Nonsense mRNA suppression via nonstop decay
- University of California, Santa Cruz, United States
- Stanford University School of Medicine, United States
Figures
Figure 1 with 1 supplement
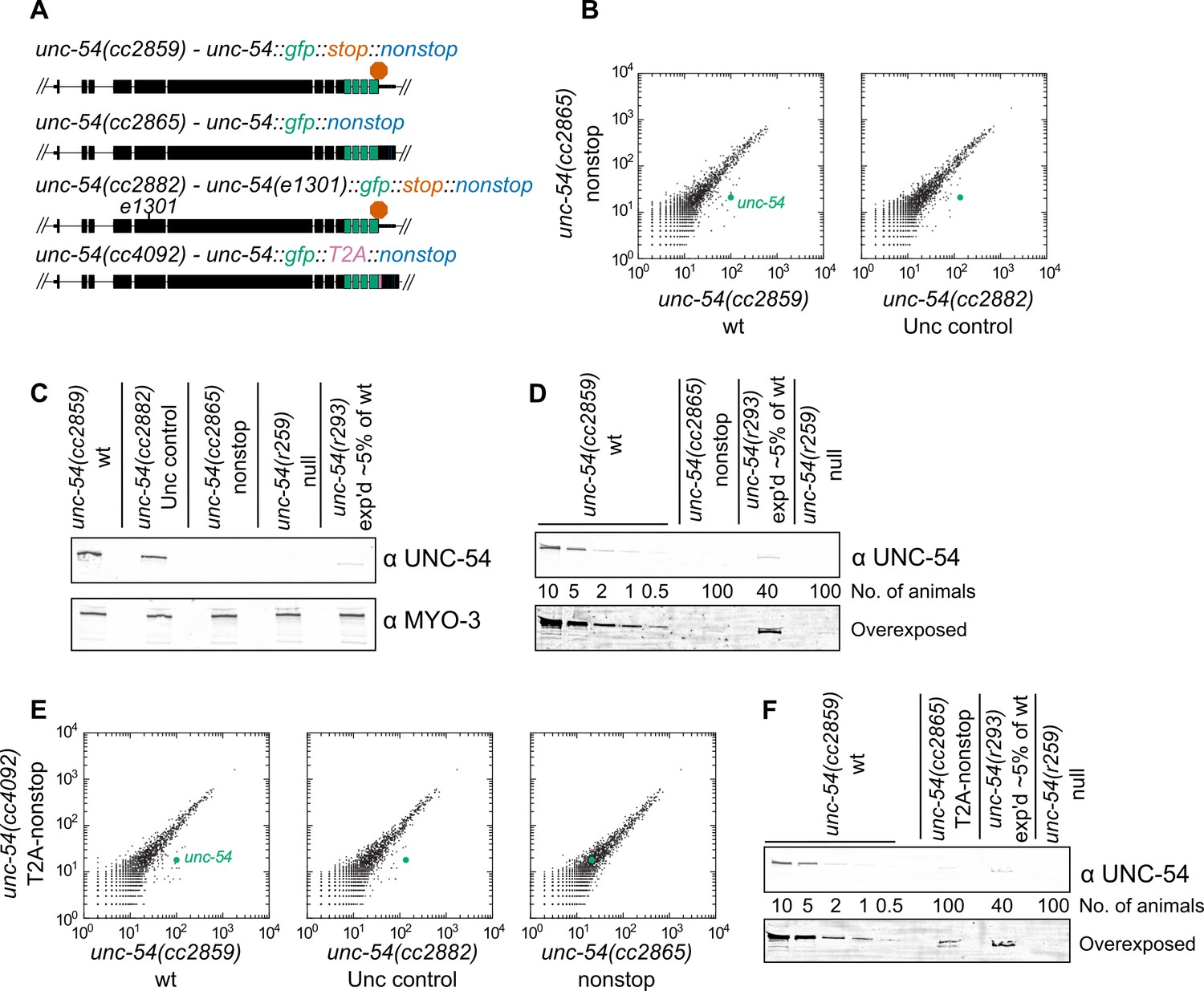
Loss of all stop codons is detrimental to mRNA and protein expression in C.
elegans. (A) Diagram of alleles made via CRISPR/Cas9 and analyzed here. cc2859 is an ancestor to the three other alleles. e1301 is a temperature-sensitive Unc mutation, which was used throughout this figure as a control for Unc effects; T2A is a viral ‘stop-and-go’ peptide that releases upstream protein during translation elongation. (B) RNA-seq was performed to quantify mRNA levels in each indicated strain, with each dot representing read counts for a gene. Additional off-diagonal genes are collagens and vitellogenins, likely a secondary consequence of slow growth conferred by cc2865. (C) Immunoblot on an equal number of animals of each indicated genotype. r259 is a ~17 kb deletion spanning much of unc-54; r293 is a control allele exhibiting <5% of normal UNC-54 protein. Samples were split in half and probed for UNC-54 and MYO-3 (loading control) separately. (D) Immunoblot of UNC-54 with different titrations of animals loaded per lane. (E) RNA-seq as in (B), but with cc4092 allele. (F) Immunoblot of UNC-54 as in (D), but with cc4092 allele.
Figure 1—figure supplement 1

Results of candidate-based approach and genetic screen for nonstop suppressors.
(A) Sequence homologs of factors known to affect nonstop decay in other systems. Large deletions creating premature stop codons were made by CRISPR/Cas9 in each factor, as indicated below each locus. Location of the y54e10a.11 RING domain indicated. All mutations led to grossly wild type animals. (B) Mutations in one or more factors were crossed into the unc-54(cc2865) nonstop reporter. ‘---' indicates a lack of detectable GFP fluorescence. ‘-' indicates very weak but detectable GFP fluorescence. (C) Summary of mutations that rescue expression or do not at the nonstop unc-54 locus. Diagram of the unc-54(cc2865) locus at top, with sequence and reading frame of C-terminus below. Mutations relative to the wild type unc-54(3’UTR) are indicated in lowercase. The wild-type stop codon location (‘former stop codon’) is indicated in red. EMS mutagenesis of PD2865 recovered at least four independent isolates of the indicated CAA >TAA the second codon after the former stop codon. These animals were indistinguishable from unc-54(cc2859). No additional 3’UTR-contained codons would be expected to mutate to a stop codon (TAA/TAG/TGA) given the mutational bias of EMS (G > A and C > T). CRISPR/Cas9 was used to shift the frame by a base (indicated in blue), leading to translation termination upstream of the poly(A) site, but did not rescue the Unc phenotype nor restore GFP expression. An additional screen with the unc-54(cc2865) reporter in the y54e10a.11(cc2862) background yielded a GGG(Gly)>GAG(Glu) mutation in GFP (not pictured), creating a splice site and a frameshift. These animals were non-Unc, but GFP negative.
