Ribozyme-catalysed RNA synthesis using triplet building blocks
- Cambridge Biomedical Campus, United Kingdom
Figures
Figure 1 with 2 supplements
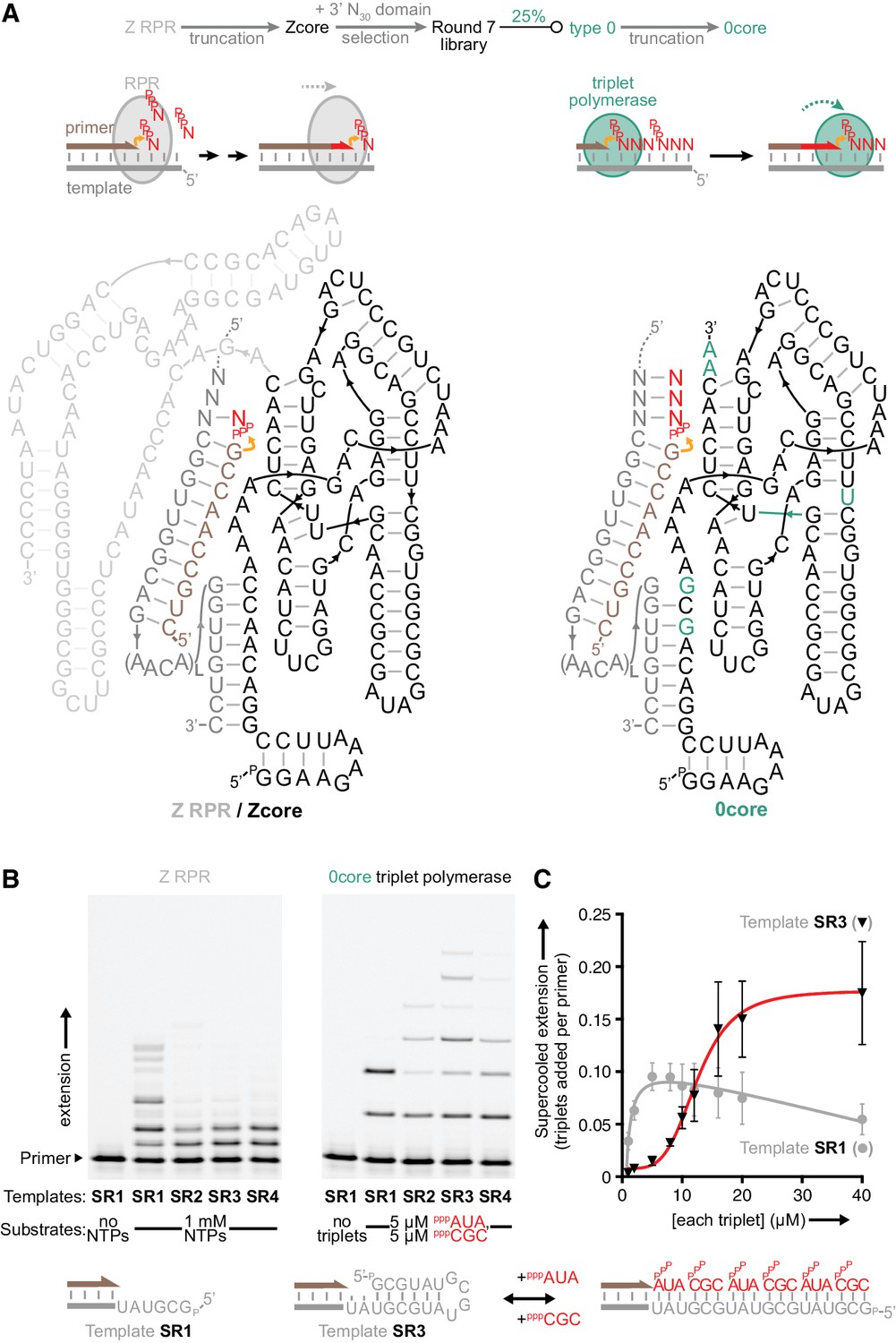
Monomer polymerisation and triplet polymerisation.
(A) Scheme outlining initial derivation of a triplet polymerase activity from a mononucleotide polymerase ribozyme via directed evolution. Z RPR truncation effects are shown in Figure 1—figure supplement 1, the selection cycle is outlined in Figure 1—figure supplement 2, and the selection conditions of rounds 1–7 are listed in Figure 1—source data 1. Below, modes of action and secondary structures of the mononucleotide polymerase ribozyme (Z RPR) and a triplet polymerase ribozyme (0core), both depicted surrounding primer (tan)/template (grey) duplexes with a mononucleoside triphosphate (NTP) or trinucleotide triphosphate (triplet) substrate present (red). Here, the templates are hybridised to the ribozyme upstream of the primer binding site, flexibly tethered to enhance local concentration and activity (via L repeats of an AACA sequence, for example L = 5 in templates SR1-4 below). Z RPR residues comprising its catalytic core (Zcore) are black; mutations in 0core arising from directed evolution of Zcore are in teal. (B) Primer extension by the Z RPR using monomers (1 mM NTPs) or by 0core using triplets (5 μM pppAUA and pppCGC), on a series of 6-nucleotide repeat templates (SR1-4, examples below) with escalating secondary structure potential that quenches Z RPR activity beyond the shortest template SR1 (−7˚C ice 17 days, 0.5 μM/RNA). Extension by the triplet polymerase ribozyme 0core can overcome these structure tendencies up to the longest template SR4. (C) Triplet concentration dependence of extension using templates SR1 (grey circles) and SR3 (black triangles) by 0core (pppAUA and pppCGC, 0.1 μM of primer A10, template and ribozyme, −7˚C supercooled 15 days, ± s.d., n = 3); shown below is a model of cooperative triplet-mediated unfolding of template SR3 structure to explain the sigmoidal triplet concentration dependence (red curve) of extension upon it. Numerical values are supplied in Figure 1—source data 2.
-
Figure 1—source data 1
Selection conditions of rounds 1–7.
- https://doi.org/10.7554/eLife.35255.006
-
Figure 1—source data 2
Triplet concentration-dependent extension values.
- https://doi.org/10.7554/eLife.35255.007
Figure 1—figure supplement 1
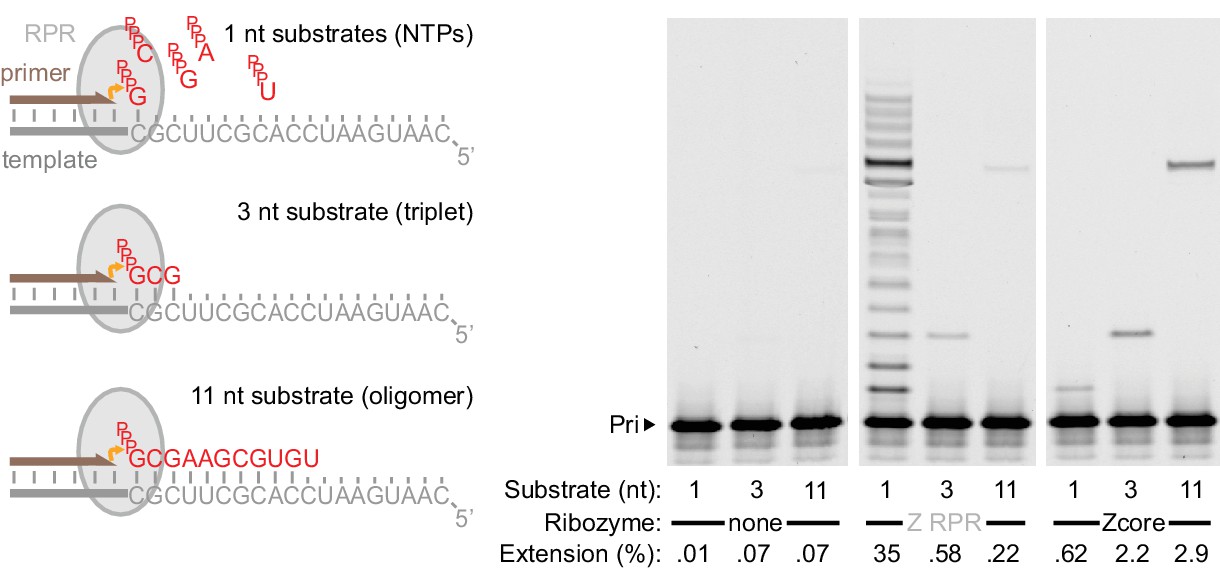
Templated ligase activity from a mononucleotide polymerase.
Extension of primer A11 on template HTI by the Z RPR (Wochner et al., 2011) and a 3’ truncation of it (Zcore, Figure 1a) at −7˚C in ice for 8 days (0.5 μM of each RNA), adding as substrates either a long oligonucleotide (‘11 nt’, pppGCGAAGCGUGU at 0.5 μM), a triplet (‘3 nt’, pppGCG at 5 μM), or NTPs (‘1 nt’, at 1 mM each). Gel densitometry was used to calculate the percentage of primer extended (shown below each lane). Z RPR has only a very limited template ligation activity (slightly exceeding background nonenzymatic ligation [Rohatgi et al., 1996]). In contrast, 3’ truncation (which abolished mononucleotide polymerase activity) accommodated and enhanced templated ligation.
Figure 1—figure supplement 2
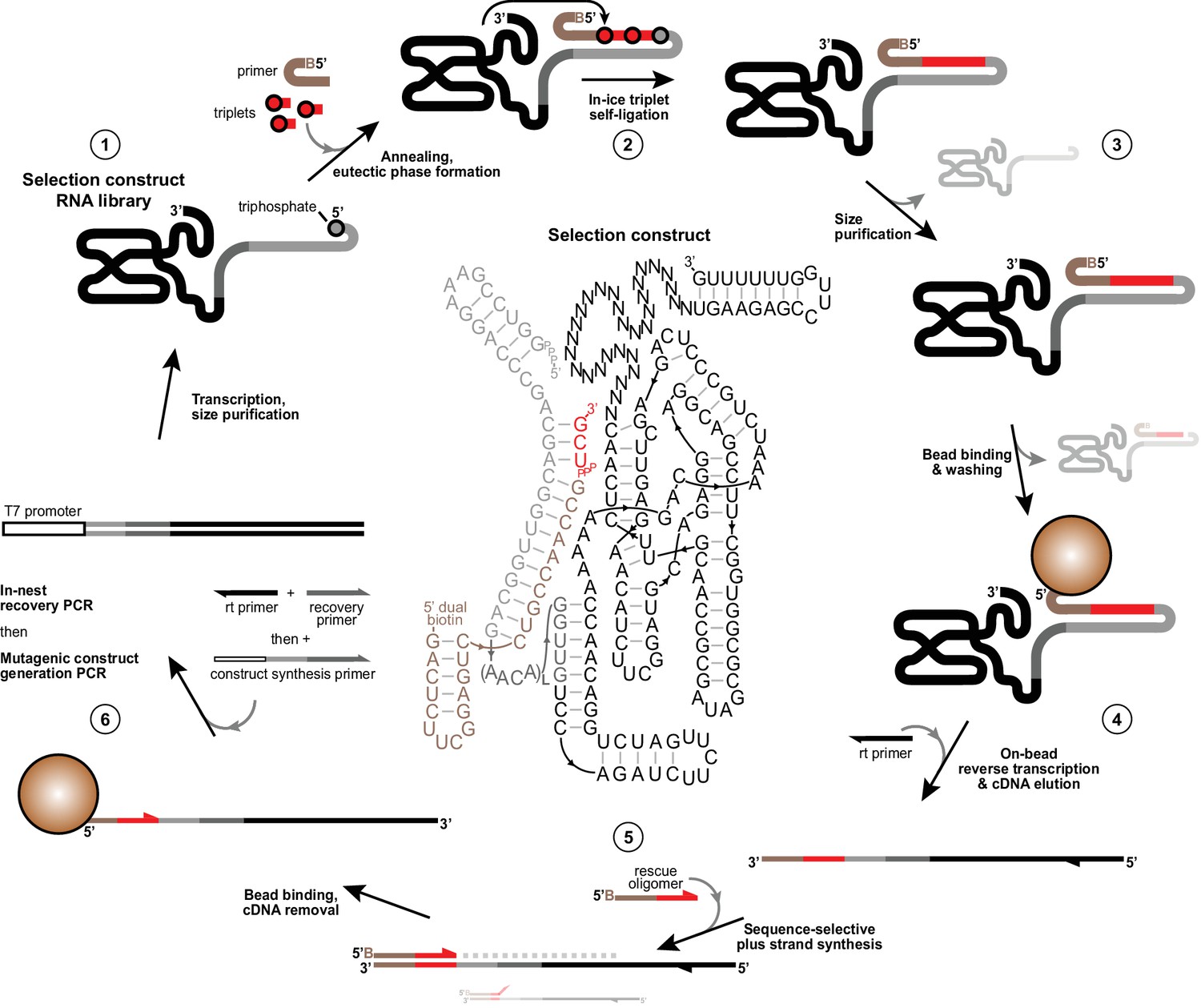
Selection scheme for in vitro evolution of triplet polymerase activity.
