Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells
- Harvard Medical School, United States
- Tsinghua University, China
- Howard Hughes Medical Institute, United States
Figures
Figure 1 with 3 supplements
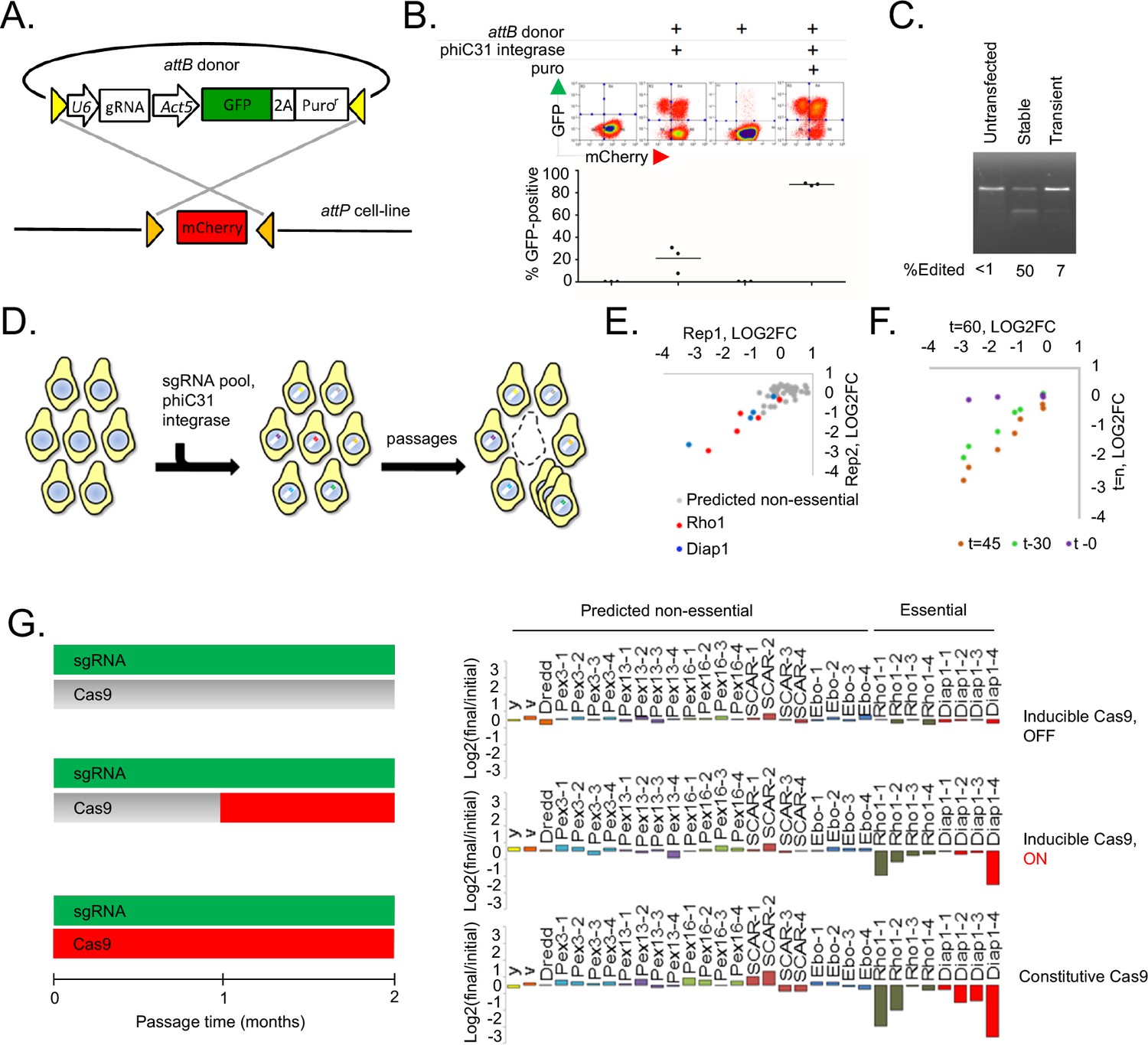
A novel method for introducing highly complex DNA libraries using phiC31 recombination.
