Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface
- Jeroen Claus

- Gargi Patel
- Flavia Autore
- Audrey Colomba
- Gregory Weitsman
- Selene Roberts
- Michael Hirsch
- Francesca Collu
- Roger George
- Elena Ortiz-Zapater
- Paul R Barber
- Boris Vojnovic
- Yosef Yarden
- Marisa L Martin-Fernandez
- Angus Cameron
- Franca Fraternali
- Tony Ng
- Peter J Parker
- The Francis Crick Institute, United Kingdom
- Kings College London, United Kingdom
- Brighton and Sussex University Hospitals, United States
- Rutherford Appleton Laboratory, United Kingdom
- King’s College London, Guy’s Hospital, United Kingdom
- University College London, United Kingdom
- Cancer Research UK and Medical Research Council Oxford Institute for Radiation Oncology, United Kingdom
- Weizmann Institute of Science, Israel
- Queen Mary University of London, United Kingdom
- Guy’s Hospital King’s College London School of Medicine, United Kingdom
- King’s College London, Guy’s Campus, United Kingdom
Figures
Figure 1 with 2 supplements
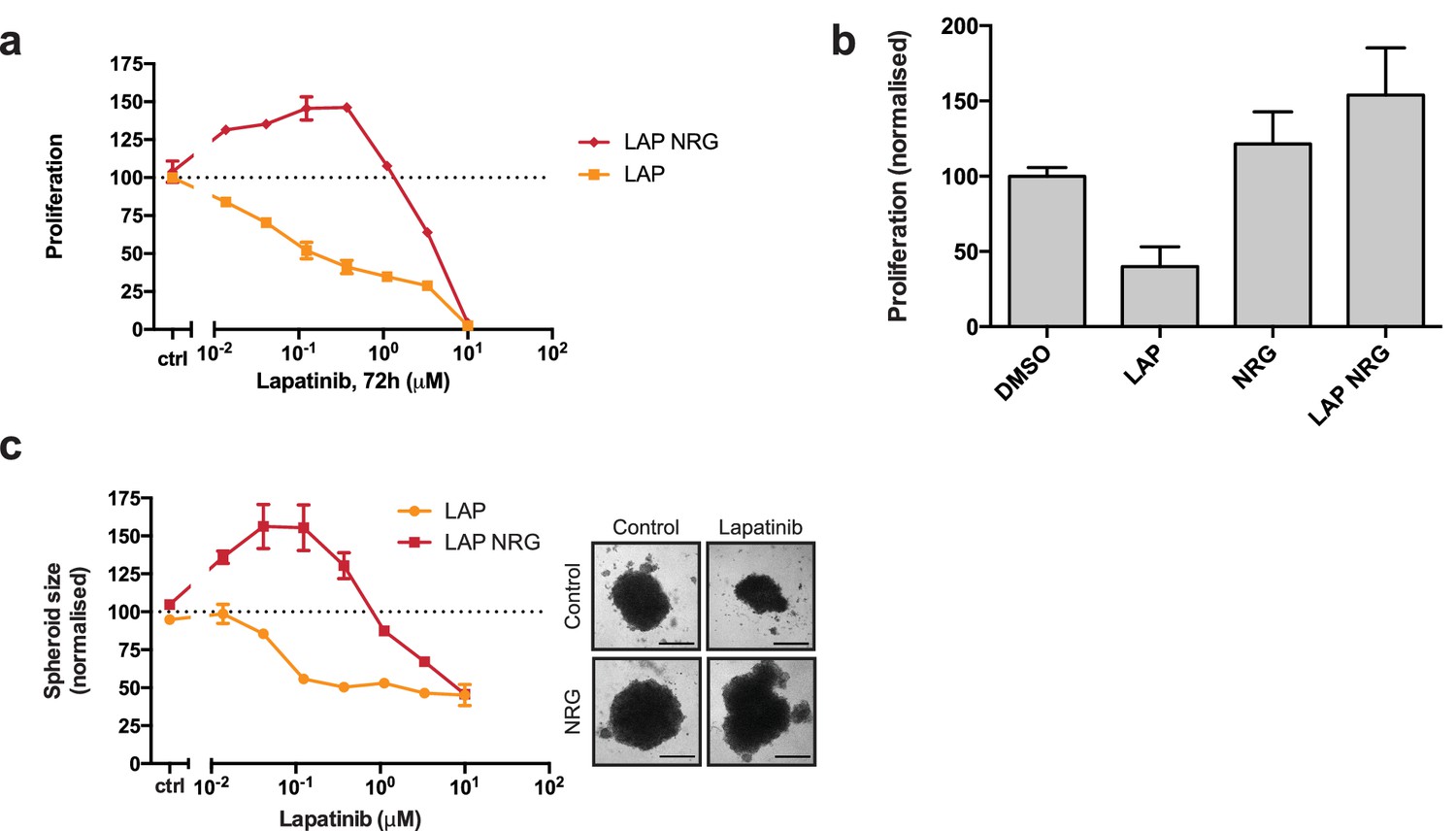
Lapatinib and NRG have synergistic effects on SKBR3 growth in 2D and 3D culture systems.
(a) CellTiter-Glo proliferation assay of SKBR3 cells after treatment for 72 hr with a range of lapatinib concentrations ± 10 nM NRG. (b) Cell counting assay of SKBR3 cells treated for 72 hr with DMSO or 250 nM lapatinib ±10 nM NRG, before quantification of cell number on a Vi-CELL counter. (c) Quantification of SKBR3 3D spheroid area after 8 days of treatment with a range of lapatinib concentrations ± 10 nM NRG, with representative bright field micrographs. Scale bars 0.5 mm. All proliferation data represented as mean ±SEM of three independent experiments each performed in quadruplicate. Corresponding data and statistics available as Figure 1—source data 1.
-
Figure 1—source data 1
Numerical data and statistics relating to Figure 1.