Illustration of the in vitro evolution strategy to enrich iterative templated ligase activity from libraries of selection constructs. Centre, an example selection construct is shown comprising Zcore with a 3’ 30 nt random domain followed by a fixed hairpin-forming reverse transcription site (1). This construct is flexibly tethered at its 5’ in cis (Tagami et al., 2017; Attwater et al., 2010) (via L repeats (3-8) of a flexible AACA linker) to a template region (grey) bound to 5’-dual biotin modified primer (tan). This construct requires successive polymerisation of two pppUCG triplet substrates (red) in −7˚C ice followed by self-ligation to its triphosphorylated 5’ terminus to attach itself to the biotinylated primer (2). Active triplet polymerases are then identified and size-purified by denaturing PAGE to deplete unreacted ribozymes and primer (3), then captured on streptavidin beads and subjected to a denaturing wash to ensure covalent linkage to primer. Remaining dual-purified bead-linked ribozymes were then reverse transcribed (4). cDNAs were eluted with NaOH and neutralised, before acting as templates for ‘+’ strand DNA synthesis from 5’ biotinylated ‘rescue’ oligonucleotides that only prime correctly on the complete synthesised sequence (5), ensuring that constructs that deviated from the specified templated triplet polymerization fail to prime on cDNA preventing their ‘+’ strand synthesis. ‘+’ strands were then captured on fresh streptavidin beads, stripped of cDNAs, reamplified and diversified to generate selection construct for a new round of selection (6). See Materials and Methods for protocol details and Figure 1—source data 1, Figure 2—source data 1, Figure 4—source data 1 and Supplementary file 3 for oligonucleotides used in selections.
Figure 2 with 2 supplements
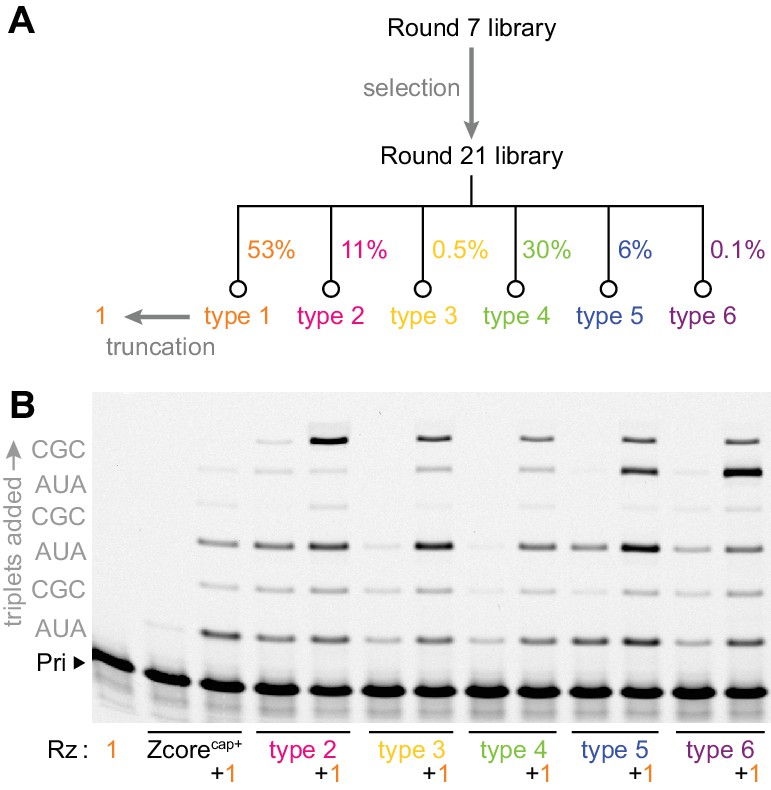
Emergence of cooperativity during in vitro evolution.
(A) Composition of the round 21 selection pool as a % of total pool sequences; selection conditions of rounds 8–21 are listed in Figure 2—source data 1. Secondary structures of ribozyme type 1–6 archetypes are shown in Figure 2—figure supplement 1, and comparison of their activities to that of the polyclonal selection pool is shown in Figure 2—figure supplement 2. (B) Primer extension with triplets by these emergent triplet polymerase ribozyme types 2–6 (‘Rz’, alongside the starting Zcore ribozyme with ‘cap+’ sequence from selection, see Figure 3a), alone or with added truncated type 1 (+1, see Figure 3a) which boosted their triplet polymerase activities (0.5 μM Rzs/A10 primer/SR3 template, 5 μM pppAUA and pppCGC, −7˚C ice 16 hr).
-
Figure 2—source data 1
Selection conditions of rounds 8–21.
- https://doi.org/10.7554/eLife.35255.011
Figure 2—figure supplement 1
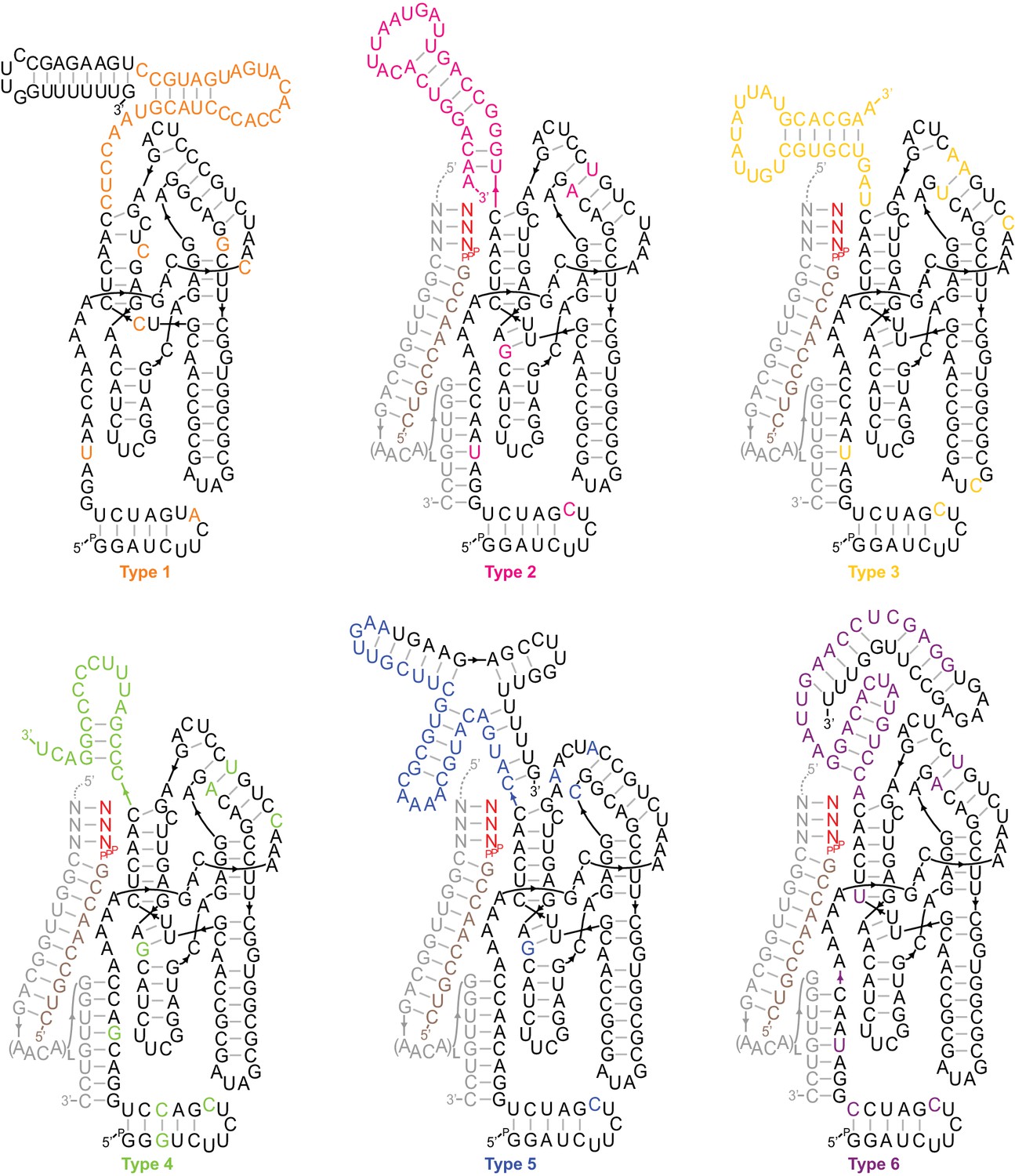
Secondary structures of type 1–6 ribozymes.
The archetypes shown are active truncations of the most common/enriched variants of each type. Types 2–6 are depicted hybridised to a flexibly tethered primer (tan)/template (grey)/substrate (red) duplex analogous to the selection construct (Figure 1—figure supplement 2), supporting extension on the SR1-4 templates (Figure 2b). Coloured bases deviate from Zcore or derive from the random sequence region. Type 1, the most common type in the output, displayed a high degree of sporadic mutation in the catalytic core domain, consistent with a loss of catalytic function. Shown here are the four most common core mutations, which were each present in >45% of type 1 sequences.
Figure 2—figure supplement 2

Clonal versus polyclonal activity.
Type 1 and 2 RNAs, and the indicated polyclonal ribozyme pools late in the selection, were transcribed as selection constructs generated with Tri8AUAM (then annealed to primer A10). Their abilities to incorporate eight pppAUA triplets then self-ligate in this selection construct context were compared (left, 0.1 μM each RNA, 2 μM pppAUA, in −7˚C ice for 5 days; right, 0.2 μM each RNA, 2 μM pppAUA and pppAUG3’d, in −7˚C ice for 9 days). Left: Despite being the most active type (Figure 2b), type two activity fell far short of the round 19 pool polyclonal activity. Right: The incorporation of wobble pairing 3’-deoxy ‘terminator’ triplet pppAUG3’d (see results section on fidelity, yielding a faster-migrating band than incorporation of the cognate pppAUA triplet) was decreased sharply after round 19, as calculated by densitometry and averaging amongst the ligation junctions in each lane, indicative of enrichment of higher fidelity triplet polymerases in the selection pool.
Figure 3 with 2 supplements
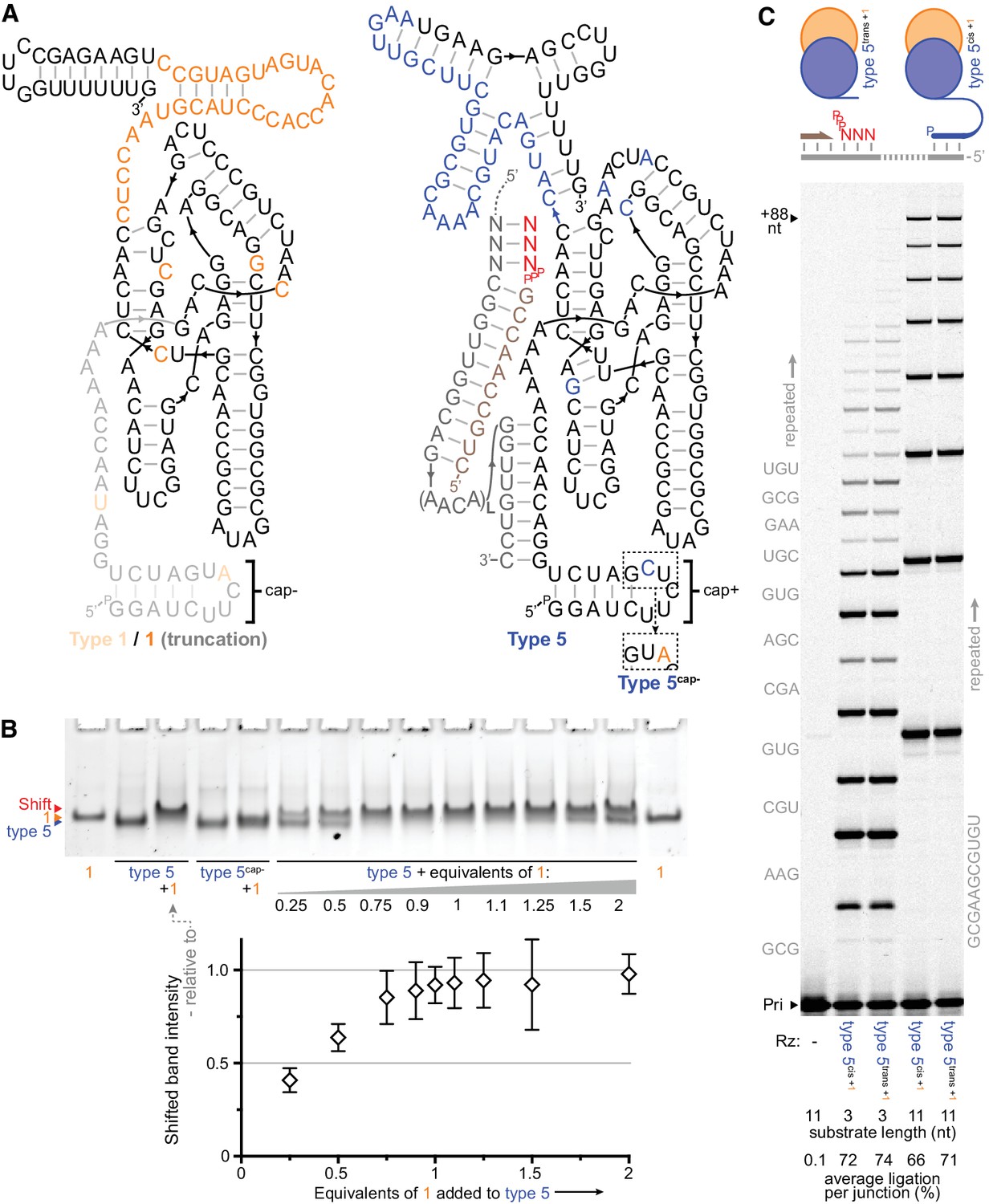
Heterodimer formation and behaviour.