Figure 2 with 3 supplements
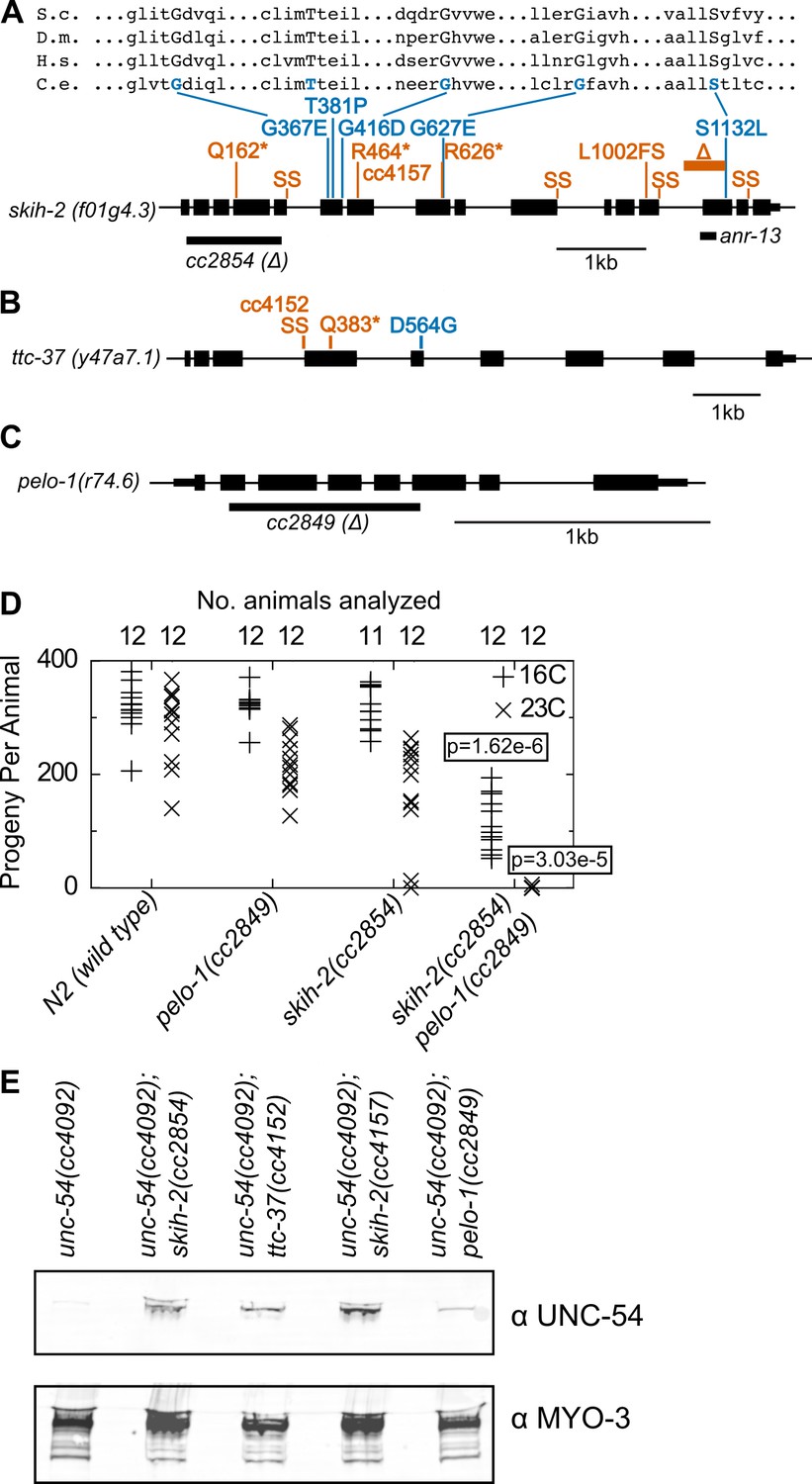
Protein factors required for nonstop mRNA decay.
(A) Diagram of the skih-2 gene, with mutations identified in the cc4092 screen. Red indicates mutations expected to prematurely terminate SKIH-2 protein synthesis. SS is a mutation of a splice site. Blue indicate missense mutations, with multiple sequence alignment of these residues with skih-2 homologs above. L1002FS is a frameshift mutation. ‘S.c.’ is S. cerevisiae; ‘D.m.’ is D. melanogaster; ‘H.s.’ is H. sapiens and ‘C.e.’ is C. elegans. A ~ 1 kb deletion introducing a premature stop codon made by CRISPR/Cas9 and used in subsequent characterization is indicated below the gene. See also Figure 1—figure supplement 1, Figure 2—figure supplement 1, Figure 2—figure supplement 2. (B) Diagram of the ttc-37 gene, a ski3 homolog, with mutations identified in the cc4092 screen. Coloring as in (A). (C) Sequence homolog and candidate ortholog of dom34/pelota. ~700 bp deletion made by CRISPR/Cas9 and used in subsequent characterization is indicated below the gene. See also Figure 1—figure supplement 1, Figure 2—figure supplement 3. (D) Brood size analysis for the indicated strains. Each symbol represents brood size of one animal, with total number of animals analyzed indicated at top. See Materials and methods. p-Value from Mann Whitney U test compared with any other strain at the same temperature. (E) Immunoblot on an equal number of animals of the indicated genotypes.