- https://doi.org/10.7554/eLife.32271.008
Figure 1—figure supplement 1
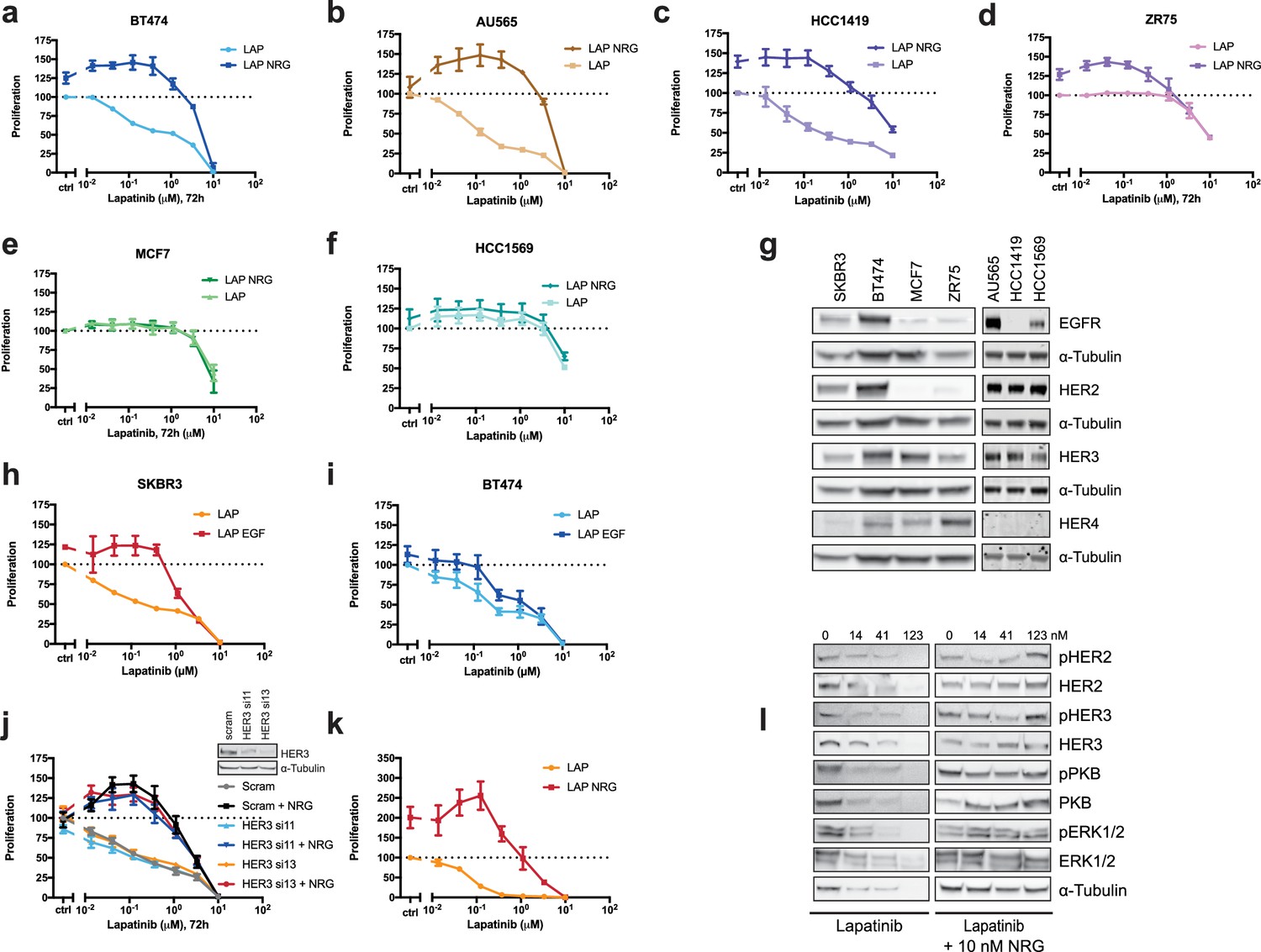
Effects of lapatinib and NRG on breast cancer cell proliferation.
(a–f) BT474, AU565, HCC1419, ZR75, MCF7, and HCC1569 breast cancer cell lines were assayed for proliferation in the response to lapatinib ±10 nM NRG as described in Figure 1a. BT474 and AU565 represent lapatinib-sensitive lines. HCC1419 and ZR75 represent partially-sensitive lines. MCF7 and HCC1569 represent lapatinib-insensitive lines. (g) Western blot analysis of endogenous EGFR family protein levels in SKBR3, BT474, AU565, HCC1419, ZR75, MCF7 and HCC1569 cell lines. (h–i) SKBR3 and BT474 cells were treated for 72 hr with a titration of lapatinib ±10 nM EGF, after which proliferation was measured using CellTiter-Glo. (j) CellTiter-Glo proliferation assay of SKBR3 cells with transient siRNA knockdown of HER3 using single oligonucleotides. Western blot denotes knockdown efficiency of HER3 si11 and HER3 si13 oligonucleotides. (k) CellTiter-Glo endpoint analysis of proliferation of SKBR3 spheroid cultures after 8 days of lapatinib ±NRG. (l) Western blot analysis of SKBR3 spheroid cultures in conditions matched to Figure 1c, Figure 1—figure supplement 1k. One representative example of three independent experiments is shown. All proliferation data represented as mean ±SEM of three independent experiments each performed in triplicate, except for (j), which represents six independent experiments each performed in triplicate. Corresponding data and statistics available as Figure 1—figure supplement 1—source data 1.
-
Figure 1—figure supplement 1—source data 1
Numerical data and statistics relating to Figure 1—figure supplement 1.
- https://doi.org/10.7554/eLife.32271.005
Figure 1—figure supplement 2

The irreversible inhibitor neratinib does not show synergistic growth under ligand co-treatment conditions.
(a) SKBR3 cells were treated for 72 hr with DMSO or 250 nM neratinib ±10 nM NRG, before quantification of cell number on a Vi-CELL counter (b) Quantification of spheroid area after 8 days of treatment with a titration of neratinib. Representative bright field micrographs of SKBR3 cell 3D spheroids. Scale bars 0.5 mm. (c) CellTiter-Glo endpoint analysis of spheroid cultures from (b). (d) Western blot analysis of cell signalling in SKBR3 spheroids after 8 days of treatment. All proliferation data represented as mean ±SEM of three independent experiments each performed in triplicate. All western blot shows a representative example of three independent experiments. Corresponding data and statistics available as Figure 1—figure supplement 2—source data 1.
-
Figure 1—figure supplement 2—source data 1
Numerical data and statistics relating to Figure 1—figure supplement 1.