(A) Secondary structures of the most common type 1 and type 5 clones from the selection, with in vitro-selected 3’ domain and core mutations coloured orange (type 1) or blue (type 5). 5’ truncation of type 1 (faded, including its putative primer/template interacting region), yielding the minimal type 1 variant ‘1’, maintained its enhancement activity (see Figure 3—figure supplement 1). The effects of transplanting the indicated 5’ hairpin ‘cap+’ element from type 5 to other ribozymes are shown in Figure 3—figure supplement 2. The inset shows type 5 ‘cap+’ hairpin element alteration to ‘cap–’ (yielding type 5cap-). (B) Gel mobility shift characteristic of complex formation resulting from mixing of type 5 ribozyme with type 1 RNA (equimolar, or with the indicated equivalents). Type 5cap- loses this shift and its susceptibility to type 1 activity enhancement (Figure 3—figure supplement 1). Below, shifted band intensities with increasing type 1 addition are plotted (quantified relative to the indicated type 5+1 lane intensities, n = 4 ± s.d.), signifying 1:1 heterodimer formation; numerical values are supplied in Figure 3—source data 1. (C) Type 1 enhancement allows type 5 variants to synthesise long RNAs using triphosphorylated oligonucleotide (11 nt) or short triplet (3 nt) substrates (Sub, 3.6 or 5 μM each, substrate sequences in grey beside lanes; 0.4 μM primer A11/template I-8, 2 μM each Rz, −7˚C ice for 16 days). This activity is independent of template tethering (Wochner et al., 2011), as comparable synthesis is achieved by versions of type 5 whose 5’ regions allow or avoid hybridisation to the template (type 5cis or type 5trans respectively, schematic above, sequences in Supplementary file 1). The average extent of ligation at the end of the reaction amongst all junctions in a lane is shown beneath each lane.
-
Figure 3—source data 1
Relative intensities of shifted bands when varying type 1 equivalents.
- https://doi.org/10.7554/eLife.35255.015
Figure 3—figure supplement 1

Parameters of type 1 activity enhancement.
(A) PAGE of primer extension reactions comprising combinations of type 5 and type 1 variants (0.2 μM primer A10, template SR3, and type 5 variant, here annealed together and combined with 0.2 μM of separately-annealed type 1 variant after buffer addition; −7˚C in ice for 63 hr, 2 μM each pppAUA, pppAUG3’d, pppCGC). Full-length type 1 enhanced the activity of type 5 as did 5’ truncated type 1 (‘1’, Figure 3a) without its duplex-interacting A-minor motif (Shechner et al., 2009) and its ‘cap−’ sequence (originally present on some type 1 sequences in the selection). Replacing the type 5 ribozyme’s selected 5’ ‘cap+’ sequence with ‘cap-’ (type 5cap-) abolished its enhancement by type 1 RNA. (B) Stoichiometry of type 1 enhancement of type 3 activity (0.5 μM primer A10, template SR3, 2 μM each pppAUA, pppGUA3’d, pppCGC in −7˚C ice for 20 hr); quantification of average extension per primer in each lane is consistent with the 1:1 stoichiometry implied by type 1’s ~ 50% abundance in the round 21 selection pool.
Figure 3—figure supplement 2
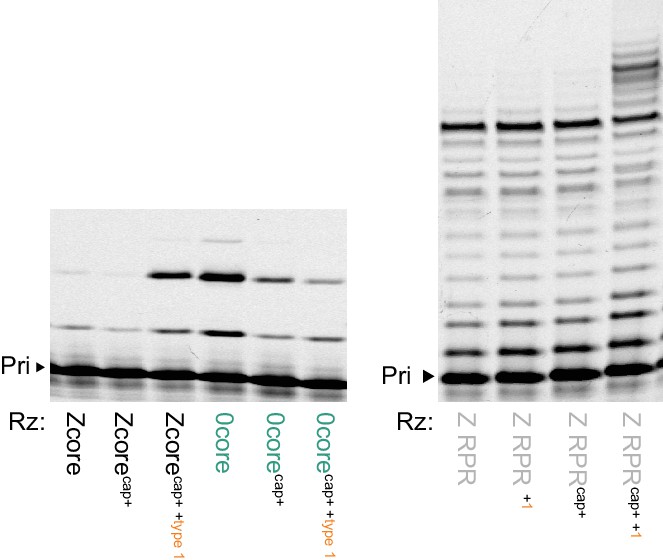
Type 1 enhancement of parental ribozymes.
Left, transplanting the conserved 5’ ‘cap+’ sequence from types 2–6 allows the Zcore but not the 0core ribozyme triplet polymerase activity to be enhanced by type 1; shown are PAGE of extensions of primer A10 on template CCCMisAUG by the indicated ribozyme cores (5 μM of pppAUG and pppCCC, 0.5 μM of each RNA with type 1 annealed and added separately, −7˚C in ice for 22 hr). Right, RNA polymerase activity of Z RPR using NTPs is enhanced by type 1 addition, but only when modified with the 5’ ‘cap+’ sequence (Z RPRcap+), extending primer A10 on tethered template HTI (0.5 μM of each RNA, 4 mM of each NTP, at 4˚C for 68 hr).
Figure 4 with 1 supplement
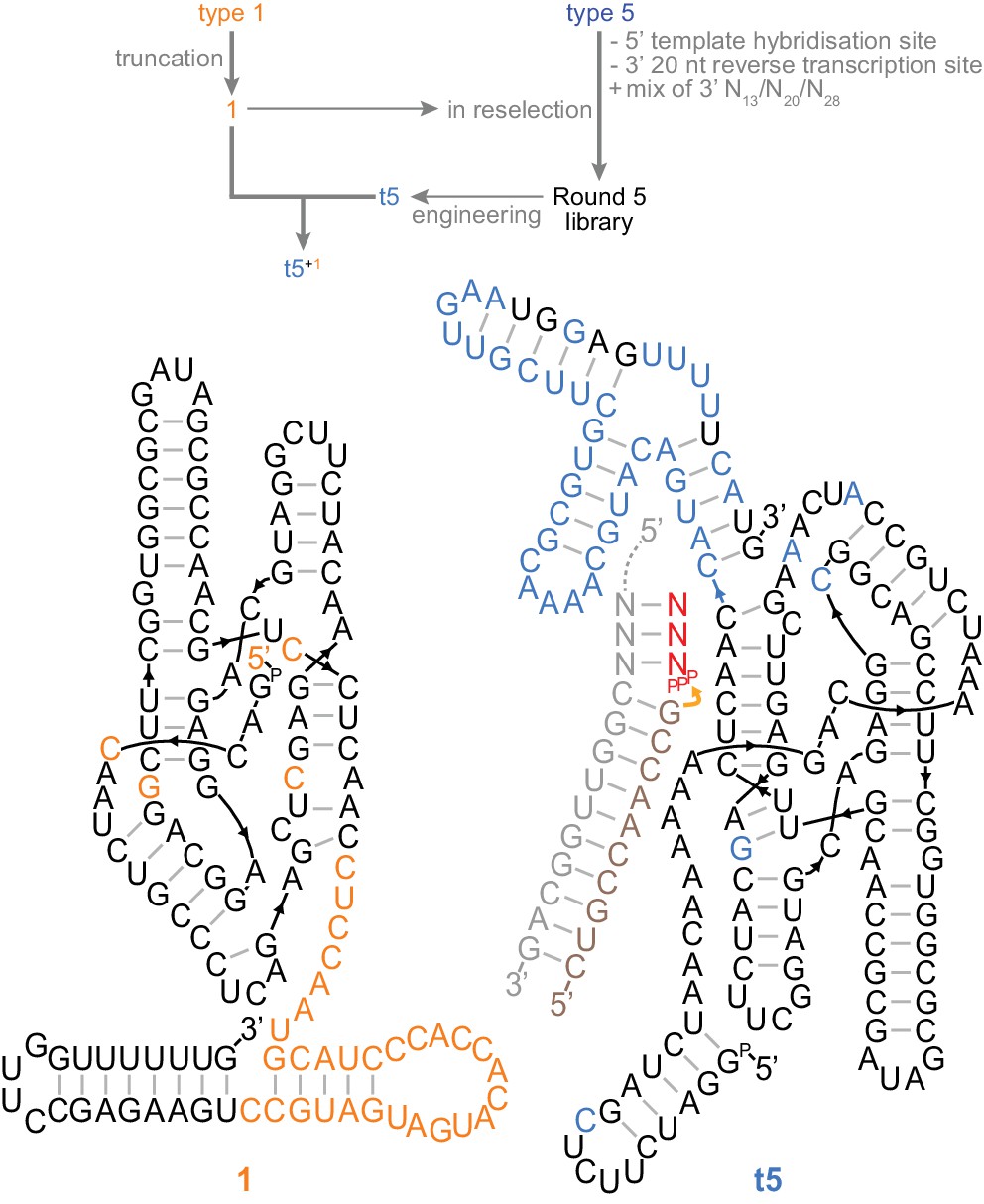
A trans-acting heterodimeric triplet polymerase.
Top, scheme outlining derivation of the final t5+1 triplet polymerase archetype from the type 1 and type 5 RNAs (shown in Figure 3a) by reselection using the conditions in Figure 4—source data 1. Below, the secondary structure of this ribozyme heterodimer, 135 nt (1) and 153 nt (t5) long, is depicted operating in trans on a non-tethered primer/template duplex. Type 5 3’ domain bases that re-emerged after randomisation during reselection are coloured black in the t5 3’ domain. Ribozyme development is summarized in Figure 4—figure supplement 1; all ribozyme sequences are listed in Supplementary file 1.
-
Figure 4—source data 1
Selection conditions of rounds 1–5 of the reselection.
- https://doi.org/10.7554/eLife.35255.017
Figure 4—figure supplement 1
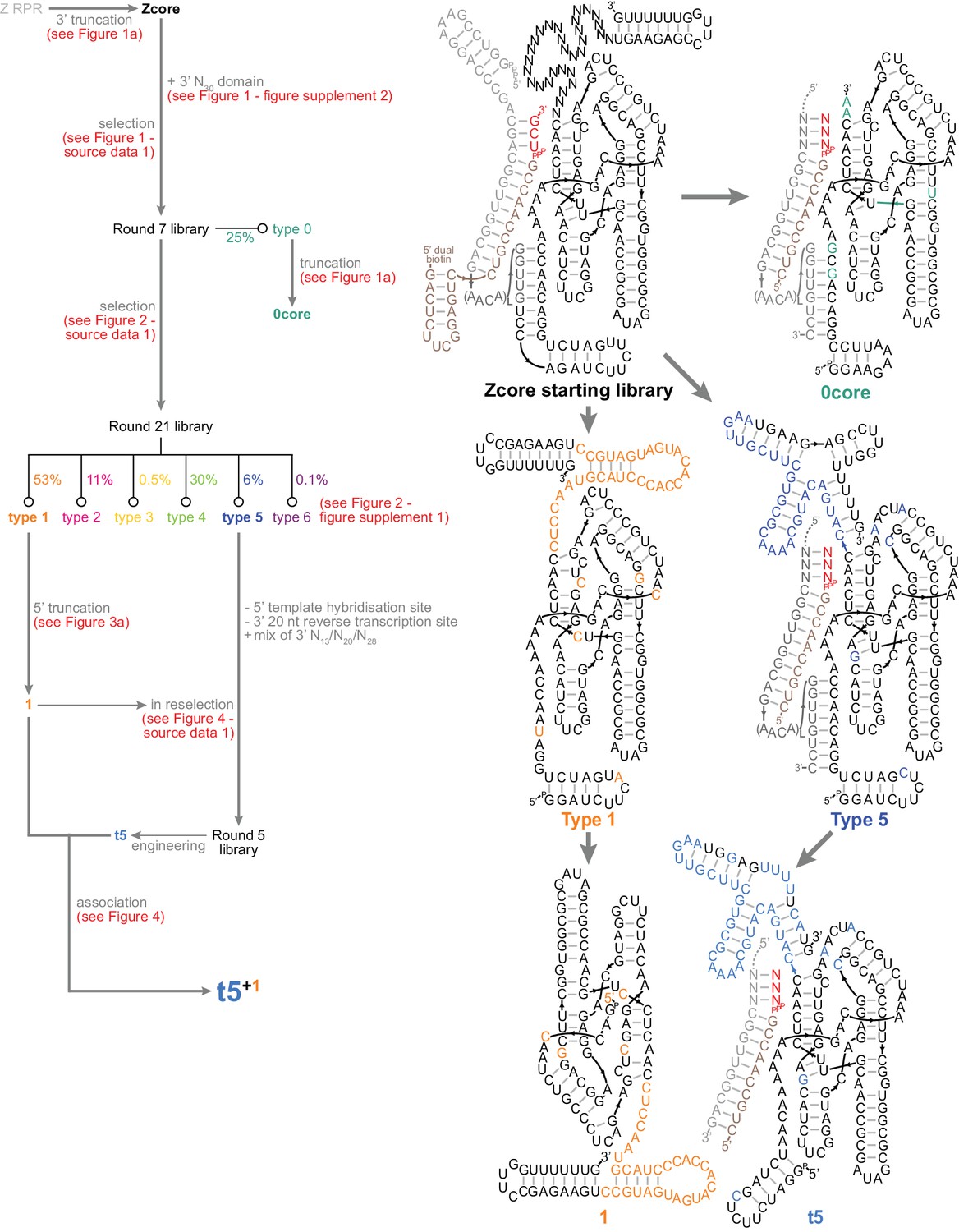
Summary of ribozyme development in this work.