Figure 2—figure supplement 1
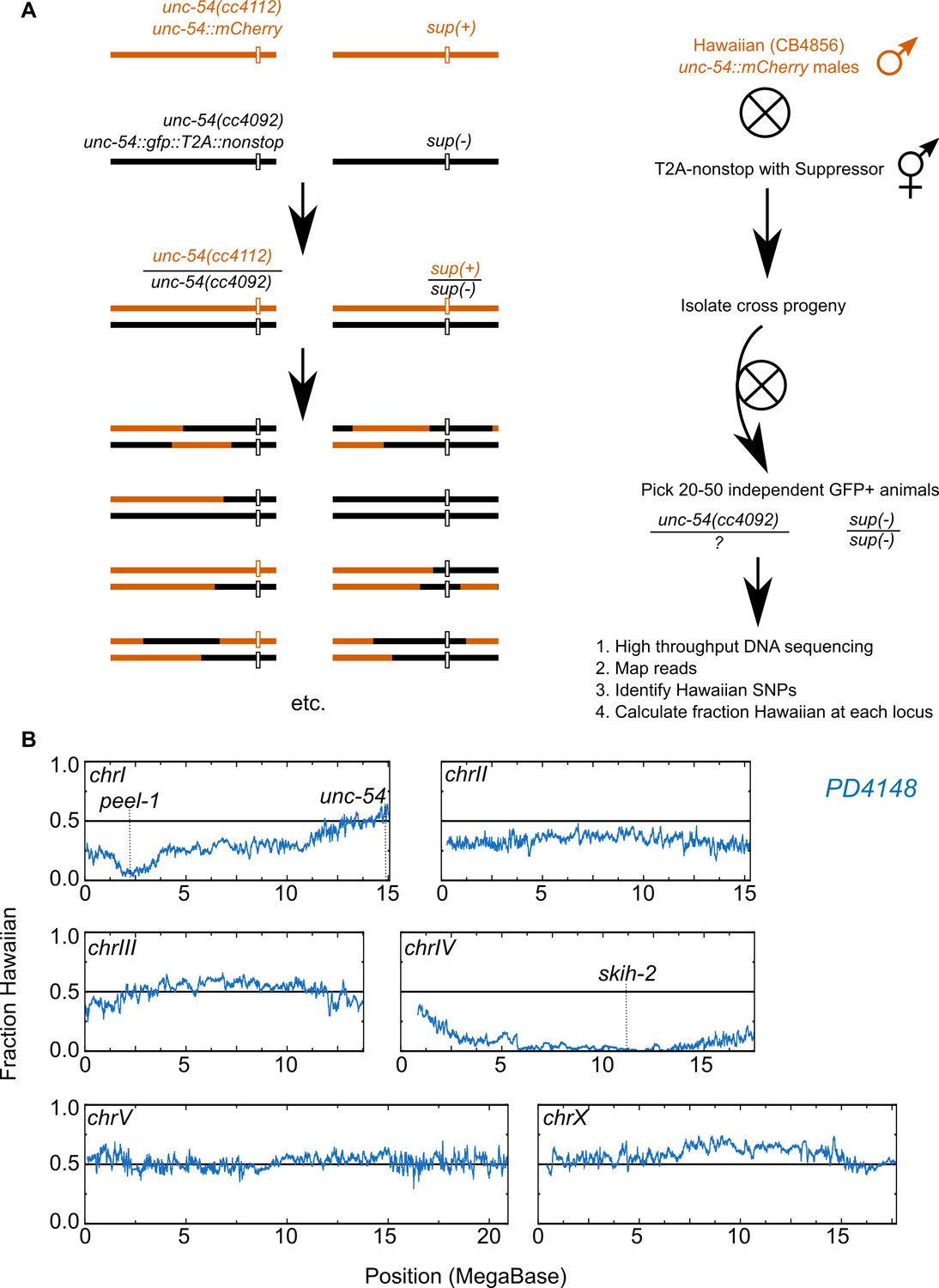
Genetic mapping of suppressor strains.
(A) Workflow schematic, see Materials and methods. (B) Example data for PD4148. The left end of chromosome I exhibits reduced Hawaiian allele frequency due to a known paternally-inherited genetic incompatibility between Hawaiian and Bristol (our ‘wild type’) C. elegans strains (Seidel et al., 2008).
Figure 2—figure supplement 2
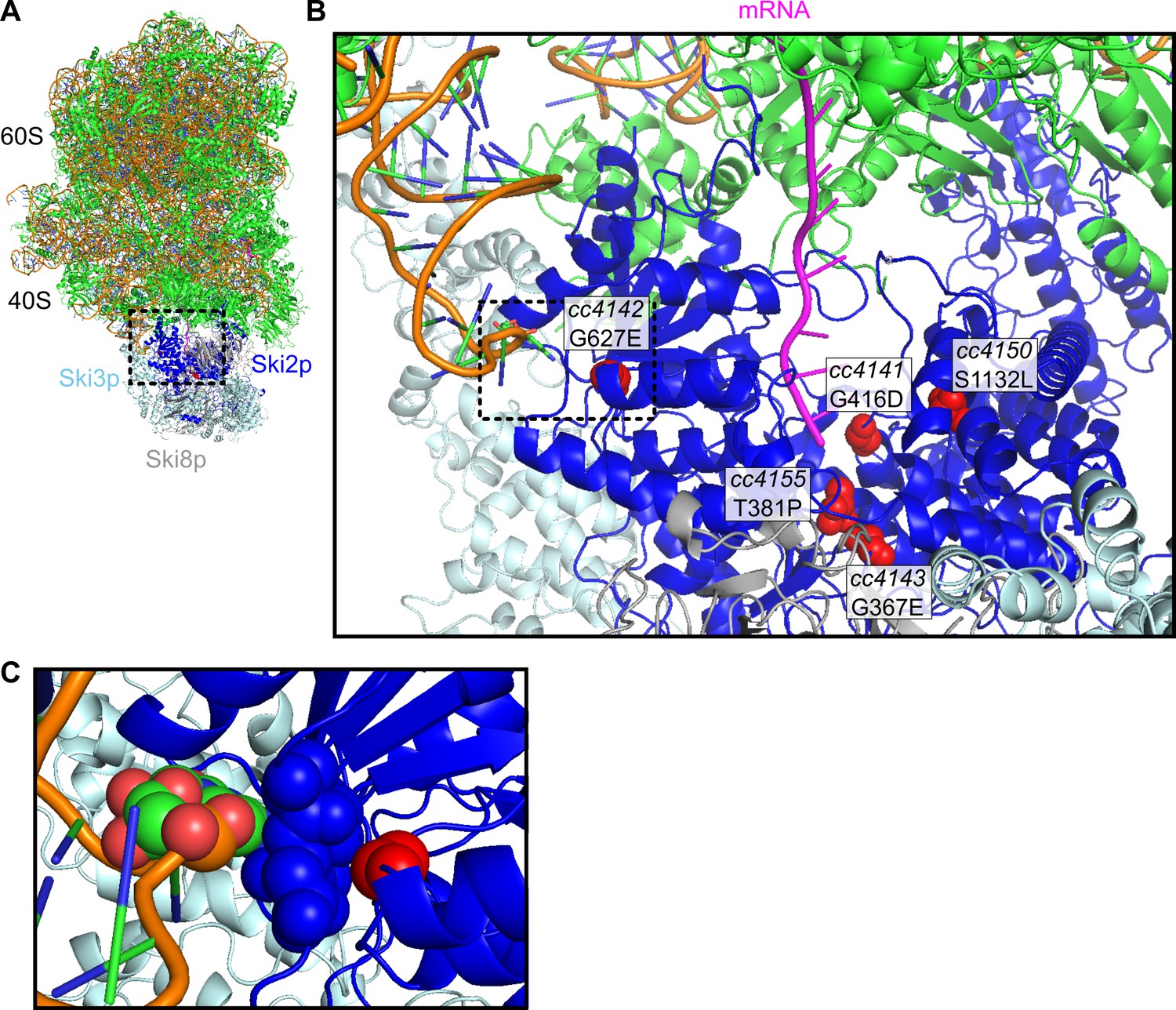
Location of missense alleles on SKI-80S structure.
