with its two helix bundles (cyan) close to the membrane surface. (3) The helix bundles insert into the membrane and unfold into two trans-membrane 85 Å β-hairpins. β-hairpins of the 42 subunits in the ring merge into one large 168-strand β-barrel, which perforates the membrane.
Video 3
Model of conformational changes in an entire ring of 42 PLY monomers from early prepore to late prepore to pore, seen from the side and from above.
https://doi.org/10.7554/eLife.23644.023
Video 4
Oblique view of PLY ring inserting into the membrane.
https://doi.org/10.7554/eLife.23644.024Tables
Table 1
Data collection and refinement statistics.
| PLYD168A (pdb-id 5aoe) | PLYΔ146/147 (pdb-id 5aof) | |
|---|---|---|
| Data collection | ||
| Beamline | PXII @ Swiss Light Source x10sa | |
| Resolution (Å) | 40–2.5 (2.6–2.5) | 40–2.45 (2.55–2.45) |
| Wavelength (Å) | 0.9786 | 0.978 |
| Space group | P21 | P 21 21 21 |
| Cell dimensions | ||
| a, b, c (Å) | 160.86 24.66 208.35 | 24.73 163.5 207.8 |
| α, β, γ (°) | 90, 90.26, 90 | 90, 90, 90 |
| Total reflections | 316531 (30235) | 137706 (13089) |
| Unique reflections | 59222 (5685) | 29722 (2937) |
| Multiplicity | 5.3 (5.3) | 4.6 (4.5) |
| Completeness (%) | 99 (100) | 91 (94) |
| Mean I / σI | 8.1 (1.4) | 9.1 (1.2) |
| Wilson B-factor | 46.75 | 45.05 |
| Rpim | 0.08 (0.593) | 0.07 (0.607) |
| CC* | 0.998 (0.887) | 0.999 (0.792) |
| Refinement | ||
| Reflections used in refinement | 59207 (5683) | 29718 (2936) |
| Reflections in test set | 2962 (285) | 1486 (147) |
| Rwork/Rfree (%) | 22.25/24.98 (32.9/33.7) | 20.87/23.44 (33.23/34.37) |
| CC(work)/CC(free) | 0.946/0.934 (0.769/0.737) | 0.955/0.930 (0.626/0.567) |
| Average B-Factor (Å2) | 72.5 | 64.3 |
| No. atoms in AU | 7878 | 4008 |
| Protein | 7724 | 3856 |
| Water | 154 | 152 |
| r.m.s. deviations: | ||
| Bond lengths (Å) | 0.003 | 0.03 |
| Bond angles (°) | 0.72 | 0.69 |
| Ramachandran favored (%) | 96 | 96 |
| Ramachandran allowed (%) | 3.6 | 3.1 |
| Ramachandran outliers (%) | 0.5 | 0.8 |
-
Note: Values for the highest resolution shell are shown in parentheses
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin
eLife 6:e23644.
https://doi.org/10.7554/eLife.23644




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}