- https://doi.org/10.7554/eLife.32271.007
Figure 2 with 2 supplements
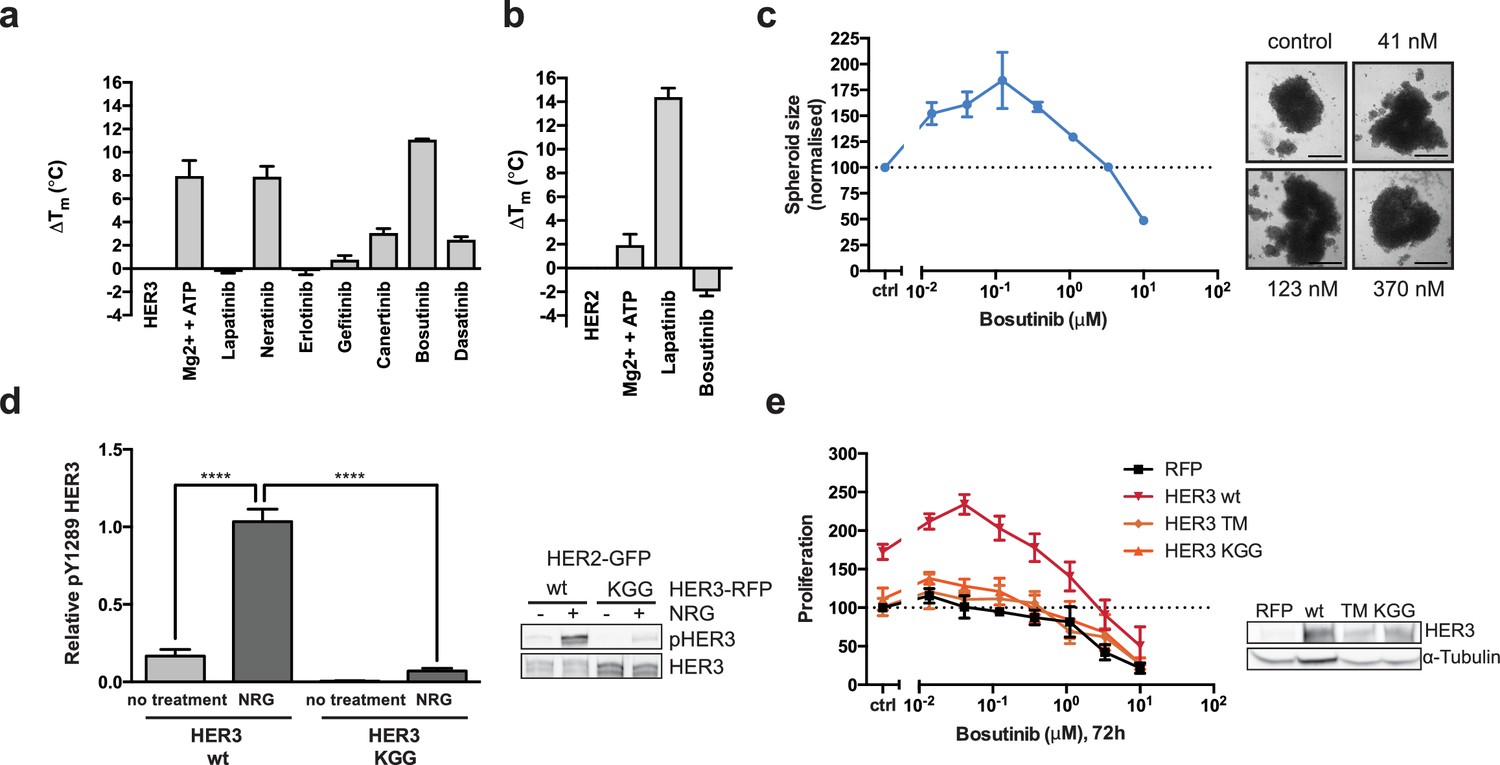
HER3 ATP-binding pocket occupation is necessary and sufficient to drive SKBR3 cell growth.
(a) In vitro TSA binding assay of HER3 with selected kinase inhibitors. (b) In vitro TSA binding assay of HER2 with lapatinib and bosutinib. (c) Quantification of spheroid size after eight days of treatment with a titration of bosutinib with representative bright field micrographs of SKBR3 cell spheroids after eight days of bosutinib treatment. Scale bars signify 0.5 mm. (d) Transient co-transfection of MCF7 cells with HER2wt-GFP and HER3wt-RFP or HER3KGG-RFP. Cells were serum starved for one hour, followed by 10 nM NRG or vehicle for ten minutes. HER3 phosphorylation on Y1289 was measured by Western blot and analysed by densitometry relative to total HER3. (e) SKBR3 cells were transfected with RFP empty vector, HER3wt-RFP, HER3T787M-RFP or HER3KGG-RFP. 72 hr of bosutinib treatment was initiated 24 hr post-transfection. Proliferation was measured using CellTiter-Glo. TSA data represented as mean ±SEM of (a) two independent experiments each performed quadruplicate, or (b) three independent experiments each performed in at least quadruplicate. Proliferation data represented as mean ±SEM of three independent experiments each performed in at least triplicate. Western blot data shown as mean ±SD for three independent experiments. Western blot quantifications analysed by one-way ANOVA. ****p≤0.0001 Corresponding data and statistics available as Figure 2—source data 1.
-
Figure 2—source data 1
Numerical data and statistics relating to Figure 2.
- https://doi.org/10.7554/eLife.32271.013
Figure 2—figure supplement 1
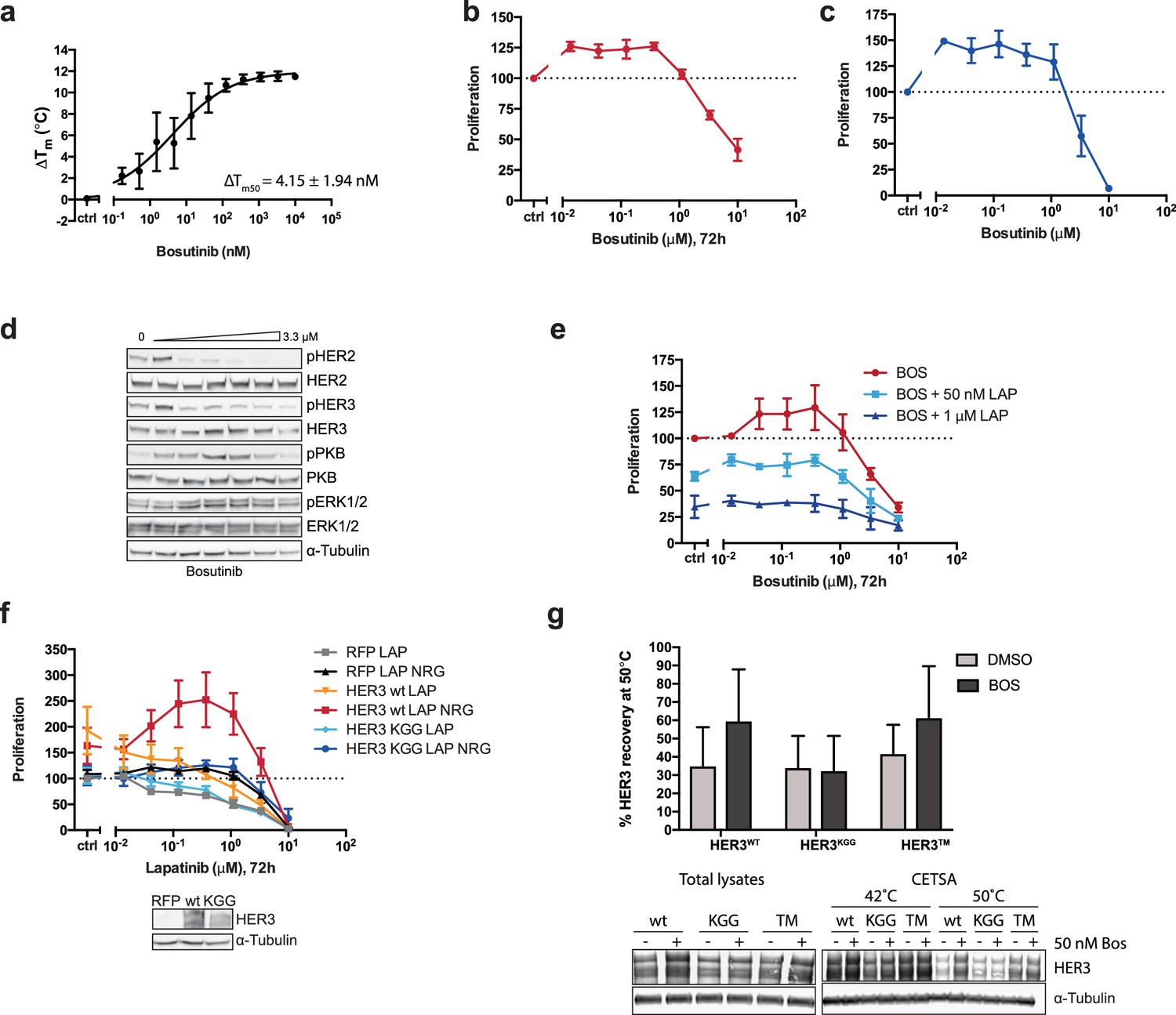
The effects of HER3 ATP-binding pocket occupation on drug-induced cell proliferation.