(A) Cryo-EM structure of S. cerevisiae SKI-80S complex, as reported in Schmidt et al. (2016). Ribosomal proteins in green, and rRNA backbone in orange. Ski2p is in blue, with residues homologous to identified missense residues in C. elegans highlighted in red. (B) Zoom in of Ski2p-ribosome-mRNA interface. One monomer of Ski8p and part of Ski2p were hidden to enable viewing of homologous residues around the mRNA (magenta). (C) A single residue, homologous to the cc4142 site, was close to the helix 16 of the small subunit rRNA.
Figure 2—figure supplement 3

Multiple sequence alignment of C.elegans PELO-1 and sequence homologs.
Sequences of dom34/pelota homologs were M. musculus (AAH57160.1), D. melanogaster (NP_476982.1), H. sapiens (BAG51633.1), Aeropyrum pernix (3WXM_B), C. elegans (R74.6/PELO-1), and S. cerevisiae (Dom34p). Multiple sequence alignment generated with Clustal Omega, and residues highlighted in blue/red indicate conserved and/or functional residues based on prior studies (Passos et al., 2009; Kobayashi et al., 2010).
Figure 3 with 1 supplement
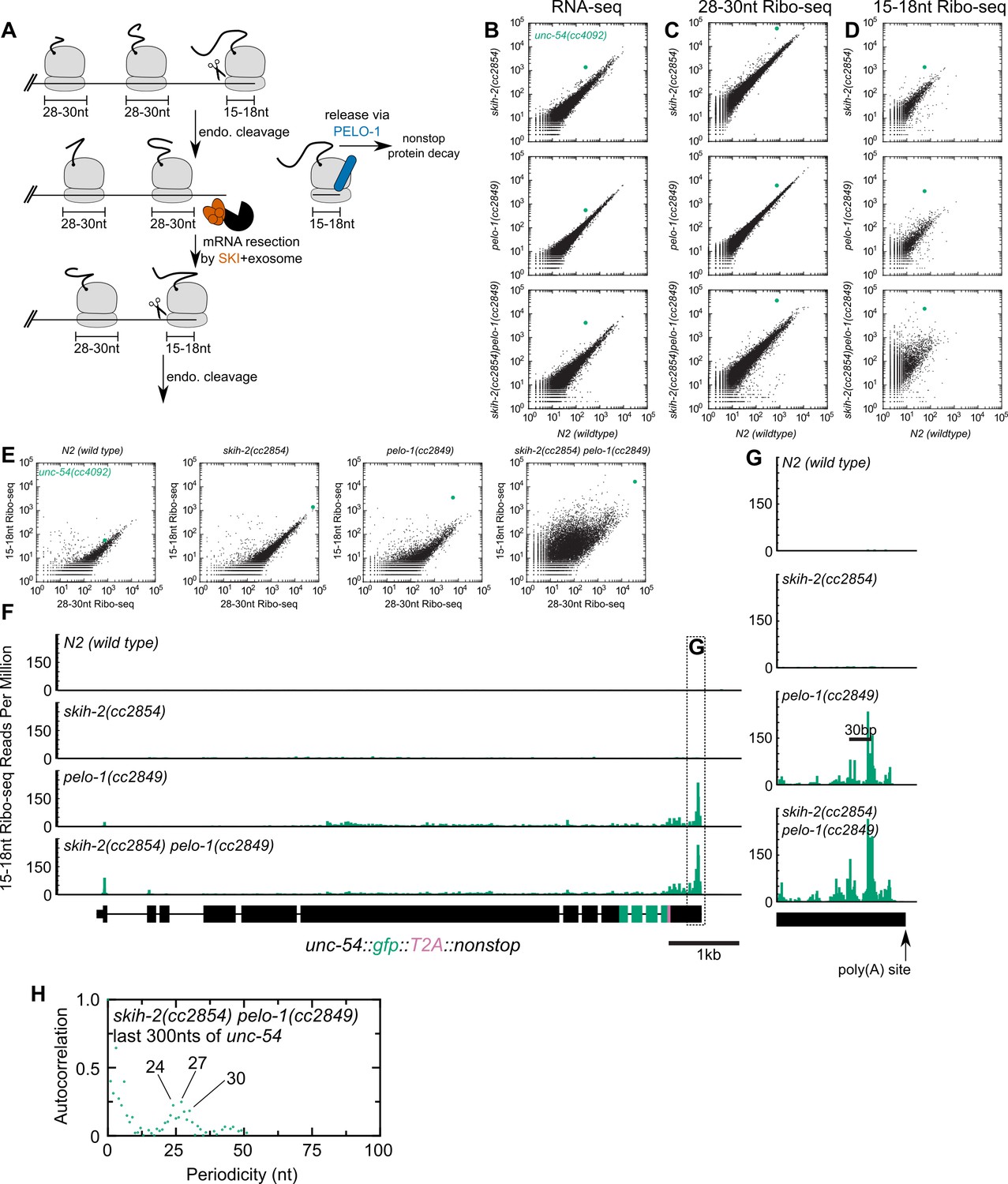
Analysis of gene expression during nonstop mRNA decay.