Left, map of triplet polymerase selection and engineering. Ribozyme sequences are listed in Supplementary file 1. Right, secondary structures of the central ribozymes in this work.
Figure 5 with 2 supplements
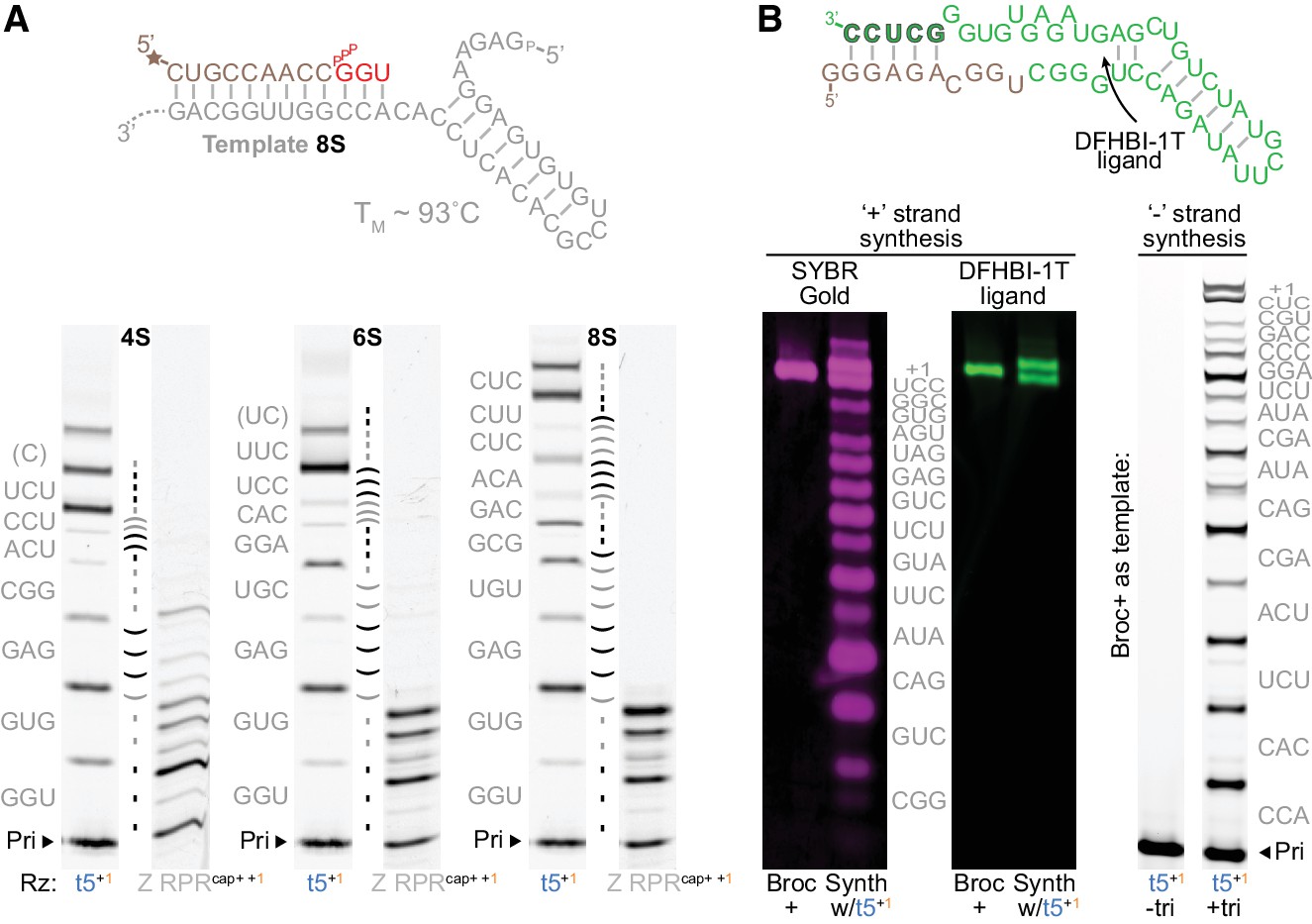
Triplet-mediated structured and functional template copying.
(A) Extension on three structured hairpin templates (4S, 6S, 8S) with increasing stability. Top, the mfold-predicted (Markham and Zuker, 2005) structure and TM of the most stable 8S template; below, primer extensions on these templates by t5+1 triplet polymerase (with 5 μM each encoded triplet) or type 1-enhanced Z polymerase ribozyme (Z RPRcap+ +1, with 1 mM each NTP) (2 μM ribozyme, 0.5 μM 4S, 6S or 8S template and primer A9, −7˚C ice 25 days). The self-complementary region in each template is indicated between each pair of lanes (shaded by triplet), with the encoded triplet substrate sequences at the left (in grey, with 5’ template overhangs in brackets). Syntheses using different substrate compositions and concentrations are shown in Figure 5—figure supplement 1 and Figure 5—figure supplement 2. While all hairpin templates are robustly copied by t5+1, synthesis by the Z RPR is completely arrested by the 6S and 8S hairpins. (B) Synthesis of the broccoli aptamer. The native secondary structure is shown above (Tan: bases from ‘+’ strand synthesis primer. Green: bases from triplets. Outlined green: primer binding site for the ‘−’ strand synthesis). Below left, t5+1-catalysed synthesis of fluorescent broccoli aptamer, run alongside standard (Broc+, synthesized by in vitro transcription), and stained for RNA with SYBR Gold (magenta) or folded with DFHBI-1T ligand (green fluorescence) (2 μM t5+1, 1 μM BBrc10/TBrc, 5 μM each triplet (in grey), −7˚C ice 22 days). Below right, ‘−’ strand synthesis on Broc + standard (0.5 μM without ligand in ribozyme extension buffer, 0.5 μM FBrcb6 primer, 2 μM t5+1, 5 μM each triplet (in grey), −7˚C ice 38 days). t5+1 is able to synthesise both full-length functional (fluorescent) Broccoli ‘+’ and encoding ‘–’ strands.
Figure 5—figure supplement 1
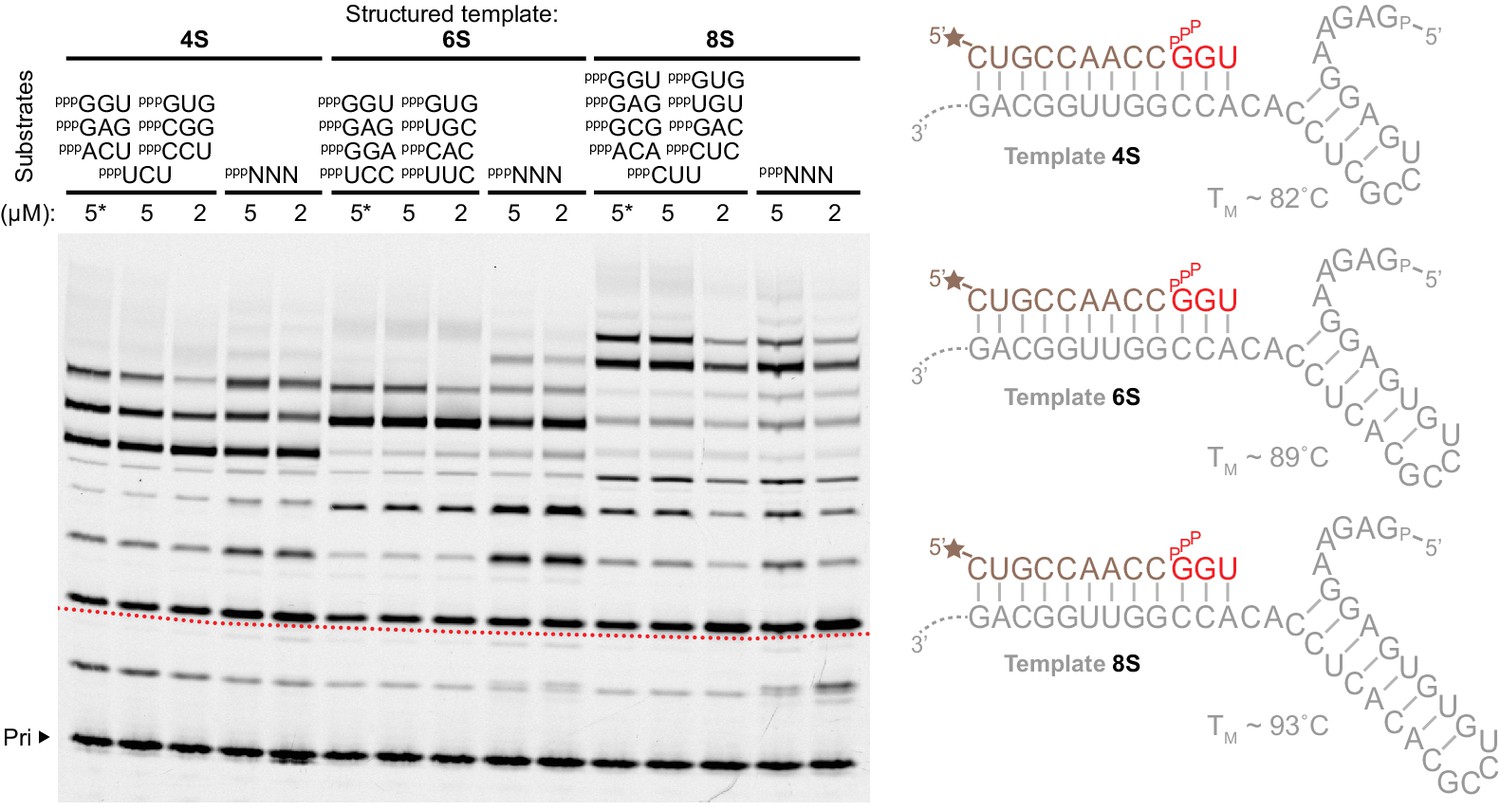
Substrates for structured template copying.
Primer extension reactions are shown using sets of defined triplet substrates (above the lanes), or random pools of all 64 triplets (pppNNN), each at the indicated concentrations on three templates with progressively longer, more stable secondary structure elements (right, TMs estimated using [Zuker, 2003]) (0.5 μM primer A9, 0.5 μM 4S, 6S, 8S templates, 2 μM t5+1,–7˚C ice for 25 days). Structure invasion and primer extension across the structured template region (beginning at the dotted red line on the image) occurred in all cases (with increased efficiency at higher substrate concentrations for the 8S template (5 µM)), including extensions where template secondary structures were allowed to form in the absence of triplets (*, triplets added last after 30 min supercooled at −7˚C).
Figure 5—figure supplement 2

Substrate concentration dependence of structured template copying.
Extension on the 8S structured template with a defined set of complementary triplets (left) or dinucleotides (right), at the indicated substrate concentrations (0.5 μM primer A9, template 8S, 2 μM t5+1, –7 ˚C ice for 25 days). Left, quantification of the extent of extension into the structured template region below each lane (measured by the fraction of third triplet addition to primers already extended by two) recapitulates a sigmoidal relationship with triplet concentration. Right, dinucleotides (like mononucleotides) exhibit negligible extension into the template hairpin structure.
Figure 6 with 5 supplements
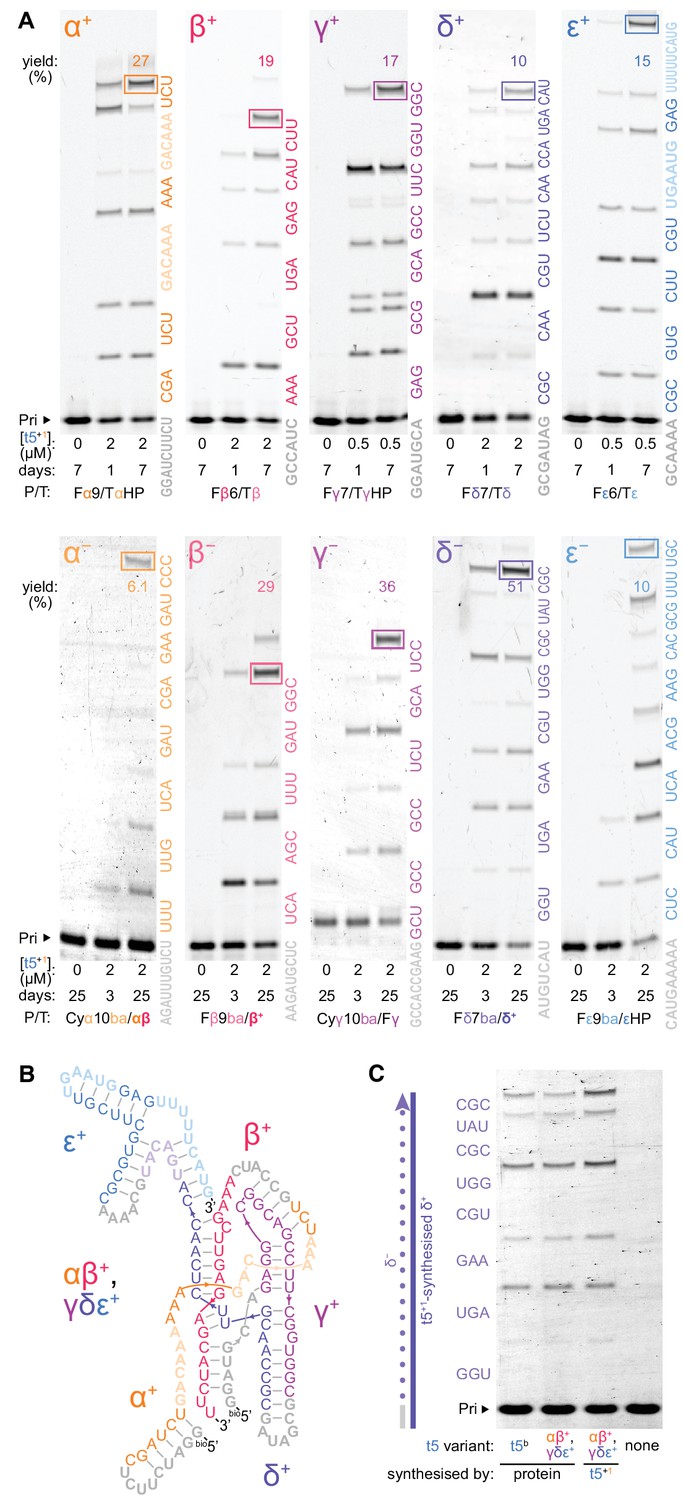
Ribozyme self-synthesis and assembly of its own catalytic domain.