(a) TSA of HER3 kinase domain and a titration of bosutinib shows a ΔTm50 of 4.15 ± 1.94 nM. (b) SKBR3 cells were treated with a range of bosutinib concentrations for 72 hr and proliferation was measured using CellTiter-Glo (c) CellTiter-Glo endpoint quantification of spheroid cultures from Figure 2b. (d) Western blot analysis of spheroid cultures treated as in (c). (e) 2D proliferation of SKBR3 cells using a titration of bosutinib ±lapatinib (50 nM or 1 μM) for 72 hr. (f) SKBR3 cells were transiently transfected with vector-RFP, HER3wt-RFP, or HER3KGG-RFP and treated with lapatinib ±10 nM NRG for 72 hr. (g) CETSA analysis of bosutinib binding to HER3wt, HER3KGG, or HER3T787M. Lysates of COS7 cells ectopically expressing HER3-RFP were treated with DMSO or 50 nM bosutinib, after which samples were split and matching samples incubated at either 42°C or 50°C. Western blot analysis shows HER3 recovery at 50°C compared to 42°C. Data in (a-f) presented as mean ±SEM of three independent experiments each performed in triplicate. Western blot data in (d) show a representative example of three independent experiments. Data in (g) presented as mean ±SD of four independent experiments. Corresponding data and statistics available as Figure 2—figure supplement 1—source data 1.
-
Figure 2—figure supplement 1—source data 1
Numerical data and statistics relating to Figure 2-figure supplement 1.
- https://doi.org/10.7554/eLife.32271.011
Figure 2—figure supplement 2
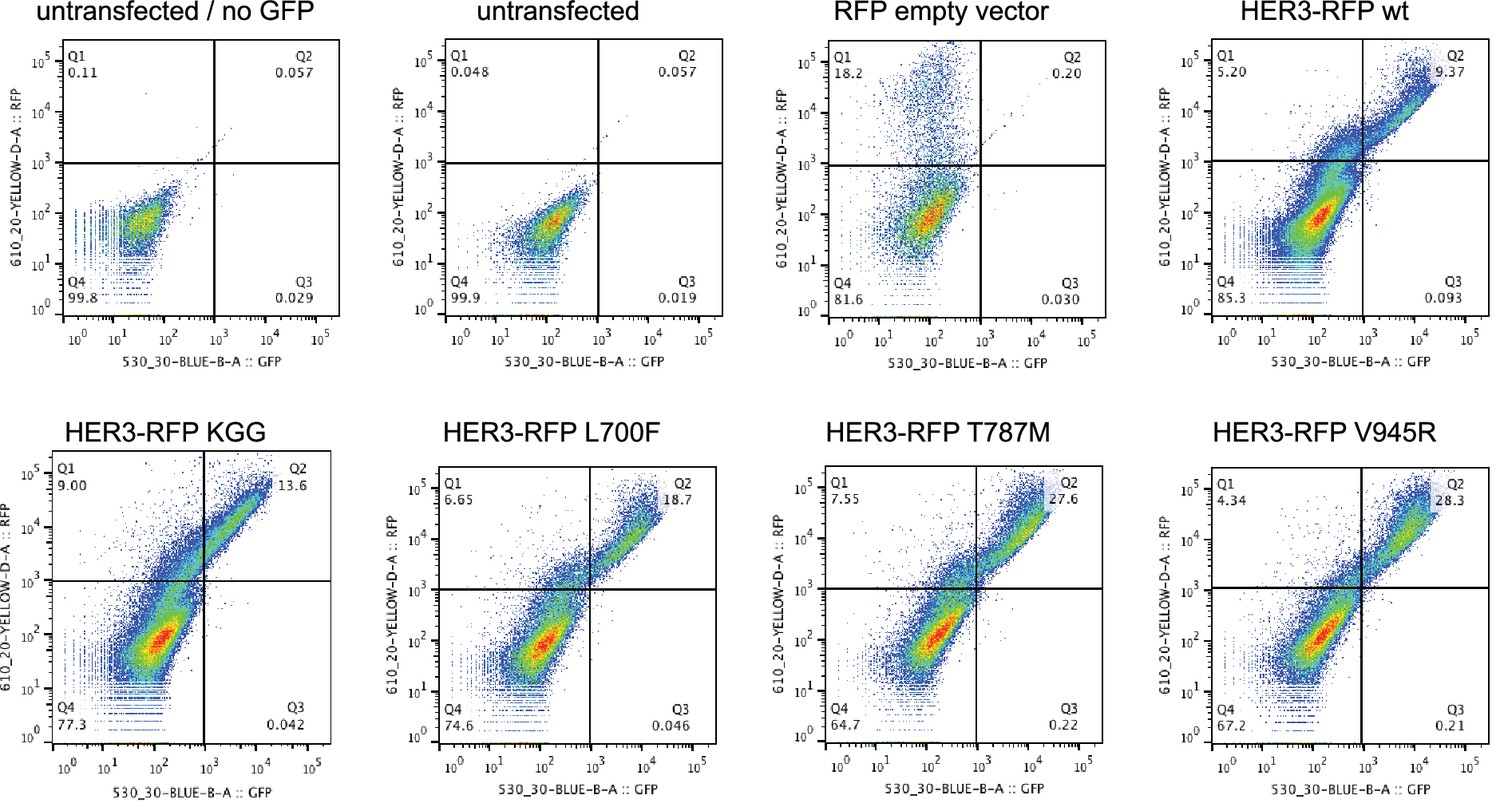
Cell surface expression of HER3 mutants.
Flow cytometric analysis of membrane localisation of all HER3-RFP constructs used in this study. Live SKBR3 cells were stained with GFP-conjugated anti-HER3 to show the combination of transfected and endogenous HER3 on the membrane. All HER3-RFP constructs show membrane localisation, as represented by the top right quadrants. Representative flow cytometry plots from one of two independent experiments.