(A) Model for nonstop decay, as described in the text. Briefly, ribosomes elongate to the end of an mRNA with no stop codons. This triggers endonucleolytic cleavage at the 5’edge of the ribosome by an unknown endonuclease. The resulting 3’end is a substrate for SKI (red) and the 3’>5’ exosome, while the downstream ribosome is subjected to PELO-1-dependent rescue (blue) and nonstop protein decay. An upstream ribosome elongates to the 3’edge of the mRNA and the process repeats itself. Three ambiguities were difficult to represent in the model: (i) SKI can bind the 40S and the 3’>5’ exosome and the order of binding events is not clear. (ii) It is unclear whether PELO-1 acts before or after the first endonucleolytic cleavage. (iii) It is unclear whether endonucleolytic cleavages occur successively, or simultaneously (e.g. cleavage may occur only once multiple ribosomes stall at the 3’end). (B) RNA-seq to quantify transcript levels. Each mutant strain is on the y-axis, and wild type is on the x-axis. Each dot represents read counts for a different gene, with unc-54(cc4092) locus highlighted (green). (C) Same as (B) for 28-30nt ribosome footprints. (D) Same as (B) for 15-18nt ribosome footprints. (E) Scatter plots of short (15-18nt) versus normal length (28-30nt) ribosome footprints for each strain background. This is an alternative way of displaying the data from (C) and (D). (F) Gene plot diagram of 15-18nt Ribo-seq reads mapping to unc-54(cc4092), displayed according to where the 5’end of a read maps. Read counts are displayed per million uniquely-mapping reads. Dotted line shows area of focus in (G). (H) Pearson’s autocorrelation of 15-18nt Ribo-seq reads mapping within 300nts upstream of the poly(A) site of unc-54(cc4092). Similar results were observed with 100 or 200nts upstream of the poly(A) site. A biological replicate of these libraries yielded very similar results.
Figure 3—figure supplement 1
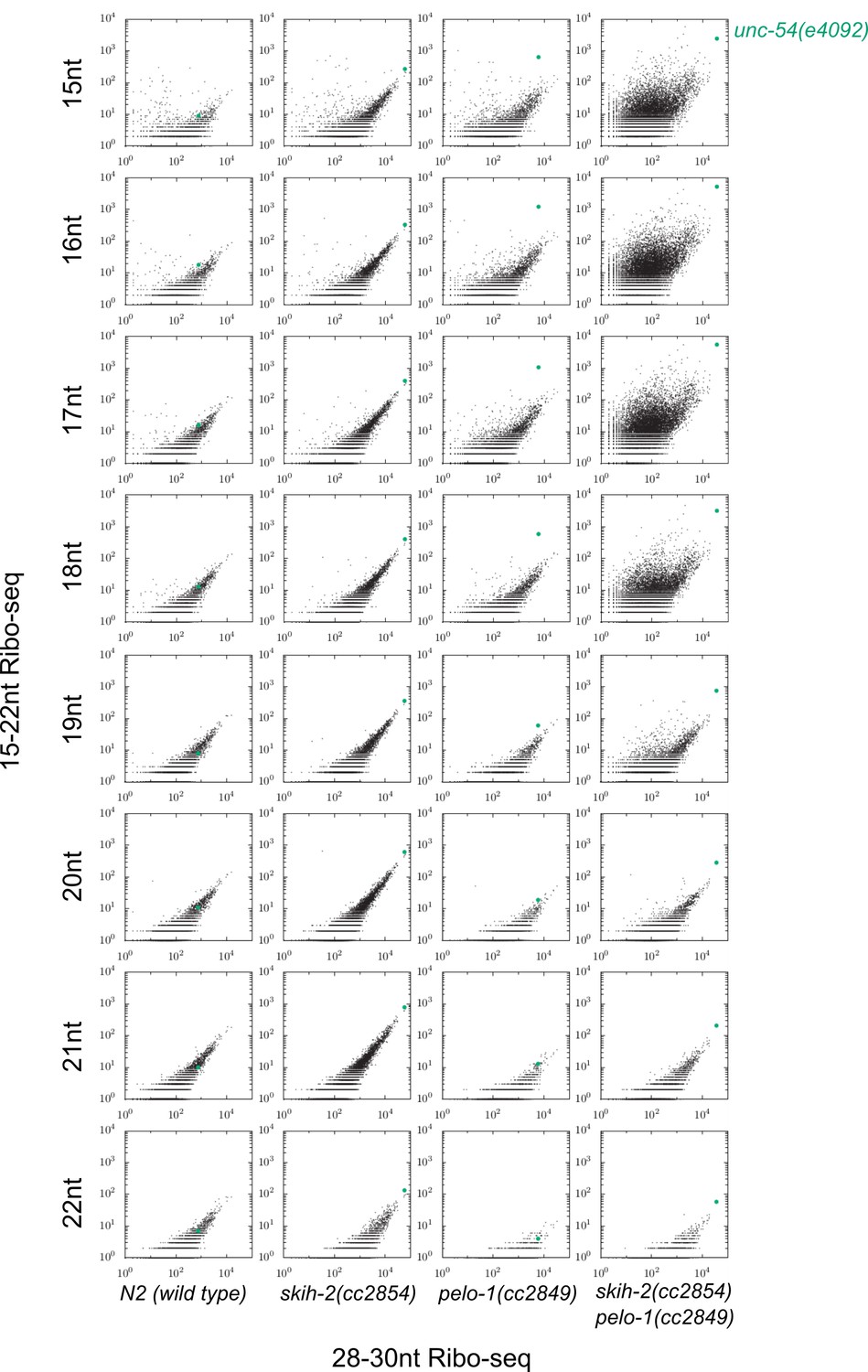
Justification for use of 15-18nt read lengths in Ribo-seq.
Plots as displayed in Figure 3E, with each read length between 15 and 22 as a separate row, and each column a different mutant background. The unc-54(4092) locus exhibited the greatest pelo-1-dependent increase of short ribosome footprints when read lengths were restricted to 15-18nt. A biological replicate yielded similar results, with an even weaker effect for 19nt fragments.