(A) t5+1-catalysed syntheses of the five catalytic domain ‘+’ and ‘−’ segments via triplet extension of primers (grey) in −7˚C ice. Triplets are coloured by segment and shown alongside the lanes; longer oligonucleotide substrates (faded) were provided for α+ and ε+ syntheses to combat ribozyme-template pairing as shown in Figure 6—figure supplement 2 and Figure 6—figure supplement 3. The triplets were supplied at 5 μM (α+ to ε+), 10 μM (β− to ε−), or 20 μM (α−) each, with 0.5 μM primer/template (P/T; 1 μM for Fβ6/Tβ) and oligonucleotides equimolar to template sites. Use of substrates of more heterogenous compositions and lengths is shown in Figure 6—figure supplement 4 and Figure 6—figure supplement 5, respectively. Densitometry gave yields of full-length products (boxed, by % of total primer), and a geometric mean of the final extents of ligation across all 70 junctions in this self-synthesis context (78%). These segment sequences derive from t5b, a t5 variant with a neutral signature mutation (Supplementary file 1). (B) Secondary structure representation of a t5 catalytic domain (αβ+/γδε+, t5b sequence), formed via non-covalent assembly of t5+1-synthesised ‘+’ strand fragments in Figure 6—figure supplement 1, coloured by segment and synthesis substrate as in (A). (C) Activity of ribozyme-synthesised αβ+/γδε+ (B), compared to protein-synthesised αβ+/γδε+ and full-length t5b equivalents. These were assayed for synthesis of a δ- strand segment on a ribozyme-synthesised δ+ template, with added in vitro transcribed type 1 (2 μM each Rz, 5 μM triplets, 0.5 μM P/T, −7˚C 0.25× ice 10 days). The ribozyme-synthesized and assembled αβ+/γδε+ ribozyme is as active as in vitro transcribed equivalents, and can efficiently utilize ribozyme-synthesized RNA (δ+) as a template.
Figure 6—figure supplement 1

Ribozyme catalytic domain self-synthesis and assembly.
Top, conditions and yields for self-synthesis and assembly of the catalytic t5 domain (as αβ+ and γδε+ fragments, Figure 6b) by t5+1. To obtain maximal amounts of fully synthesized and assembled t5b ribozyme for activity testing (Figure 6c), we implemented t5 ‘+’ strand synthesis using sequence-specified triplet substrates for segment syntheses (as shown). The synthesis scale corresponds to the limiting component present. *: Unlike other segment ligation steps, the δε ligation was carried out using freeze-thaw cycling (Mutschler et al., 2015) (2.5 hr at −30˚C, 21 hr at −7˚C, then 0.5 hr at 37˚C), which modestly improved yield. Below, scheme of t5+1-catalysed t5 synthesis and assembly reactions. PAGE separations of syntheses (xt) alongside purified reference segments and fragments (in bold) were stained with SYBR Gold to quantify boxed full-length products. These were excised for use in subsequent assembly steps (illustrated by dashed black arrows). Synthetic schemes of colour-coded segments are shown beside corresponding lanes, denoting ‘+’ strand synthesis (bold primer and dashes) on ‘–’ strand template. Fully synthesized segments are shown as bold lines. Assembly reactions involve use of fully synthesized segments as the final substrate in a primer extension reaction (α + β, γ + δε) or direct templated ligation of synthesized segments (δ + ε). The αβ+ and γδε+ fragments associate spontaneously to form an active triplet polymerase ribozyme (Figure 6b), tested for activity in Figure 6c. The synthesised segment sequences were derived from the t5b variant of t5, which differs by one neutral signature mutation from t5 in the α segment, and by another neutral signature mutation from t5a in the ε segment (mutations highlighted in Supplementary file 1); these neutral mutations, not present in the t5 and t5a used to synthesise the segments (above), allowed verification of the synthetic origin of the product fragments by sequencing, which revealed the correct signature mutations in each fragment ruling out contamination of the synthesized ‘+’ strand products by the synthesizing (‘+’ strand) triplet polymerase ribozyme.
Figure 6—figure supplement 2
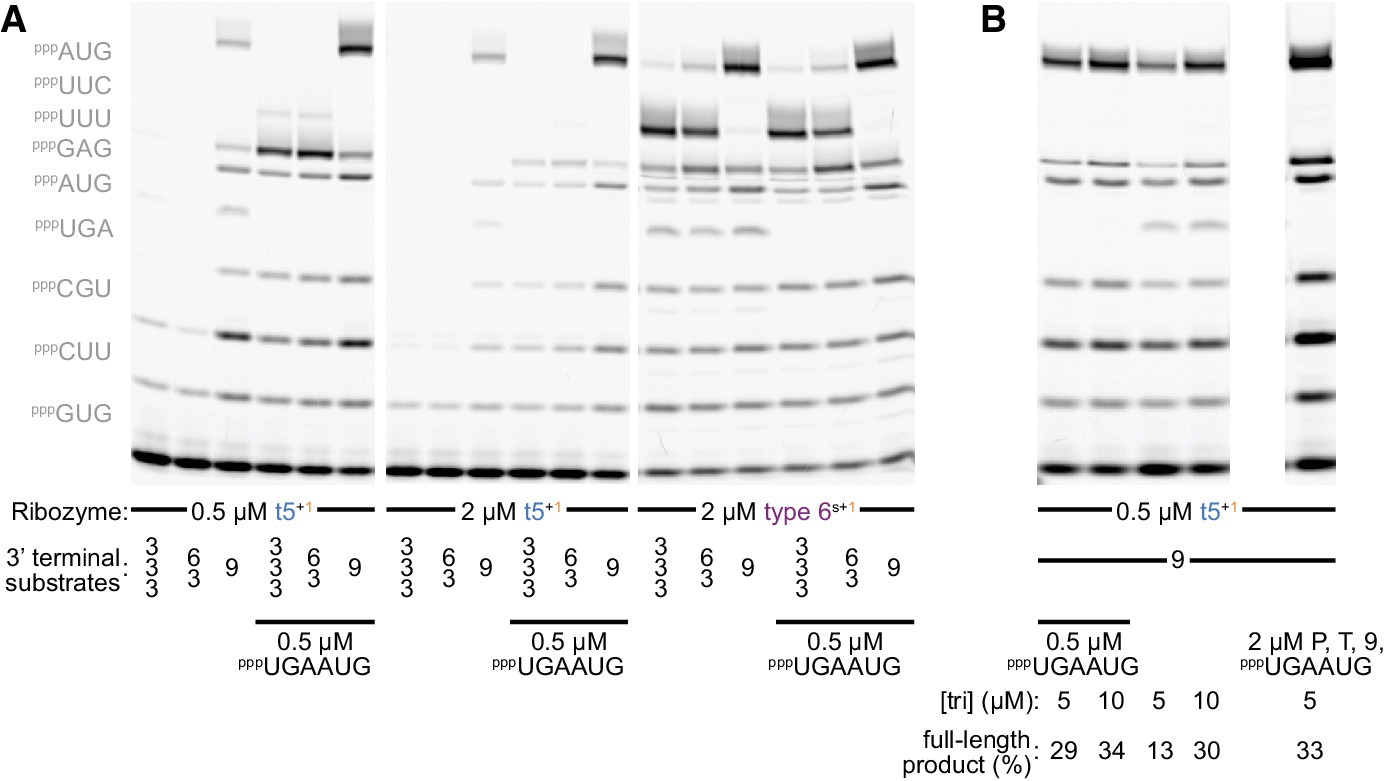
Substrate competition attenuates inhibitory ε+/ε- pairing during self-synthesis.
(A) Longer oligonucleotide substrates are required to maximise synthesis of full-length ε+ segment (0.5 µM Fε9 primer/Tε template/oligonucleotide substrate and 5 µM each triplet (left, omitted where equivalent oligonucleotide substrate was present), 13 days in −7˚C ice). Replacing two triplets with a preformed hexanucleotide substrate (pppUGAAUG) boosts full-length ε+ product synthesis by t5+1 (left panel), but not by the type 6s+1 ribozyme - with a different accessory domain (see Figure 2—figure supplement 1, sequence in Supplementary file 1) - which synthesizes ε+ independent of this hexanucleotide (right panel, allowing some full-length ε+ synthesis with only triplets). This likely reflects unfavourable competition between the t5 ribozyme’s own ε+ domain and triplet substrates for complementary pairing to the ε- RNA used as template. Consistent with this hypothesis, increasing t5+1 concentrations with this template is counterproductive (middle panel). For both ribozymes, using a preformed nonanucleotide substrate pppUUUUUCAUG at the end of the template boosted full-length product over use of a pppUUCAUG hexanucleotide +pppUUU triplet, or just the three constituent triplets (S = 9 vs. 6/3 vs. 3/3/3); this is not due to the individual triplet sequences as t5+1 can efficiently incorporate both pppUUU and pppUUC (see Figure 5b, Figure 6a). (B) Doubling triplet concentrations to 10 µM can attenuate t5+1’s requirement for the pppUGAAUG hexanucleotide substrate during ε+ synthesis. This suggests that at higher triplet concentrations they successfully compete with this part of t5 for ε- template hybridisation (0.5 µM Fε9 primer/Tε template/longer oligonucleotide substrates, 13 days in −7˚C ice). With a fourfold excess of primer/template duplex and substrates (right lane), the amount of full-length segment generated exceeds the ribozyme added, evidence of multiple turnover of full-length ε+ product.
Figure 6—figure supplement 3
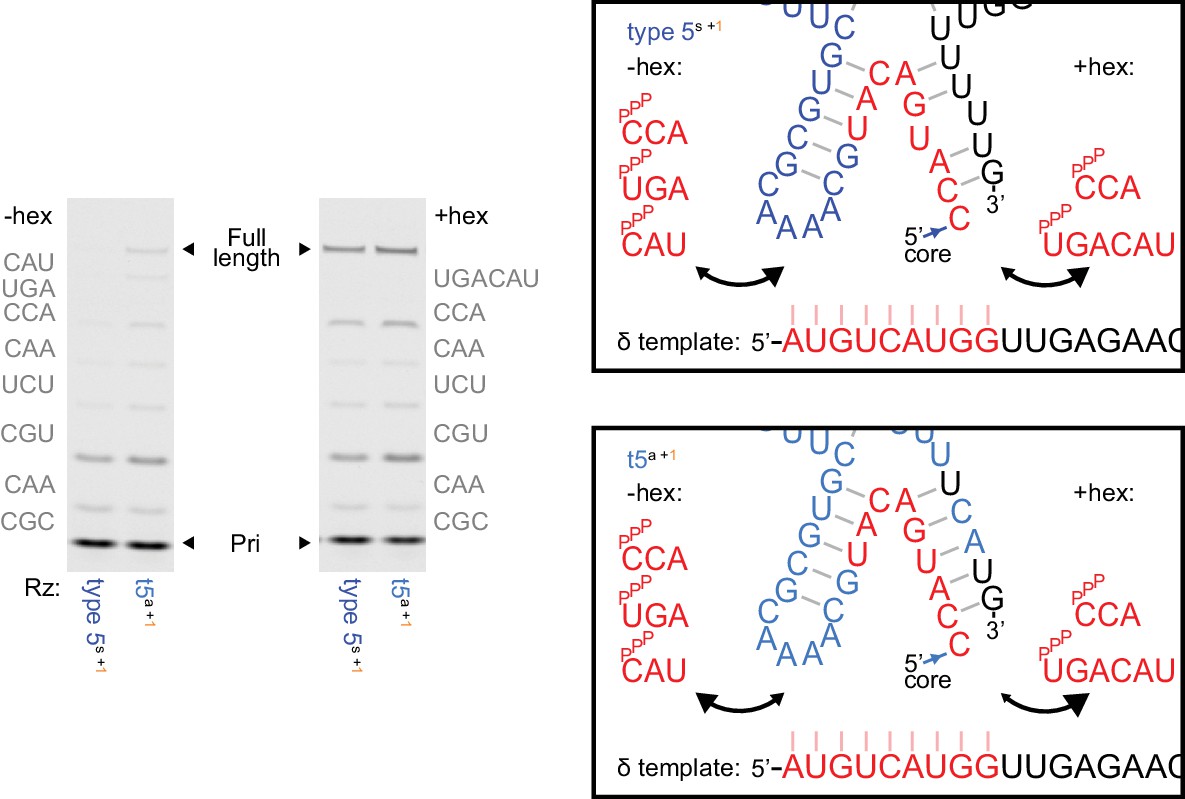
Ribozyme stabilisation attenuates inhibitory δ+/δ- pairing during self-synthesis.