Figure 3
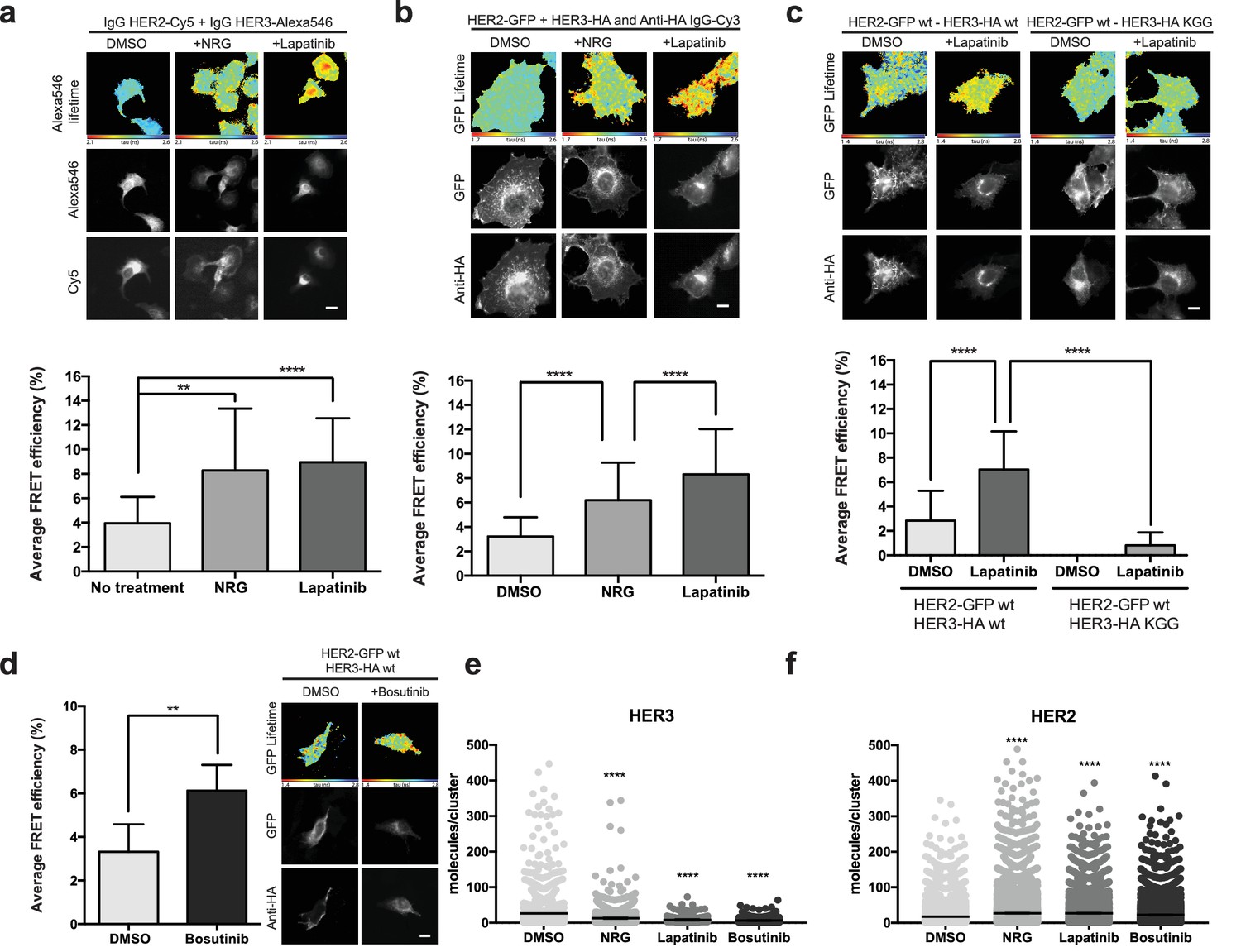
Inhibitor-induced HER2-HER3 heterotypic interactions.
(a) FRET-FLIM analysis of endogenous HER2-HER3 association in SKBR3 cells, serum starved for 1 hr, and stimulated with 6.7 nM NRG for 15 min, or inhibited with lapatinib (10 µM) for 1 hr, prior to fixation and staining with IgG anti-HER2-Cy5 and IgG anti-HER3-Alexa546 overnight, at 4°C. (b) MCF7 cells were transfected with vectors encoding HER2wt-GFP and HER3wt-HA. Cells were incubated as in (a) and stained with anti-HA antibody conjugated to Alexa-546 (controls treated with vehicle). (c) MCF7 cells were transfected with vectors encoding HER2wt-GFP and HER3wt-HA or HER3KGG-HA. Cells treated with lapatinib (10 µM) for 1 hr, prior to fixation and staining with anti-HA antibody conjugated to Alexa-546. (d) SKBR3 cells were treated with bosutinib (50 nM, 1 hr), and stained as in (b). (e, f) Molecules/cluster measurements from STORM data taken of SKBR3 cells labelled with HER2Affibody-Alexa488 and HER3Affibody-Alexa647 or NRG-Alexa647 ±14 nM lapatinib or 41 nM bosutinib. Cumulative FRET-FLIM histograms show average FRET efficiency from three independent experiments. **p≤0.01; ****p≤0.0001 Scale bars 5 μm. Clustering data represent mean combination of two independent experiments with each measuring >1000 clusters. Clustering data presented as mean with 95% CI. Corresponding data and statistics available as Figure 3—source data 1.
-
Figure 3—source data 1
Numerical data and statistics relating to Figure 3.