(A) phiC31 attP-attB recombination strategy. S2R+/PT5 cells containing attP sites (gold) flanking mCherry were recombined with attB donor (pLib6.4) containing attB sites (yellow) flanking U6 promoter for sgRNA expression and GFP-2A-Puro expression cassette. (B) Recombination efficiency measured by flow-cytometry. Transfected cells were and grown with or without puromycin as indicated and passaged for 60 days. Graphs reflect total percentage of stable integrants (GFP+/total). N = 3. (C) Cells stably or transiently transfected to express Cas9 or control vector were each additionally transfected with an sgRNA targeting the Dredd allele followed by editing efficiency assay (T7E1) at the Dredd locus. (D) Scheme for pooled screens containing a library of integrating sgRNA expression vectors. (E) Dropout of essential-gene targeted sgRNAs from a minipool of 31 sgRNAs. Two replicates of PT5 or PT5/Cas9 cells transfected with sgRNAs targeting Rho1 (red) or Diap1 (blue) and additional sgRNAs targeting eight genes predicted to have non-essential functions (grey) were passaged with puromycin for 60 days and sgRNA abundance was measured using next-generation sequencing. Graph shows log2(fold-change) of each sgRNA in cells expressing Cas9 divided by sgRNAs in cells not expressing Cas9. (E) Optimizing passage time for dropout measurements. sgRNA abundance was detected from cells transfected as in (D) but analyzed initially, after 30 days, or after 45 days, and log2 fold-changes were compared to those at 60 days. (G) Left: Schematic of experiment to test effect of inducible versus constitutive Cas9 activity. Right: Dropout efficiencies from pooled screens using inducible versus constitutive Cas9 and a mixture of sgRNAs targeting either essential genes or those predicted to be non-essential. Vertical axis reflects log2(fold-change) for each sgRNA. Shown are means of two independent replicates.
Figure 1—figure supplement 1

Copper induction is not required in PT5/MT-Cas9 cells to give maximal gene editing efficiency.
Control S2R+/PT5 cells or cells stably transfected with metallothioein-promoter-driven Cas9 (MT-Cas9) were additionally transiently transfected with Dredd-targeted sgRNA followed by editing efficiency assay (T7E1) at the Dredd locus after 4 days.
Figure 1—figure supplement 2
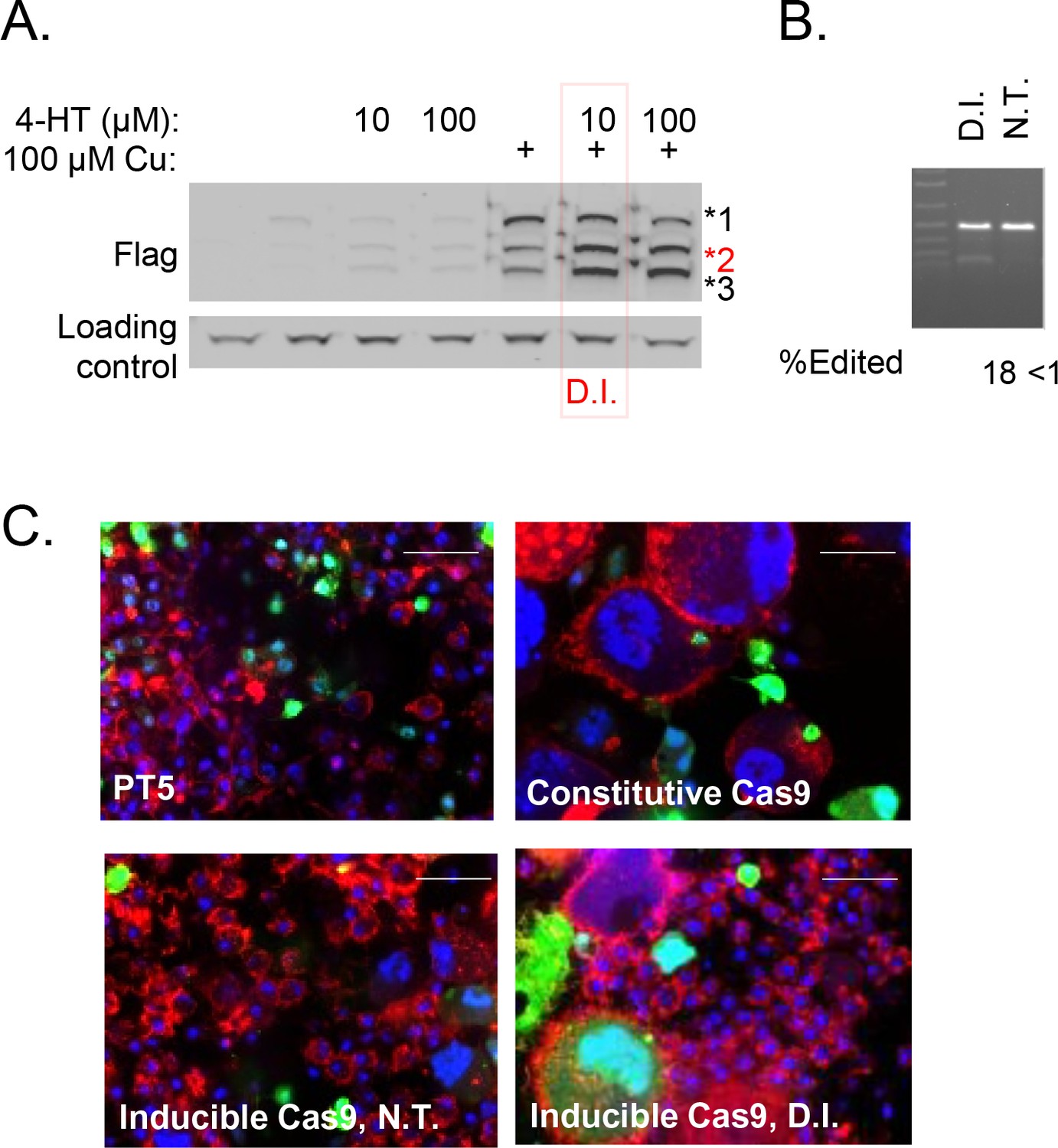
Validation of Cas9 induction system in Drosophila S2R + cells.