Left, δ+ synthesis using 5 μM of each complementary triplet substrate (−hex) or 1 μM of pppUGACAU (+hex) replacing the final two triplets (0.25 μM primer Fδ7/template HTδ, 0.5 μM each ribozyme, 6 days in −7˚C ice). Type 5 with the initially isolated ε domain (type 5s) needs hexamer substrate for δ+ synthesis, but with the reselected ε domain (t5a) does not. See Supplementary file 1 for ribozyme sequences. Right, model of substrate/ribozyme competition for template binding. The region at the end of the δ segment exhibits irregular pairing in the initially isolated ε domain (above), and appears vulnerable to δ- template pairing during δ synthesis with triplet substrates. Reselection strengthened base-pairing in the corresponding ε domain stem (below), replacing a G-U wobble with a cognate G-C pair and a U.U mispair with a cognate U-A pair, stabilizing the δ - ε domain junction and potentially reducing competition with triplets during δ segment synthesis.
Figure 6—figure supplement 4

Ribozyme segment synthesis with random substrate pools.
For maximum self-synthesis yield, we had used specific triplet substrate sets. Here we compare synthesis by 0.5 µM t5+1 of the five t5 ‘+’ segments (30 days in −7˚C ice) using specific triplets vs. random triplet pools or reduced G-content random triplet pools. Reactions included 5 µM of each triplet in specific triplet sets (‘tri’, as in Figure 6a ‘+’ syntheses), random pppNNN, or a low-G pppNNN. The low-G pppNNN had the same overall triplet concentration as pppNNN, but individual triplets were five-fold less common for each constituent G (~10/2/0.4/0.08 µM for 0/1/2/3 Gs per triplet; some primer/template (P/T) concentrations were reduced here to ensure excess substrate over template). Synthesis is compared with and without some longer oligonucleotide substrates (indicated below, equimolar to template sites, replacing the corresponding triplets in ‘tri’ substrate mixes). Importantly, for all ‘+’ strand segments, full-length products are generated using pppNNN with yields approaching those obtained when using specific triplets (calculated by densitometry, above the gel); intriguingly the low-G pppNNN pools often gave superior yields, possibly helped by the higher concentrations of weaker-binding AU-rich triplets therein.
Figure 6—figure supplement 5
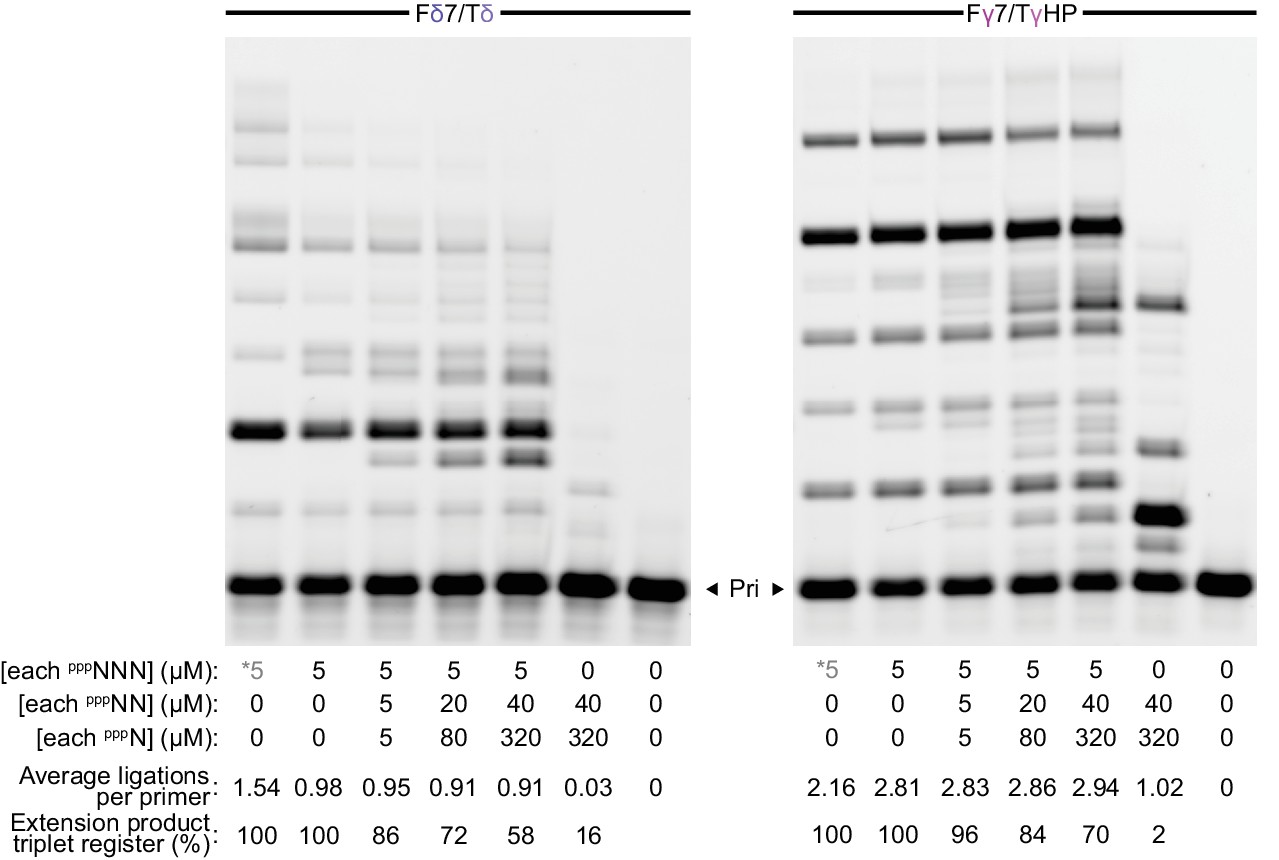
Ribozyme segment synthesis with mixed length substrate pools.
δ+ (left) or γ+ (right) syntheses were performed (using 0.5 µM each of t5+1 and the primer/templates above, 15 days in −7˚C ice) with their constitutive triplet sets alone (*, see Figure 6a) or the random-sequence substrate mixes indicated. Introduction of dinucleotide and mononucleotide substrates (pppNN, pppN i.e. NTPs) decreases full-length product band intensity but not ligations performed (here quantified whilst assuming each extension product was formed from the fewest substrates possible), with increasing numbers of extension products deviating from the starting triplet register. Dinucleotides and mononucleotides appear poor substrates in the absence of triplets.
Figure 7
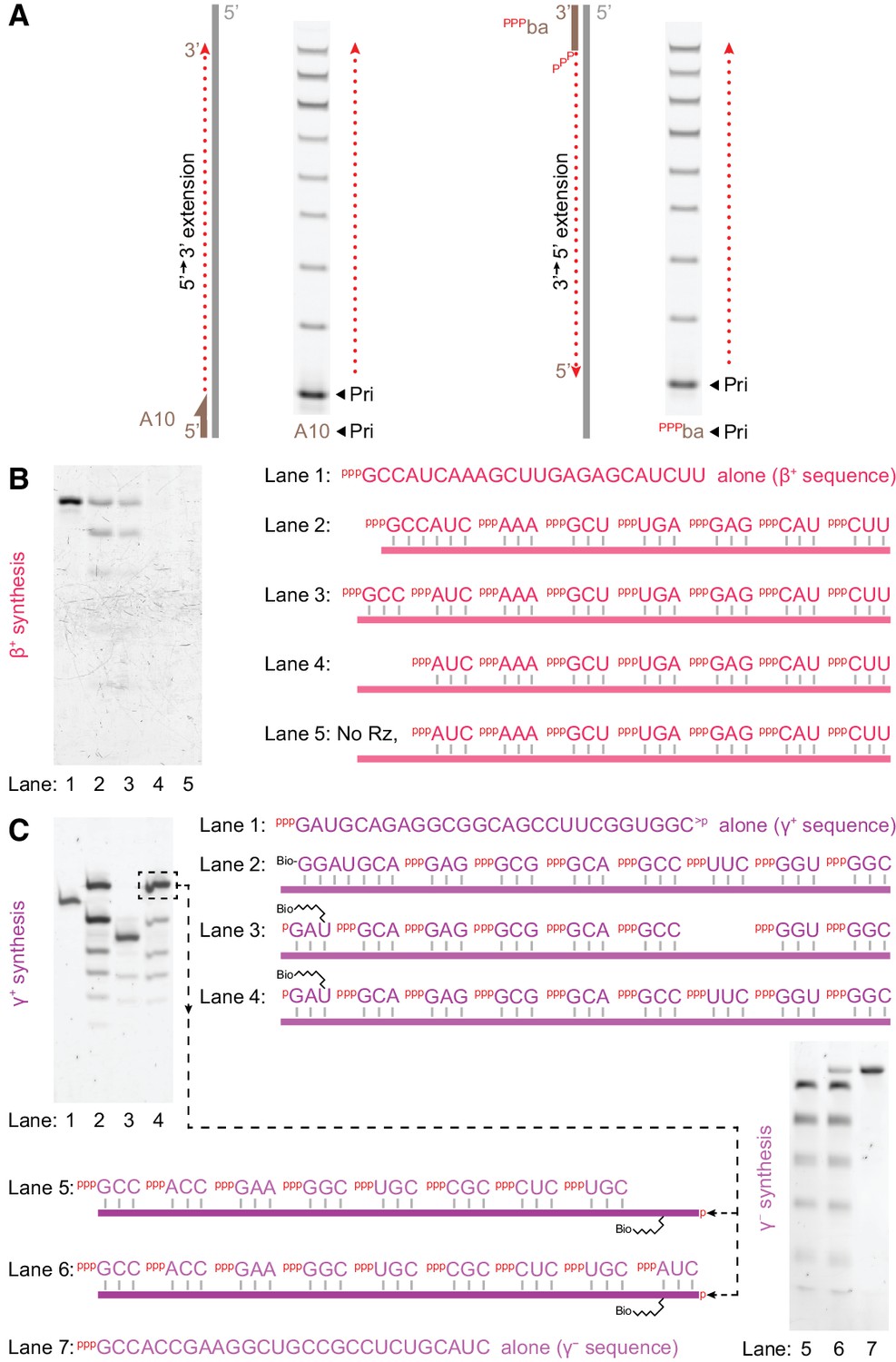
Triplet-initiated template sequence copying.
(A) Extension by t5+1 of fluorescein-labelled primers bound to either the 3’ (A10) or 5’ (pppba) ends of a template (T8GAA, 10 μM pppGAA, 0.5 μM/RNA, −7˚C ice 69 hr), demonstrating extension in either 5’−3’ (A10) or 3’−5’ (pppba) directions. (B) Synthesis of β+ on Tβ template via t5+1-catalysed polymerisation of the substrates indicated on the right (2 μM t5+1, 5 μM each triplet, 0.5 μM template (lane 2: 0.5 μM hexanucleotide), −7˚C ice 9 days). Extension products in lanes 2–5 were eluted from template, PAGE-separated and SYBR-Gold stained alongside in vitro transcribed full-length segment control (lane 1). Lane 3 shows full-length synthesis of β+ segment from triplets alone. (C) Triplet-based replication of the γ segment. Top left, synthesis of γ+ from the indicated substrates (1 μM t5+1, 5 μM each triplet, 2 μM TγHP template (lane 2: 2 μM Bioγ7 primer), −7˚C ice 7 days). Biotinylated extension products in lanes 2–4 were isolated from template, PAGE-separated and SYBR-Gold stained alongside in vitro transcribed staining marker (Mγ+m1, lane 1). This indicated 10% yield (per template) of full-length synthesis of γ+ segment from triplets alone (the final band in lane 4), which was purified for use as a template in γ - segment synthesis (bottom right, 1 μM t5+1, 5 μM each triplet, 0.05 μM template, with 0.05% Tween-20, –7˚C ice 27 days). Extension products in lanes 5 and 6 were eluted from template, PAGE-separated and SYBR-Gold stained alongside in vitro transcribed full-length segment control (Mγ -m1, lane 7). This indicated 6% yield (per template) of full-length synthesis of γ - segment from triplets alone (the final band in lane 6).
Figure 8 with 3 supplements
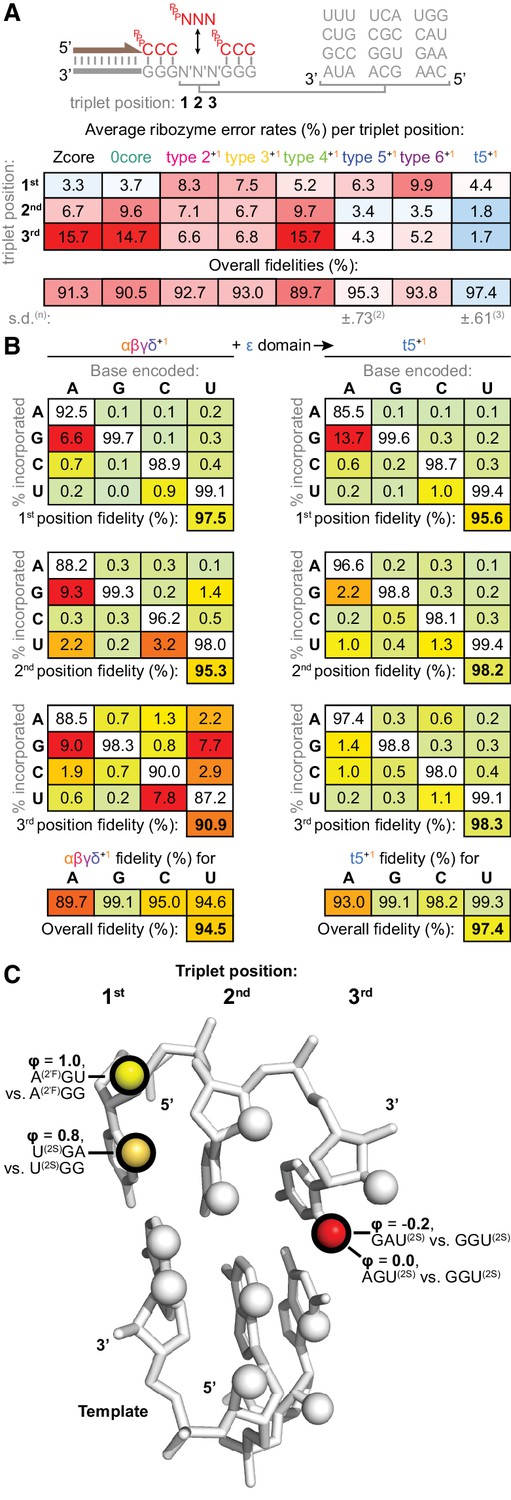
Fidelity of ribozyme-catalysed triplet polymerisation.