- https://doi.org/10.7554/eLife.32271.015
Figure 4 with 2 supplements
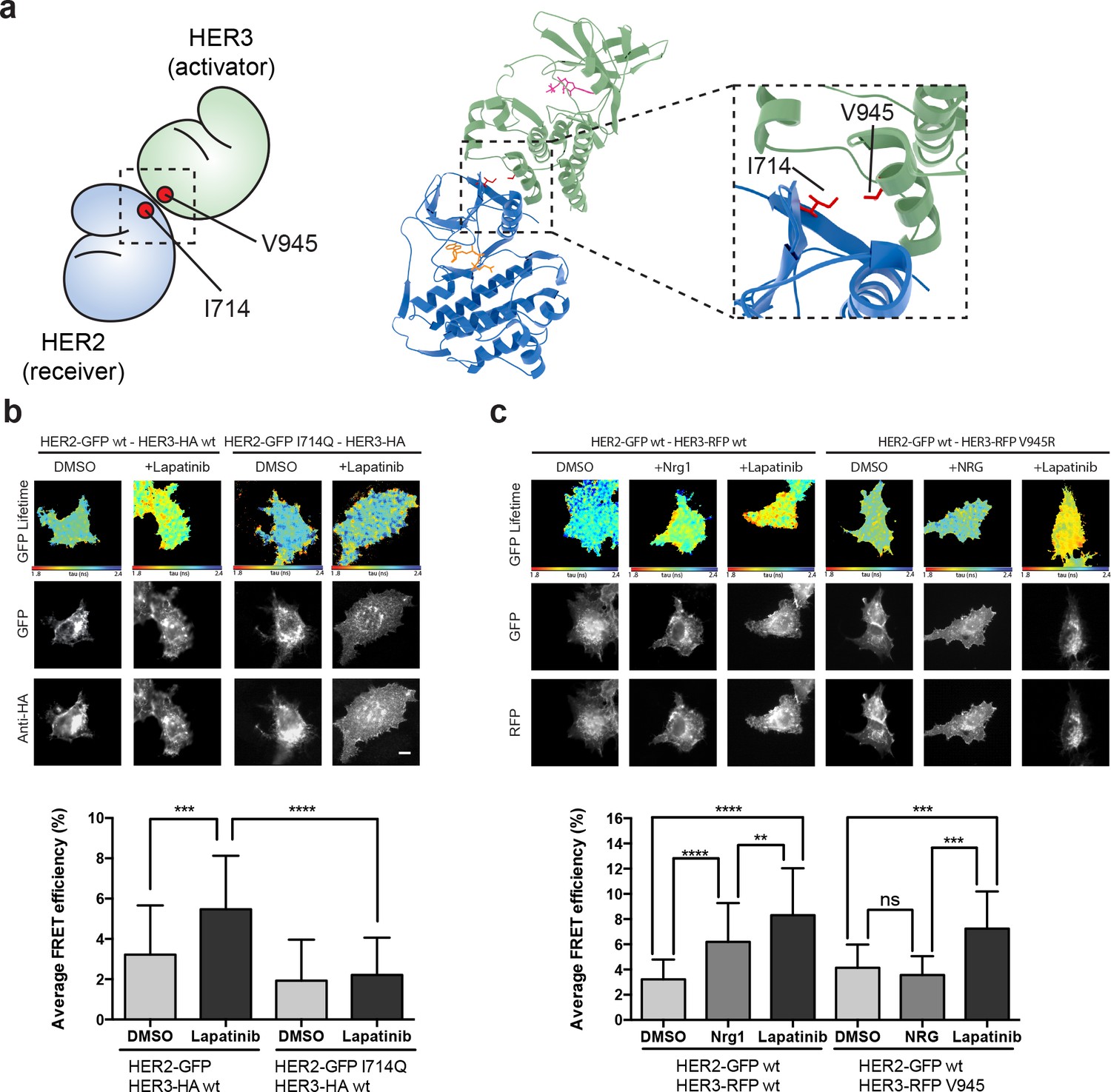
The lapatinib-induced HER2-HER3 dimer is distinct from the active, asymmetric HER2-HER3 dimer orientation.
(a) Schematic representation and molecular model of HER2-HER3 active, asymmetric kinase domain dimer orientation. Inset denotes interaction interface. (b) MCF7 cells were transfected with vectors encoding HER2wt-GFP or HER2I714Q-GFP and HER3wt-HA. Cells were treated as described in Figure 3 and HER2-HER3 association was measured by FRET-FLIM. (c) MCF7 cells were transfected with vectors encoding HER2-GFP and HER3wt-RFP or HER3V945R-RFP. Cells were incubated as described above, and treated with DMSO, lapatinib or NRG prior to fixation. Data represents mean ±SEM. *p≤0.05; **p≤0.01, ***p≤0.001; ****p≤0.0001 by one-way ANOVA. Scale bars represent 5 μm. Corresponding data and statistics available as Figure 4—source data 1. Molecular model for the interaction in (a) available as Figure 4—source data 2.
-
Figure 4—source data 1
Numerical data and statistics relating to Figure 4.
- https://doi.org/10.7554/eLife.32271.020
-
Figure 4—source data 2
PDB structure file of molecular interaction model in Figure 4a.
- https://doi.org/10.7554/eLife.32271.021
Figure 4—figure supplement 1
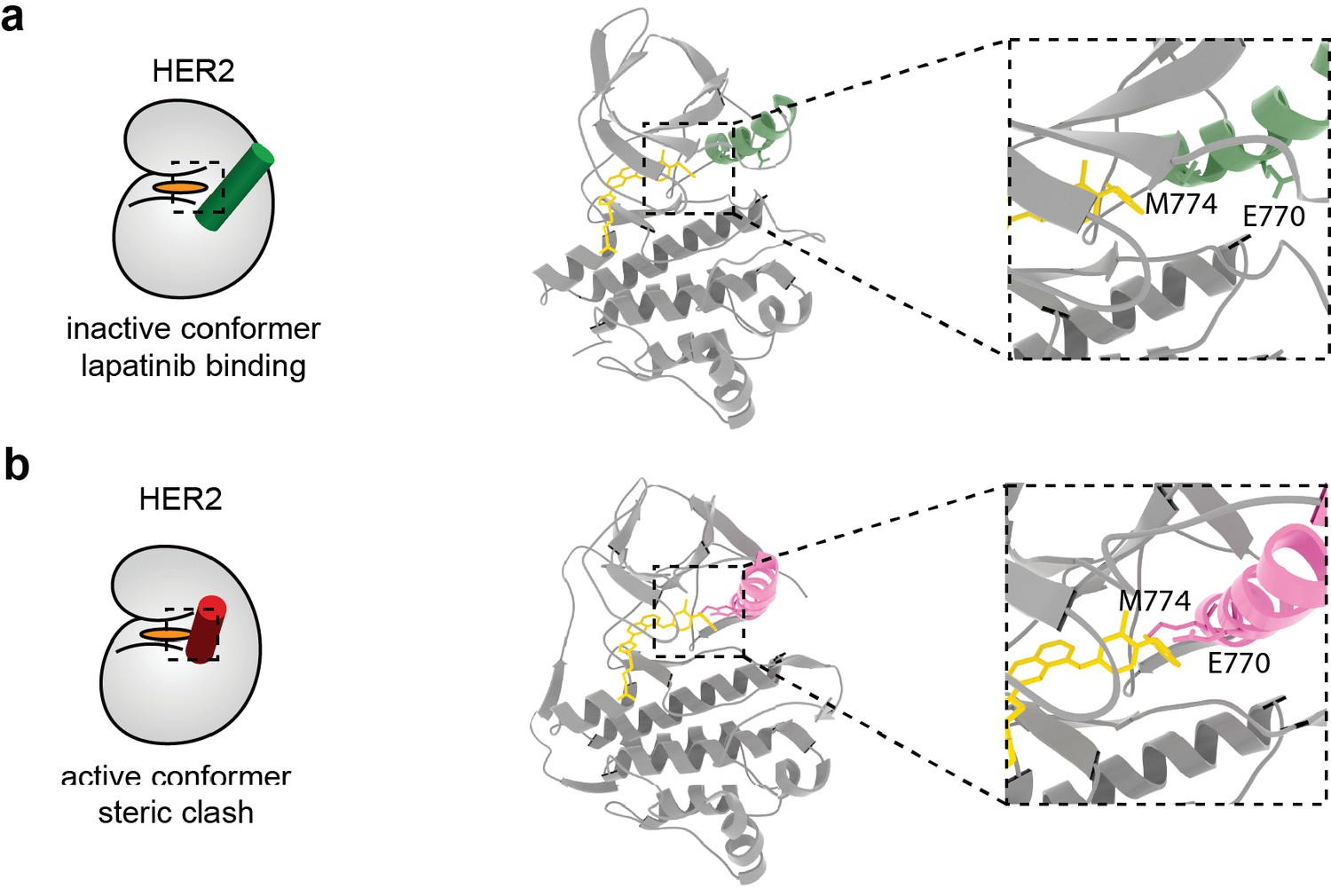
Model of lapatinib binding in HER2 inactive and active conformations shows a potential steric clash.