Figure 4 with 1 supplement
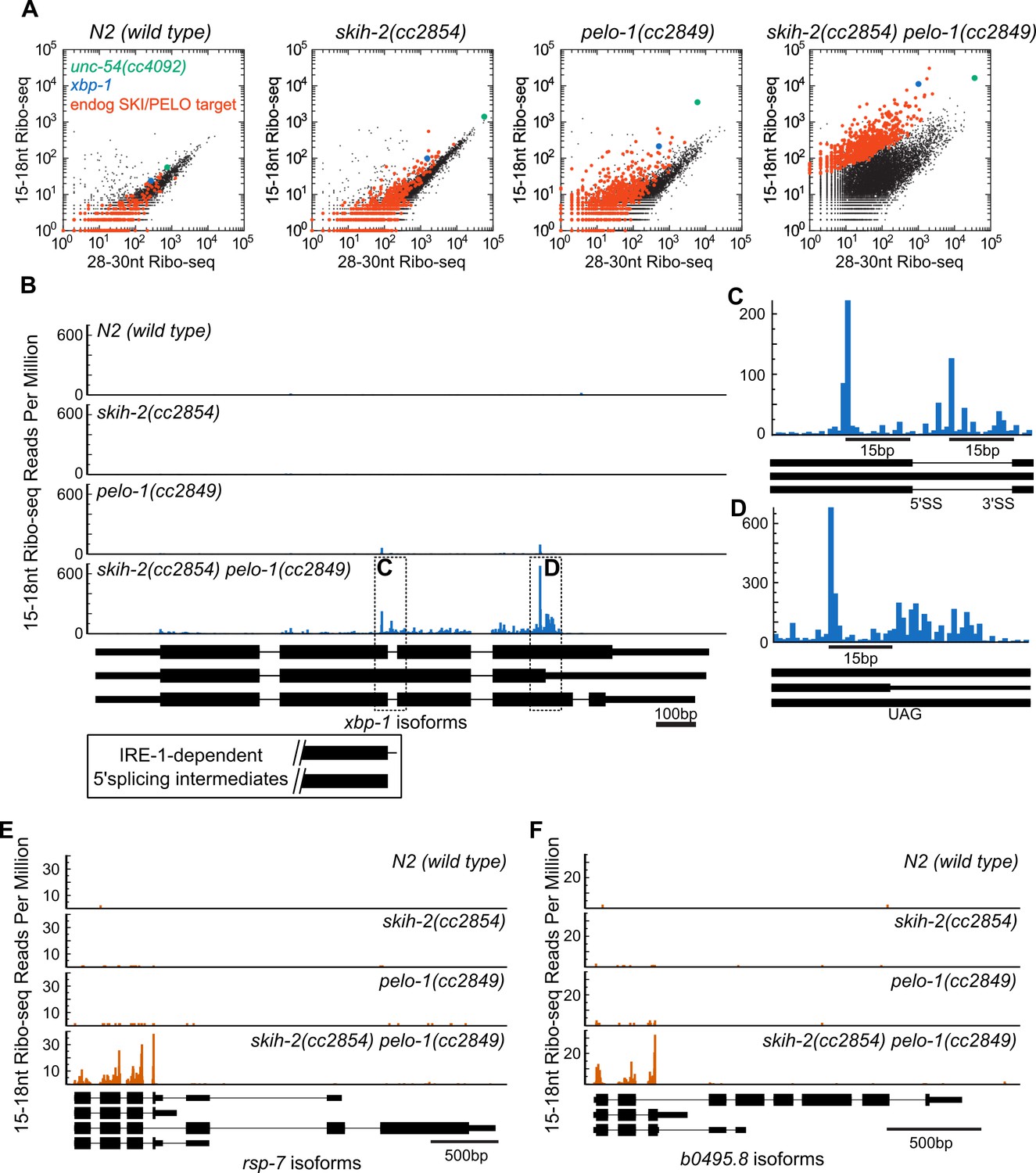
Hundreds of endogenous SKI/PELO targets.
(A) Same data as in Figure 3E, with genes exhibiting consistently elevated 15-18nt Ribo-seq reads highlighted (red). One of these genes is xbp-1 (blue), which is displayed in (B). (B) Gene plot diagram of 15-18nt Ribo-seq reads mapping to xbp-1, with reads displayed according to where their 5’ends map. Annotated xbp-1 isoforms displayed below, and possible IRE-1-dependent splicing intermediates boxed. Read counts normalized to million uniquely mapping reads, as in Figure 3F. Dotted lines show regions of interest in (C) and (D). Note difference in scale in (C). (E) Gene plot diagram for rsp-7. (F) Gene plot diagram for b0495.8.
-
Figure 4—source data 1
Gene-specific p-value for enrichment of skih-2/pelo-1-dependent 15-18nt Ribo-seq reads.
Table of gene name and p-value, where p-value is DESeq-determined enrichment of 15-18nt Ribo-seq reads in a skih-2/pelo-1 mutant relative to 28-30nt Ribo-seq reads in skih-2/pelo-1 and wild type, as well as 15-18nt Ribo-seq reads in wild-type animals. A cutoff of 2.7319e-6 was used to define significant enrichment, that is, endogenous SKI/PELO targets.