(A) Cells stably expressing intein-Cas9_S219-3XFLAG (Davis et al., 2015) treated with indicated concentration of 4-HT with or without CuSO4 as indicated were subjected to anti-Flag Western blot. 4-hydroxytamoxifen (4-HT) treatment increases proportion of cleavage product 2 (red), which represents nuclease-active Cas9 (Davis et al., 2015). 10 µM 4-HT + 100 µM CuSO4 was chosen as a double-induction (D.I.) condition to optimize inducibility of Cas9. (B) PT5/intein-Cas9_S219-3XFLAG cells, transfected with Dredd sgRNA-expression vector, were subjected to D.I. or no treatment (N.T.). Quantified T7E1 assay is shown. (C) Cells as indicated were transfected with an sgRNA encoded in pLib6.4 (expressing Actin5C promoter driven GFP) targeting Rho1 (5’-CAGCAAAGATCAGTTCCCCG-3’) and cells were fixed and stained with phalloidin (red) and DAPI (blue) to reveal F-actin and nuclear DNA and representative images are shown.
Figure 1—figure supplement 3
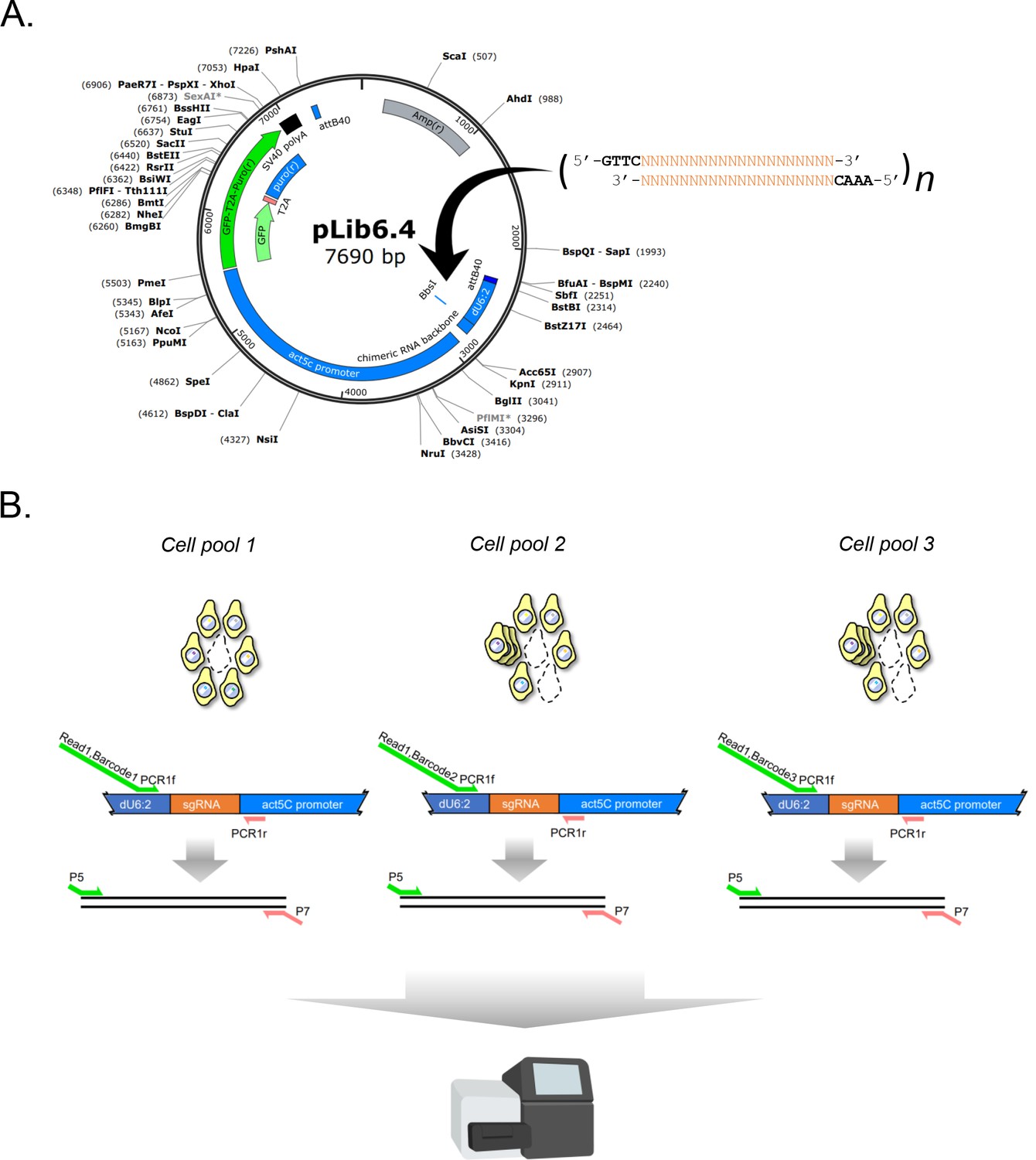
Design of sgRNA library vector and sgRNA PCR for next-generation sequencing.