(a) Lapatinib docking in HER2, with the HER2 active site displayed in the inset. Lapatinib binds the inactive conformation of HER2 where the α-C helix is in the ‘out’ position. E770 and M774 on the HER2 α-C helix highlighted. (b) Lapatinib docked into the active conformation of HER2. The α-C helix is in the ‘in’ position, causing a steric clash between E770/M774 and lapatinib. Molecular models for inhibitor docking in (a) and (b) available as Figure 4—figure supplement 1—source datas 1 and 2.
-
Figure 4—figure supplement 1—source data 1
PDB structure file of inhibitor docking model in Figure 4—figure supplement 1a.
- https://doi.org/10.7554/eLife.32271.018
-
Figure 4—figure supplement 1—source data 2
PDB structure file of inhibitor docking model in Figure 4—figure supplement 1b.
- https://doi.org/10.7554/eLife.32271.019
Figure 4—video 1
Interface view of the molecular model of an active HER2-HER3 heterodimer, with HER2I714 and HER3V945 highlighted.
https://doi.org/10.7554/eLife.32271.022
Figure 5 with 3 supplements
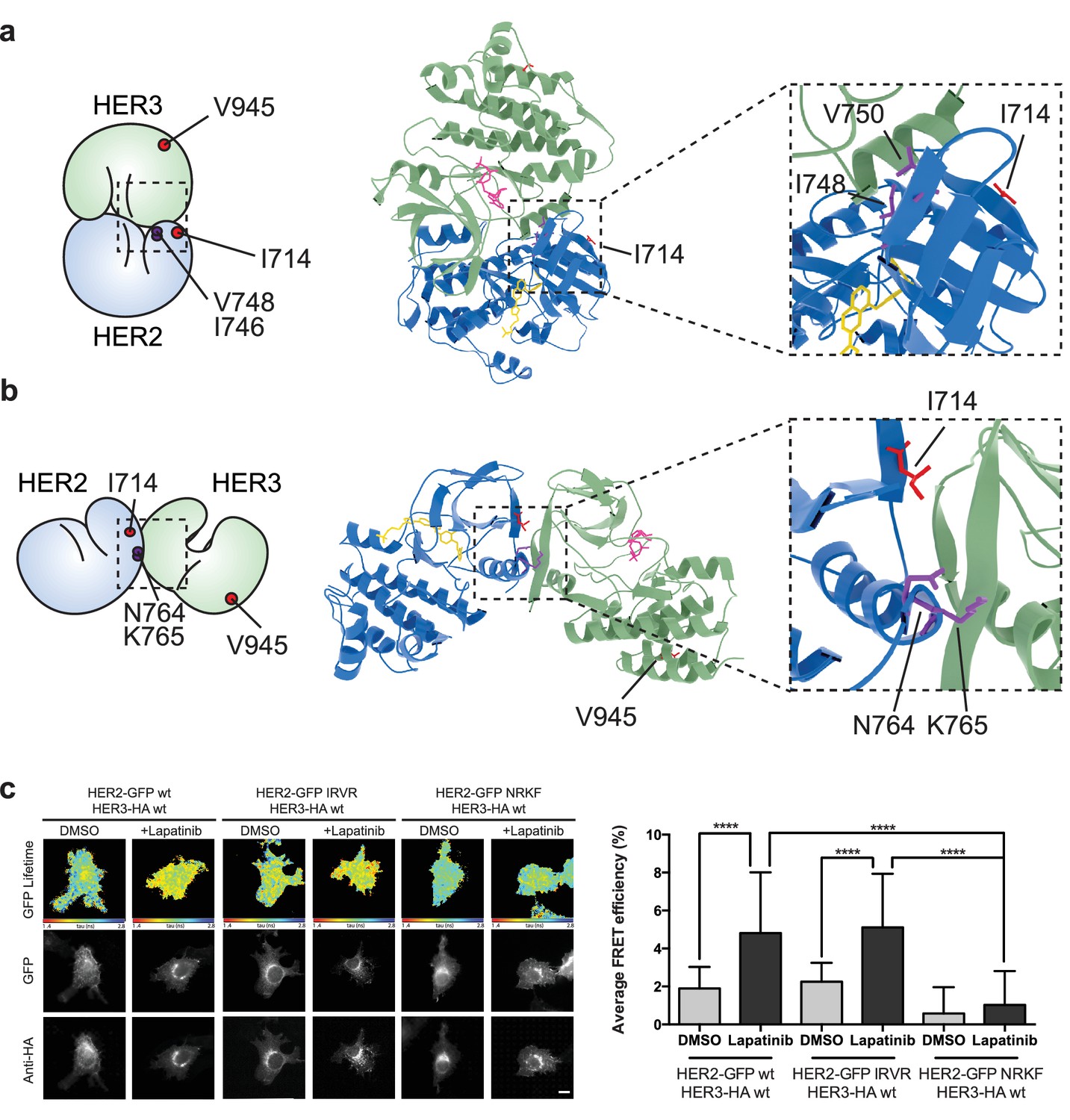
The lapatinib-induced HER2-HER3 dimer is in a symmetric orientation.
(a) Lapatinib-bound HER2 (blue, lapatinib in yellow) and ATP analogue-bound HER3 (green, AMP-PNP in pink) were modelled in an EGFR-like symmetric dimer orientation (Jura et al., 2009a). Inset highlights the interaction interface. The schematic representation shows active dimer interface residues HER2I714 and HER3V945, as well as the two residues in HER2 unique to this interface for further mutational analysis. (b) Lapatinib-bound HER2 in the HER3-like head-to-head symmetric dimer orientation (Jura et al., 2009b). Dimer-specific residues are highlighted in the schematic. (c) MCF7 cells were transfected with vectors encoding HER2wt-GFP, HER2N764R/K765F-GFP or HER2I748R/V750R-GFP and HER3wt-HA. Cells were incubated for 24 hr, and inhibited with 10 µM lapatinib for 1 hr, prior to fixation and staining with anti-HA antibody conjugated to Alexa-546. Data represented as mean ±SEM. ****p≤0.0001, as analysed by one-way ANOVA. Scale bars 5 μm corresponding data and statistics available as Figure 5—source data 1. Molecular model for the interactions in (a) and (b) available as Figure 5—source data 2 and 3. Residues marking the dimer interface of the lapatinib-induced HER2-HER3 heterodimer, in either the EGFR-like or HER3-like modelled conformations, including the per-residue solvent accessible surface area (in Å2) are available as Figure 5—source data 4.