(A) To estimate its fidelity, each ribozyme was provided with an equimolar mix of all 64 triplet substrates (pppNNN at 5 μM each) for primer extension using templates containing twelve representative trinucleotide sequences (N′N′N′). Deep sequencing of extension products identified the triplets added opposite each template trinucleotide, yielding position-specific error tendencies; the overall fidelity was calculated as a geometric mean of positional errors at each triplet position (n and s.d. of this value shown for ribozymes assayed multiple times, see Figure 8—source data 1 for analysis of collated errors). The triplet polymerases exhibit diverse fidelity profiles; fidelity profiles of other type 5 variants are shown in Figure 8—figure supplement 1. (B) Collation of error rates by base type and position for type 5 with (t5+1) and without (αβγδ+1) the ε fidelity domain. Positional and overall fidelities are calculated as geometric means (see Figure 8—source data 1); individual positional fidelities are plotted in Figure 8—figure supplement 2. (C) Schematic summary of effects of triplet minor groove modification upon the fidelity phenotype. In the depicted trinucleotide RNA duplex segment, spheres represent minor groove groups potentially available for hydrogen bonding in a sequence-general manner. For three of these groups (highlighted in black), we assayed whether their modification in substrates (2’F = 2’ fluoro, 2S = 2-thio) affected the fidelity domain’s mismatch discrimination capabilities (detailed in Figure 8—figure supplement 3, with data and calculations in Figure 8—source data 2). These groups are labelled with the fraction of fidelity phenotype retained (ϕ) when discriminating between the indicated modified substrates. Colour reflects the impact of that group’s modification upon the fidelity phenotype, with red denoting a strong disruptive effect, and yellow weak or negligible effects.
-
Figure 8—source data 1
Analysis of collated errors by ribozymes in the fidelity assay.
- https://doi.org/10.7554/eLife.35255.033
-
Figure 8—source data 2
Calculation of residual fidelity phenotypes in Figure 8c.
- https://doi.org/10.7554/eLife.35255.034
Figure 8—figure supplement 1
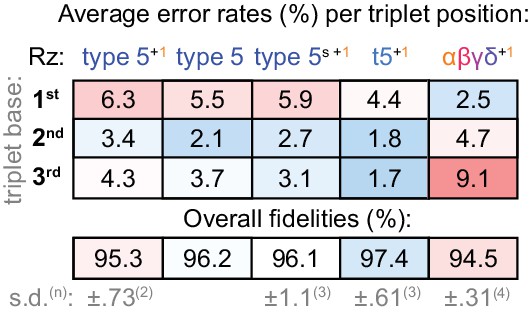
Fidelity of type 5 variants.
Positional error rates and fidelities (determined as in Figure 8a) for type 5 ribozyme variants; the overall fidelity was calculated as a geometric mean of positional errors at each triplet position (n and s.d. of this value shown for ribozymes assayed multiple times, see Figure 8—source data 1). Fidelity of the initial type 5 isolate (tethered to the assay templates) is modestly improved in the absence of type 1. However, with type 1, fidelity is improved when operating fully in trans (type5s +1). Reselection and stabilisation of the ε domain yields a further fidelity improvement (t5+1) but ε truncation (αβγδ+1) reverts the pattern of error tendencies along the triplet towards that of the starting core. See Supplementary file 1 for ribozyme sequences.
Figure 8—figure supplement 2
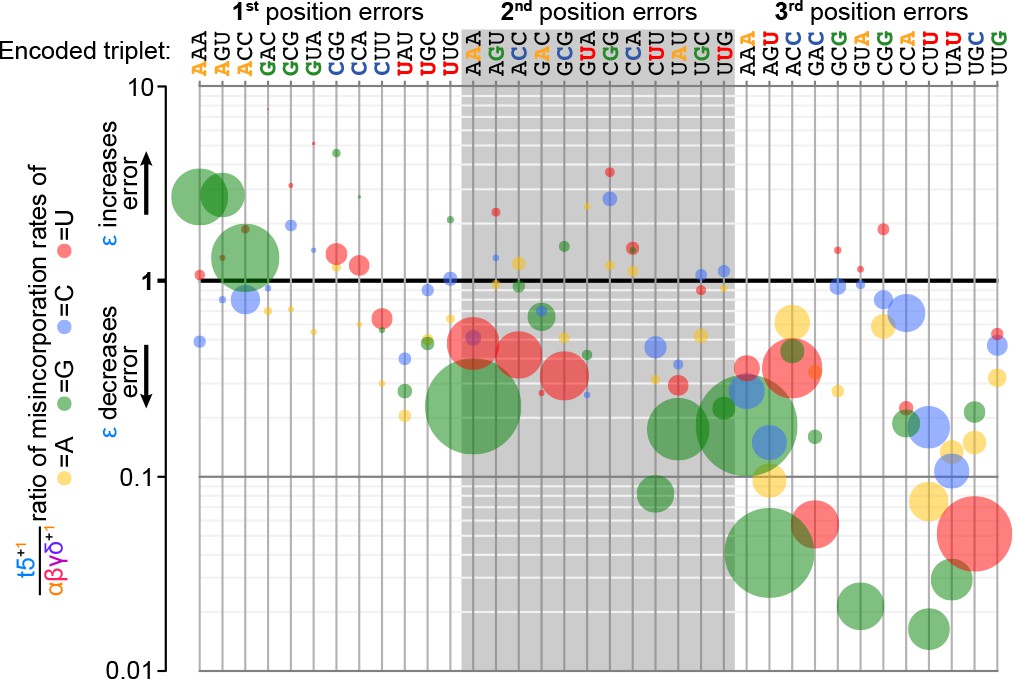
Fidelity domain influence upon template- and position-specific error rates.
At each position in each of the twelve triplet templates (top) tested in the fidelity assay (Figure 8a), three different errors are possible after analysis of sequence composition of added triplets (see Figure 8—source data 1). Errors are represented by discs, coloured by mutation (e.g. A to G: green; A to C: blue; G to C: blue). Disc size is proportional to error frequency without the fidelity domain (its occurrence using αβγδ+1). The effect of the ε fidelity domain upon these error tendencies is plotted on the y-axis (by comparison of error rates between t5+1 and αβγδ+1). Many of the most significant errors (using αβγδ+1) exhibit the greatest proportional reductions with the fidelity domain, including substantial (>10 fold) reductions in several misincorporations at the third position. Improvements in fidelity are variable but focused on the second and third triplet positions, while the first triplet position shows an increase specifically in A to G mutations, indicative of a tolerance for G-U wobble pairing.
Figure 8—figure supplement 3
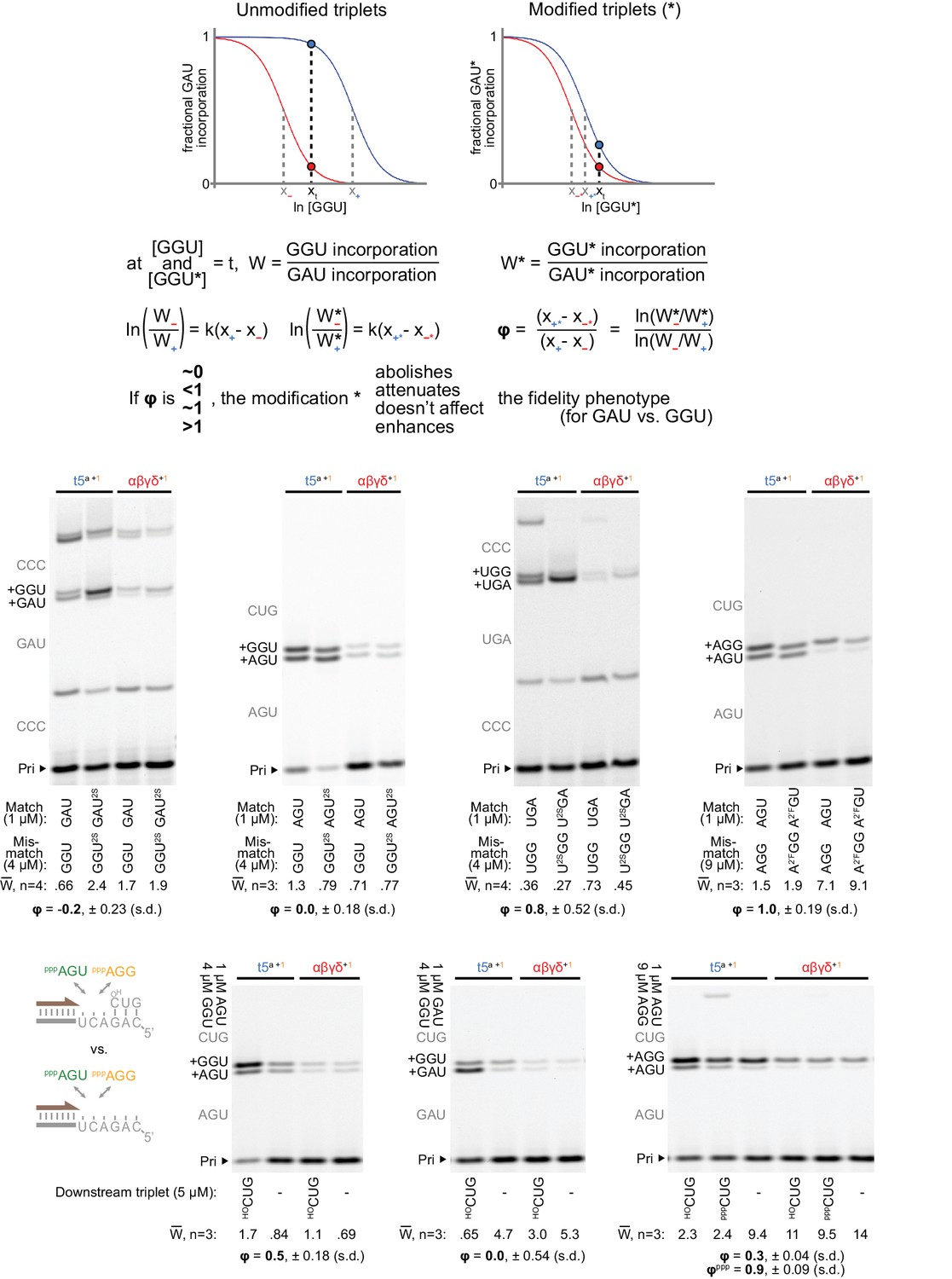
Determination of residual fidelity phenotype when using minor groove-modified substrates.
Top: framework for estimation of residual fidelity phenotype (ϕ). Shown are model logistic curves for incorporation of a triplet, for example GAU (at a fixed concentration, opposite 3’-AUC-5’), versus increasing concentrations of a mismatching triplet, for example GGU. A ribozyme ‘+’ with a fidelity function (e.g. t5a, blue curve) will incorporate equal amounts of the matched and mismatched triplets at a different concentration of the mismatched triplet compared to a ribozyme ‘−’ without a fidelity function (e.g. αβγδ, red curve). x+ and x− represent the lns of these concentrations for + and −; their separation (x+ - x−) is a proxy for the strength of the ‘+’ fidelity phenotype. We measured the relative incorporation (W) of triplet pairs by the two triplet polymerases at test concentrations (t) of mismatched triplet, chosen to maximise the difference in the ribozymes’ resulting fractional incorporations (marked by a blue dot vs. a red dot on respective curves). If a triplet modification (*, right) interferes with mismatch discrimination by ‘+’, it would shift the ‘+’ relative incorporation curve (blue) towards that of ‘−’ (red), reducing the difference in fractional incorporation. Assuming curve steepness (k) remains constant, the residual phenotype (ϕ) for that modification is described by the new separation (x+* - x−*) as a proportion of the original separation (x+ - x−); numerical values and calculations from the measurements described below are supplied in Figure 8—source data 2. Middle: measurements of ratios of incorporation vs. misincorporation (W) for unmodified and modified triplet pairs (at the indicated test concentrations) by the t5a +1 triplet polymerase compared to the ε fidelity domain-truncated αβγδ+1 (at 0.5 μM each). The expected triplet additions are in grey along the left of each gel; the average W from n independent experiments, calculated via densitometry of products containing slower-migrating G misincorporations, is shown below each lane along with the average ϕ. Primers (P), templates (T) and additional triplets were at 0.5/0.5/5 μM, incubated for three days in −7˚C ice. Left: presence of a 2-thiouracil (2SU) at the third position of the triplet abolishes the fidelity domain’s ability to discriminate against a second position wobble pair (P: A10, T: CCCMisGAU, +5 μM pppCCC). Centre-left: the same third position modification (2SU) also abolished the fidelity domain’s preference for G misincorporation at the first position (P: Fγ7, T: TγAGU, +5 μM HOCUG). Centre-right: in contrast, a 2SU at the first position of the triplet exerted no clear influence upon third position wobble discrimination (P: A10, T: CCCMisUGA, +5 μM pppCCC). Right: replacement of a 2’ hydroxyl group with a 2’ fluoro (2’F) at the first triplet position likewise had no effect upon third position mismatch discrimination (P: Fγ7, T: TγAGU, +5 μM HOCUG). Below: Measurement of fidelity phenotype in the presence or absence of a downstream triplet. Residual phenotypes indicate attenuation or abolishment of third position (left panel, P Fγ7, T TγAGU), second position (middle panel, P Fγ7, T TγGAU), and third position (right panel, P Fγ7, T TγAGU) fidelity effects from downstream triplet absence. No effects are seen (upon third position discrimination) from the presence or absence of a 5’ triphosphate on the downstream triplet (ϕppp). In the absence of a triplet bound downstream on the template, the fidelity phenotype is severely compromised, suggesting this adjacent triplet:template duplex plays a critical role in positioning the fidelity domain relative to the incoming triplet.