- https://doi.org/10.7554/eLife.33292.012
Figure 4—figure supplement 1
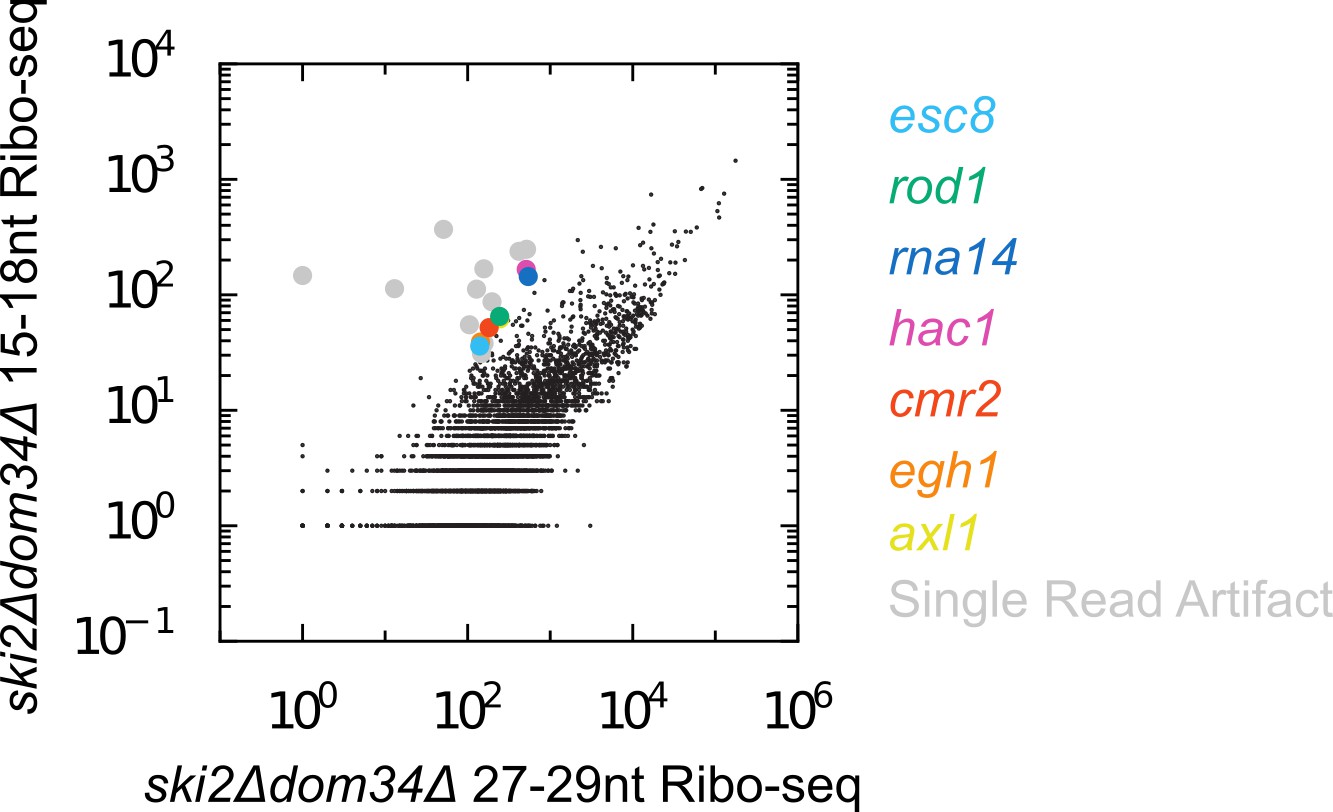
There are few endogenous SKI/PELO targets in S. cerevisiae.
Scatter plot of Ribo-seq gene counts for 15-18nt versus 27-29nt reads. In contrast to C. elegans, there are far fewer mRNAs enriched for 15-18nt Ribo-seq reads relative to longer Ribo-seq reads. We manually inspected the handful of off-diagonal genes. Most genes fell into the category of ‘artifactual’ (grey), where all 15-18nt reads mapping to the gene derived from a single, highly abundant read. Such reads may represent PCR jackpots, and/or may be derived from RNAs absent from the assembled genome sequence. A handful of genes (colored) exhibited a more believable accumulation of 15-18nt Ribo-seq reads. Among these genes is hac1, a previously identified endogenous SKI/PELO target in S. cerevisiae (Guydosh and Green, 2014). Note the size range of reads included on the x-axis (27-29nt) is slightly shorter than what we used in C. elegans (28-30nt). We chose this range because a higher fraction of 27-29nt reads are in frame in S. cerevisiae, whereas 28-30nt reads tend to be in-frame in C. elegans. Changing these size ranges (e.g. using 28-30nt for S. cerevisiae and 27-29nt in C. elegans) did not substantially alter our conclusions.
Figure 5 with 1 supplement

Ribosomes accumulate in the C-terminus and over truncated stop codons in pelo-1.
(A) Metagene plot for all genes (solid lines), focusing on the C-terminus. Reads were assigned according to where their 5’ends map. Endogenous SKI/PELO targets (Figure 4A) are also shown (dotted line). Inset shows zoom of region just upstream of stop codons. (B) Metacodon plots showing where 15-18nt Ribo-seq reads terminate relative to nucleotides of each codon. For each nucleotide of each codon, we counted the number of times a 15-18nt Ribo-seq read terminated at that position (3’ end), and normalized to positions upstream of the codon (see Materials and methods). Sense codons of similar nucleotide composition are shown as controls, and all codons in Figure 5—figure supplement 1.
Figure 5—figure supplement 1
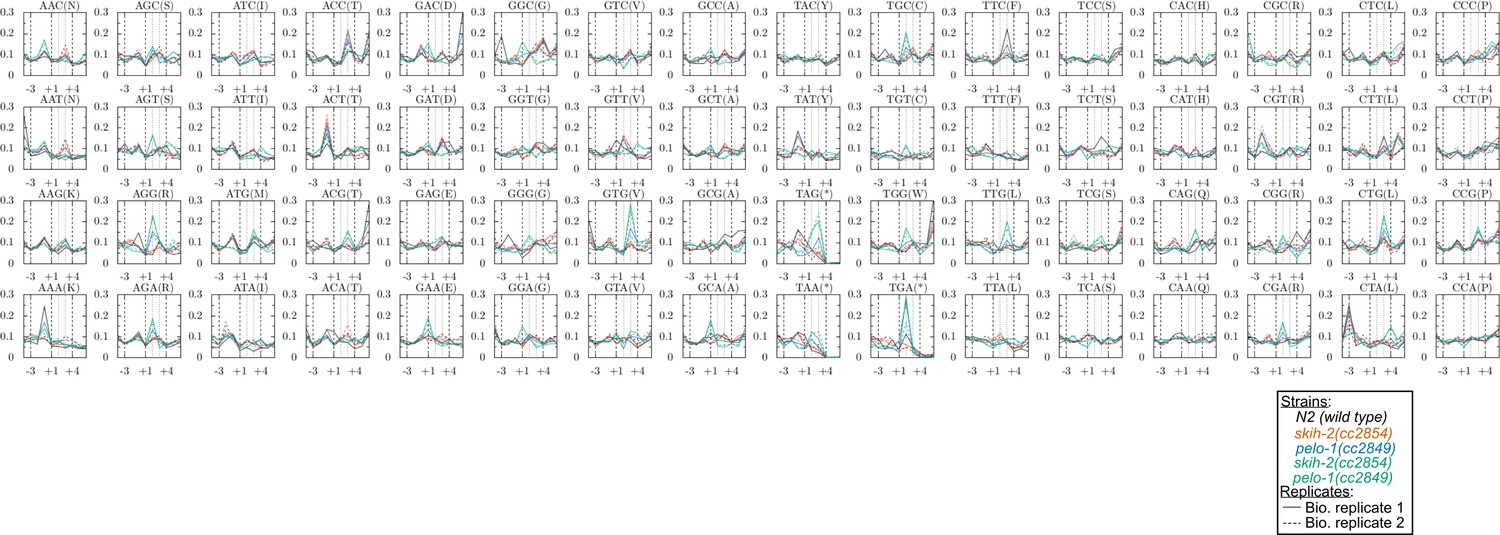
Metacodon plot of 3’end locations for all codons.