-
Figure 5—source data 1
Numerical data and statistics relating to Figure 5.
- https://doi.org/10.7554/eLife.32271.025
-
Figure 5—source data 2
PDB structure file of molecular interaction model in Figure 5a.
- https://doi.org/10.7554/eLife.32271.026
-
Figure 5—source data 3
PDB structure file of molecular interaction model in Figure 5b.
- https://doi.org/10.7554/eLife.32271.027
-
Figure 5—source data 4
Table with modelled interface residues, including the per-residue solvent-accessible surface area in Å2.
- https://doi.org/10.7554/eLife.32271.028
Figure 5—figure supplement 1

Molecular models of potential orientations of the lapatinib-induced HER2-HER3 dimer.
Molecular model of the lapatinib-induced HER2-HER3 dimer in the (a) EGFR-like and (b) HER3-like orientation with interface residues shown as sticks.
Figure 5—video 1
Interface view of the molecular model of a lapatinib-induced HER2-HER3 heterodimer in the EGFR-like conformation, with HER2I714 and HER3V945 highlighted, as well as model-specific interface residues HER2I748/V750.
https://doi.org/10.7554/eLife.32271.029
Figure 5—video 2
Interface highlight of the molecular model of a lapatinib-induced HER2-HER3 heterodimer in the HER3-like conformation, with HER2I714 and HER3V945 highlighted, as well as model-specific interface residues HER2N764/K765.
https://doi.org/10.7554/eLife.32271.030
Figure 6 with 1 supplement
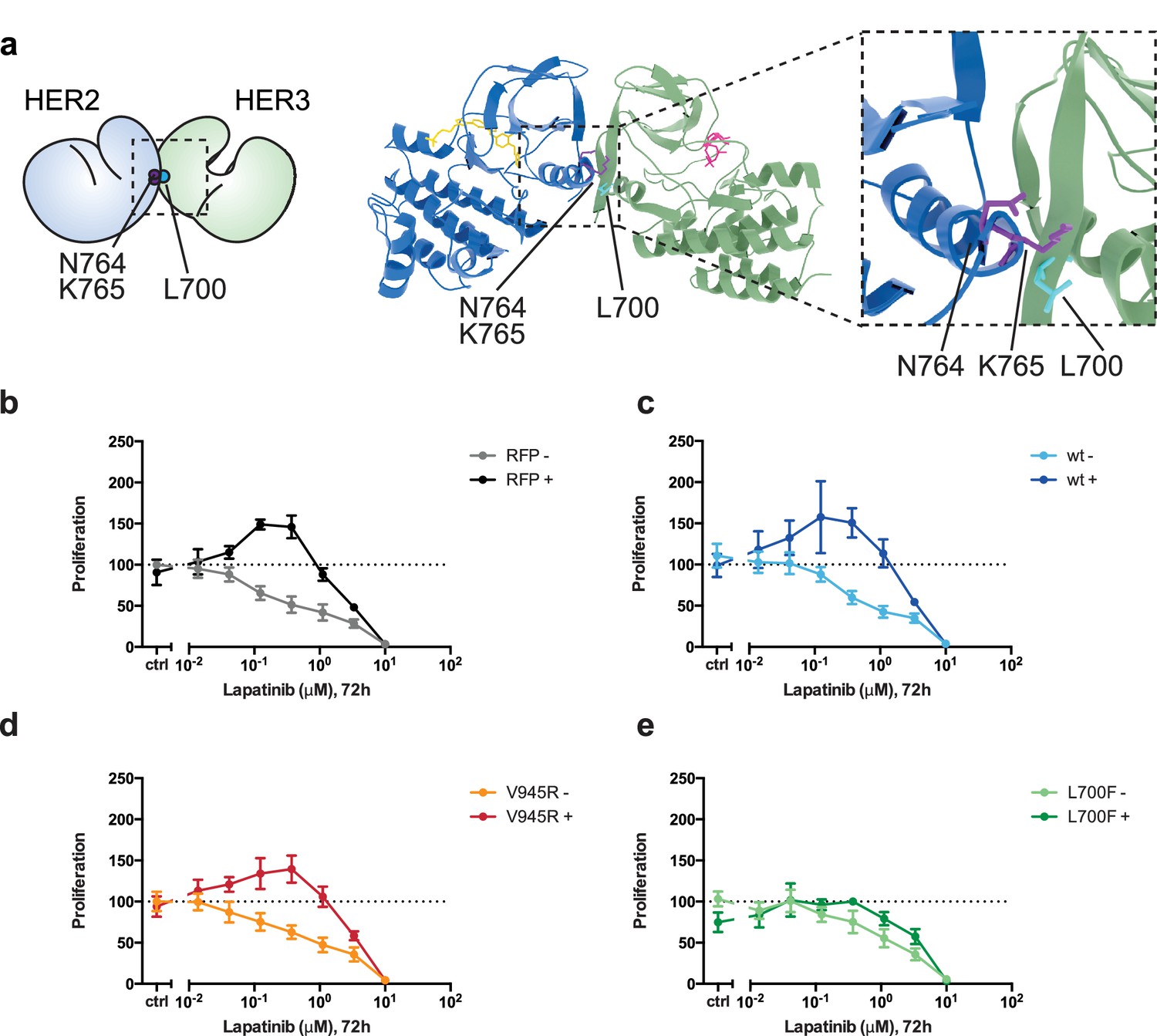
Disruption of the lapatinib-induced dimer inhibits lapatinib-NRG synergistic growth.
(a) Molecular model of the lapatinib-induced HER2-HER3 dimer with the lapatinib-dimer interface residues HER2N764 and HER2K765 highlighted (purple), and a potential reciprocal residue HER3L700F (cyan). (b–e) 2D proliferation assays of SKBR3 cells transfected with (b) RFP empty vector, (c) HER3wt, (d) HER3V945R, or (e) HER3L700F and treated with lapatinib ±10 nM NRG as before. Data represents mean ±SEM for six independent experiments, each performed in triplicate. Corresponding data and statistics available as Figure 6—source data 1.
-
Figure 6—source data 1
Numerical data and statistics relating to Figure 6.
- https://doi.org/10.7554/eLife.32271.033
Figure 6—video 1
Interface highlight of the molecular model of a lapatinib-induced HER2-HER3 heterodimer in the HER3-like conformation, with HER2I714 and HER3V945 highlighted, as well as model-specific interface residues HER2N764/K765 and HER3L700.
https://doi.org/10.7554/eLife.32271.032Additional files
-
Transparent reporting form
- https://doi.org/10.7554/eLife.32271.034
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface
eLife 7:e32271.
https://doi.org/10.7554/eLife.32271




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}