Figure 9 with 1 supplement
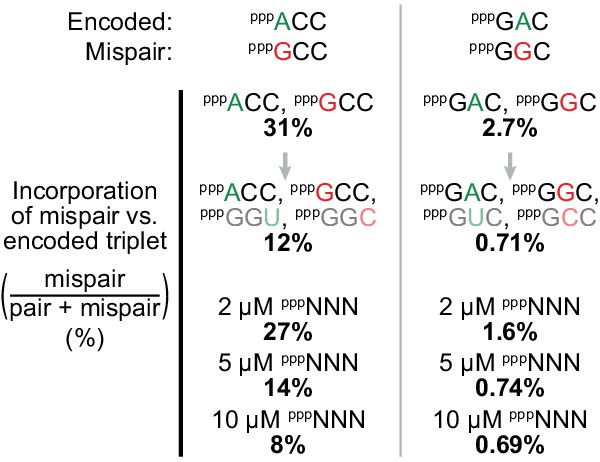
Substrate pool interactions improve triplet fidelity.
Applying the fidelity assay (Figure 8a, using t5+1) to single templates with only an encoded triplet and a mispairing one as substrates (at 5 μM each), we observed that relative mispair incorporation was proportionally reduced (by 61% (left) and 73% (right)) upon introduction of complementary triplets. Using all 64 triplets (pppNNN) has an analogous effect upon these pair/mispair comparisons with fidelity progressively improved upon increasing overall pppNNN concentrations, with examples of effects on other triplets and overall fidelity presented in Figure 9—figure supplement 1, and comprehensive error rates and ratios in Figure 9—source data 1.
-
Figure 9—source data 1
Collated error rates and ratios at different substrate concentrations.
- https://doi.org/10.7554/eLife.35255.038
Figure 9—figure supplement 1
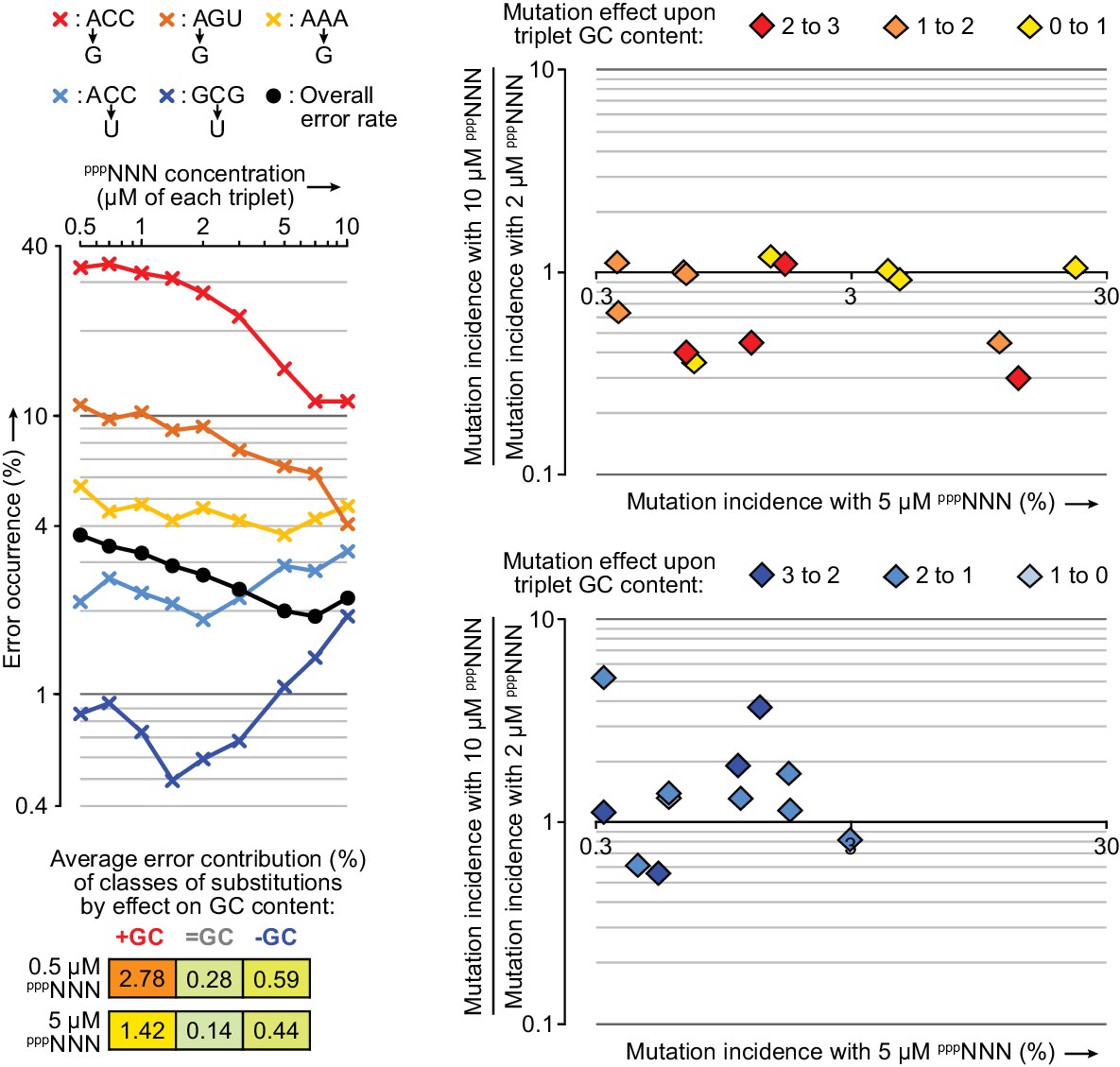
Influence of random triplet pool concentrations upon triplet misincorporation tendencies.
Left: 0.2 µM t5+1 was tested in the fidelity assay at a series of pppNNN concentrations. The observed overall error rate (black circles) in these reactions halved as pppNNN concentrations were increased to 7 µM each (after which extension activity began to reduce and error rates increase). Diverse patterns were observed in the concentration dependence of individual triplet positional errors, of which a selection are displayed here (×) coloured according to whether they raise (red-yellow) or lower (blue) the GC content of the expected triplet, and coloured more intensely for higher GC content triplets. Right: For a broader set of errors with 0.5 µM t5+1 (those with >0.3% incidence at 5 µM pppNNN, x-axes) the changes in their incidences from raising concentrations of each pppNNN from 2 to 10 µM substrate are plotted on the y-axes. The frequencies of several mutations that increase triplet GC content in GC-rich triplets are reduced at 10 µM pppNNN concentrations (top panel). Conversely, the frequencies of some mutations that reduce GC content are increased at 10 µM pppNNN concentrations (bottom panel); these suggest relative depletion of available GC rich triplets as overall triplet concentrations rise. Such pool behaviour can explain the reduction of overall error rates in ~5 µM pppNNN triplet pools, as misincorporations that increase GC content (e.g. G opposite U) are responsible for more errors than those that decrease it (bottom left). Values and ratios for a wider range of errors are supplied in Figure 9—source data 1.
Tables
Table 1
Sequencing of ribozyme-synthesised β+ segment.
Shown are the individual base fidelities (%) along the β+ sequences (top) synthesised by t5+1, using the six specific triplets (tri), or random (pppNNN) or compositionally-biased random (low-G pppNNN, see Figure 6—figure supplement 4) substrate pools, from Fβ6 primer (the first six positions at the left) with template Tβ (1 μM each RNA, 13 days −7˚C ice). For their sequencing, extension products were eluted from templates, and full-length products were gel-purified, ligated to adaptor, reverse-transcribed and PCR amplified. For compositional analysis, a small percentage of unrelated amplified products were excluded (those with >9 mutations vs. the expected β+ sequence; similar levels were excluded if a > 6 mutation threshold was applied, 0.2%/0.2–3.7%/4.2% and 3.7%/3.8% for tri & pppNNN and low-G pppNNN). These sequences mostly appeared to derive from off-target priming and extension of Fβ6 on the ribozyme in the presence of pppNNN. The sequencing of products generated from specific triplets provides an estimate of background error arising from amplification and sequencing. The final triplet constitutes an error hot-spot - likely to mutate to a more mutationally stable triplet during self-replication - exacerbated in pppNNN samples by the inability of the fidelity domain to operate in the absence of a downstream triplet (Figure 8—figure supplement 3). The geometric average of internal triplet position fidelities is used to gauge overall t5+1 fidelity during RNA synthesis. While overall fidelity drops from defined to random triplets (98.8 to 96.7%), much of this loss in fidelity can be recovered by adjusting the triplet composition to a low-G random pool, where reductions in G-U wobble pairing more than compensate for increases in rarer misincorporations opposite template C.
| G | C | C | A | U | C | A | A | A | G | C | U | U | G | A | G | A | G | C | A | U | C | U | U | Internal triplets’ average: |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 μM each tri: | 93.3 | 99.4 | 99.5 | 97.8 | 99.8 | 99.4 | 99.6 | 97.9 | 99.1 | 99.3 | 99.5 | 99.1 | 99.8 | 98.9 | 99.5 | 98.2 | 96.0 | 96.3 | 98.79 | |||||
| 10 μM each PPPNNN: | 92.8 | 97.0 | 98.8 | 99.2 | 99.5 | 99.3 | 99.1 | 97.8 | 98.6 | 99.4 | 99.1 | 98.7 | 94.3 | 81.4 | 96.6 | 97.4 | 59.8 | 42.3 | 96.65 | |||||
| 10 μM average, low-G PPPNNN: | 97.3 | 98.2 | 99.5 | 97.3 | 99.7 | 99.4 | 99.6 | 97.8 | 99.0 | 99.3 | 98.9 | 98.8 | 97.6 | 97.3 | 98.8 | 97.4 | 73.2 | 43.2 | 98.56 | |||||
Additional files
-
Supplementary file 1
Ribozyme sequences.
All sequences are written in a 5’-to-3’ direction, and generated by GMP transcription of the corresponding PCR-generated dsDNA of the sequence downstream of 5T7 sequence duplex. All transcripts were PAGE-purified. HDV ribozyme sequences (blue) were transcribed in series with reselected type 5 ribozymes and cleave themselves off during transcription (Schürer et al., 2002) to yield precise 3’ ends with 2’, 3’-cyclic phosphates; the presence of this group did not affect type 5 activity. Sequences corresponding to the 5’ ‘cap+’ or ‘cap−’ regions from the selection, presenting a target for type 1 interaction, are coloured light green, with alternative arbitrary inert 5’ hairpin-forming sequences in dark green. Single-stranded sequences capable of hybridization with sites at the 3’ (or 5’, type 5cis) ends of certain templates to endow flexible ribozyme-template duplex tethering (Wochner et al., 2011; Attwater et al., 2010) are coloured yellow. Accessory domains 3’ of the catalytic core are in bold.
- https://doi.org/10.7554/eLife.35255.039
-
Supplementary file 2
Transcription of triplets.
Each target was transcribed from oligonucleotides 5T7 (Supplementary file 3) and 5’-(var)-TATAGTGAGTCGTATTAATTTCGCGGGCGAGATCGATC-3’, where the (var) overhang encodes (is the DNA reverse complement of) the sequence indicated below. Guide yields: >15 nmol = ***, 10–15 nmol = **, 5–10 nmol = *, <5 nmol = ~. †: These transcriptions yield one main product suitable for use as markers to identify comigrating triplets with a similar G/AU/C content.
- https://doi.org/10.7554/eLife.35255.040
-
Supplementary file 3
Oligonucleotide sequences.
All sequences are written in a 5’-to-3’ direction. DNA sequences are coloured grey. RNA sequences are coloured black, with the exception of hammerhead ribozyme (brown) and HDV ribozyme (blue) sequences transcribed in series that cleave themselves off during transcription (Schürer et al., 2002) to yield precise 5’ and 3’ ends respectively. All RNAs were denaturing PAGE-purified, and DNAs were not, unless otherwise noted (‘(non)-GP’). Competing oligonucleotides, listed beneath templates, were purified using RNeasy columns (Qiagen). Primer and oligonucleotide binding sites on templates are underlined.
- https://doi.org/10.7554/eLife.35255.041
-
Transparent reporting form
- https://doi.org/10.7554/eLife.35255.042
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Ribozyme-catalysed RNA synthesis using triplet building blocks
eLife 7:e35255.
https://doi.org/10.7554/eLife.35255




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}