See Figure 5 for description.
Figure 6 with 1 supplement

Nonsense decay generates nonstop substrates.
(A) Gene plot diagram showing reads mapping to unc-54(e1092), with position of e1092 early stop codon indicated. Read counts are averaged over the entire sequence overlapping a read (not just the 5’end). All data in this figure were in the skih-2(cc2854) pelo-1(cc2849) double mutant background, abbreviated skih-2 pelo-1. (B) Zoom in of region upstream of e1092. Reads are displayed according to where their 5’ends map. (C) Genome-wide counts of Ribo-seq reads with and without smg-1(e1228), with indicated genes highlighted. ‘Endog. SKI/PELO’ are the same genes identified in Figure 4A. (D) Genome-wide distribution of 15-18nt Ribo-seq reads with and without smg-1(e1228). Solid lines are all genes, and dotted lines are endogenous SKI/PELO targets, as identified in Figure 4A. Purple is the smg-1(+) strain, black is smg-1(e1228). (E) X-axis shows the total number of 15-18nt Ribo-seq reads in a skih-2/pelo-1 double mutant with and without smg-1(e1228). Y-axis shows the fraction of those reads that come from the smg-1(e1228) mutant. Endogenous SKI/PELO targets are highlighted (red), and endogenous SKI/PELO targets with >95% of 15-18nt Ribo-seq reads derived from the smg-1(+) strain are highlighted in orange. (F) Log2 fold change in total mRNA levels as assayed by RNA-seq for genes shown in Figure 1E, using published datasets from Muir et al., 2018. Genes highlighted in blue are the 12 endogenous NMD targets identified in (Barberan-Soler et al., 2009) and described in the text. p-Value from Kolmogorov–Smirnov test relative to all genes (black line). (G) Distribution of 15-18nt Ribo-seq reads at xbp-1 locus with and without smg-1(e1228). (H) Brood size of skih-2(cc2854) pelo-1(cc2849) with and without smg-1(e1228). Brood size analysis performed as in Figure 2D and described in Materials and methods. p-Value from Mann Whitney U test. The skih-2/pelo-1 double mutant was backcrossed to the smg-1(e1228) background as a control, and smg-1(+) homozygotes made by segregating away the smg-1(e1228) allele.
Figure 6—figure supplement 1
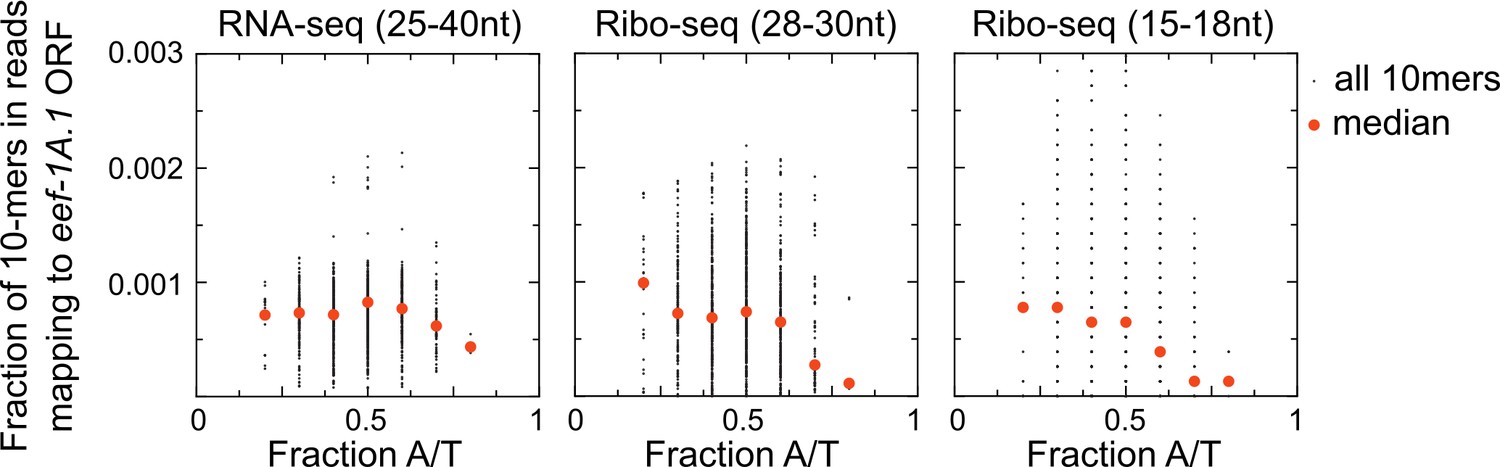
Bias against capturing A/T-rich RNAs in small RNA sequencing libraries.
Scatter plots show coverage of 10 nucleotide sequences for reads mapping to one highly expressed open reading frame (eef-1A.1 ORF) for each of three different size (and type) of library preparations. An unbiased RNA fragment capture protocol would yield approximately uniform coverage of all 10-mer sequences across an mRNA. We instead observe low coverage of A/T-rich sequences, especially in Ribo-seq libraries. This is one reason why we may have recovered few examples of untemplated adenosines in our skih-2/pelo-1 mutant libraries. Libraries shown are from the skih-2/pelo-1 mutant. Similar results were observed with other libraries, other genes, and different size kmers.
Additional files
-
Supplementary file 1
Oligos, Worm strains, and Plasmids used in this study
Each table is a separate sheet; reagents available upon request.
- https://doi.org/10.7554/eLife.33292.017
-
Transparent reporting form
- https://doi.org/10.7554/eLife.33292.018
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Nonsense mRNA suppression via nonstop decay
eLife 7:e33292.
https://doi.org/10.7554/eLife.33292




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}