(A) Restriction map of sgRNA library vector pLib6.4. Annealed oligos are ligated into BbsI/BpiI site as indicated. (B) Two-step PCR scheme and amplicon barcoding. Amplicons are amplified from separate experimental cell pools by PCR with 6 bp in-line barcoded primers. A second round of amplification adds Illumina-sequencing compatible P5 and P7 sites. Finally, the amplicons are sgRNA-concentration normalized, mixed, and subjected to next-generation sequencing.
Figure 2 with 1 supplement

Genome-wide CRISPR dropout screen in Drosophila S2R+ cells, results and metrics.
(A) CRISPR library is maintained in three distinct sublibrary groups as indicated, containing common controls. (B) sgRNA-level analysis of common controls in each group verify similar growth rates during each sublibrary screen. log2(fold-changes) of all sgRNAs representing two common controls, Rho1 (grey) and intergenic (pink). The average and standard deviation of log2(fold-changes) are shown for all individual sequences corresponding to Rho1-targeting positive controls or intergenic negative control sequences. (C) Gene-level analysis of sequential replicate screens. Log2(fold-changes) for all sgRNAs (85,558 in total) were first determined and then aggregated into a single Z-score using the maximum likelihood estimate (MLE) computational approach for each of 13,928 Drosophila genes in two independent, sequential replicates and plotted. (D) Z-score was calculated from average of replicate Log2(fold-changes), (Supplementary file 1) and these were plotted against RNAseq expression value (log(RPKM + 0.010)) (MODEncode). (E) Rank-wise false-discovery rate (FDR) of pooled CRISPR compared with arrayed RNAi (Boutros et al., 2004), original data or following re-analysis (see Materials and methods). Cumulative distribution of false-discovery error at indicated gene rank divided by the total possible false-discovery error, where ‘error’ is defined as a phenotypic assertion for any gene with RPKM <1. (A) True-positive rate (TPR) for major eukaryotic essential genes shows broader distribution of functional classes revealing fitness essentiality from CRISPR than RNAi screens. Receiver operating characteristic (ROC) curves displaying rate of discovery of components of selected essential eukaryotic complex (Kanehisa et al., 2017) as a function of FDR. Curves compare CRISPR knockout screen (this study) with reanalyzed genome-wide RNAi (Boutros et al., 2004). (G) True-positive rate (TPR) of Drosophila CRISPR screen is in a similar range to TPRs from human CRISPR screens using libraries of similar size. Comparison of true positive rate between human cell-line screens (infected with GeCKO v2) and Drosophila CRISPR screening using high-confidence RNAi hits as true positives (Lenoir et al., 2018; Sanjana et al., 2014; Boutros et al., 2004). (H) Position matrix for optimal sgRNA design based on CRISPR screen. For the top 500 genes, which were hits with <2% FDR in the CRISPR screen, hypergeometric probability of an A, C, G, or T nucleobase was calculated from strongly depleted (‘good’, LOG(p-value), above 0 on the y-axis) sgRNA designs versus unchanging (‘bad’, negative LOG(p-value), below 0 on the y-axis) sgRNA designs.
-
Figure 2—source data 1
CRISPR and RNAi screen comparisons, continued.
Table containing CRISPR or RNAi Z-scores as well as RNA expression data for all targeted genes.
- https://doi.org/10.7554/eLife.36333.009
Figure 2—figure supplement 1
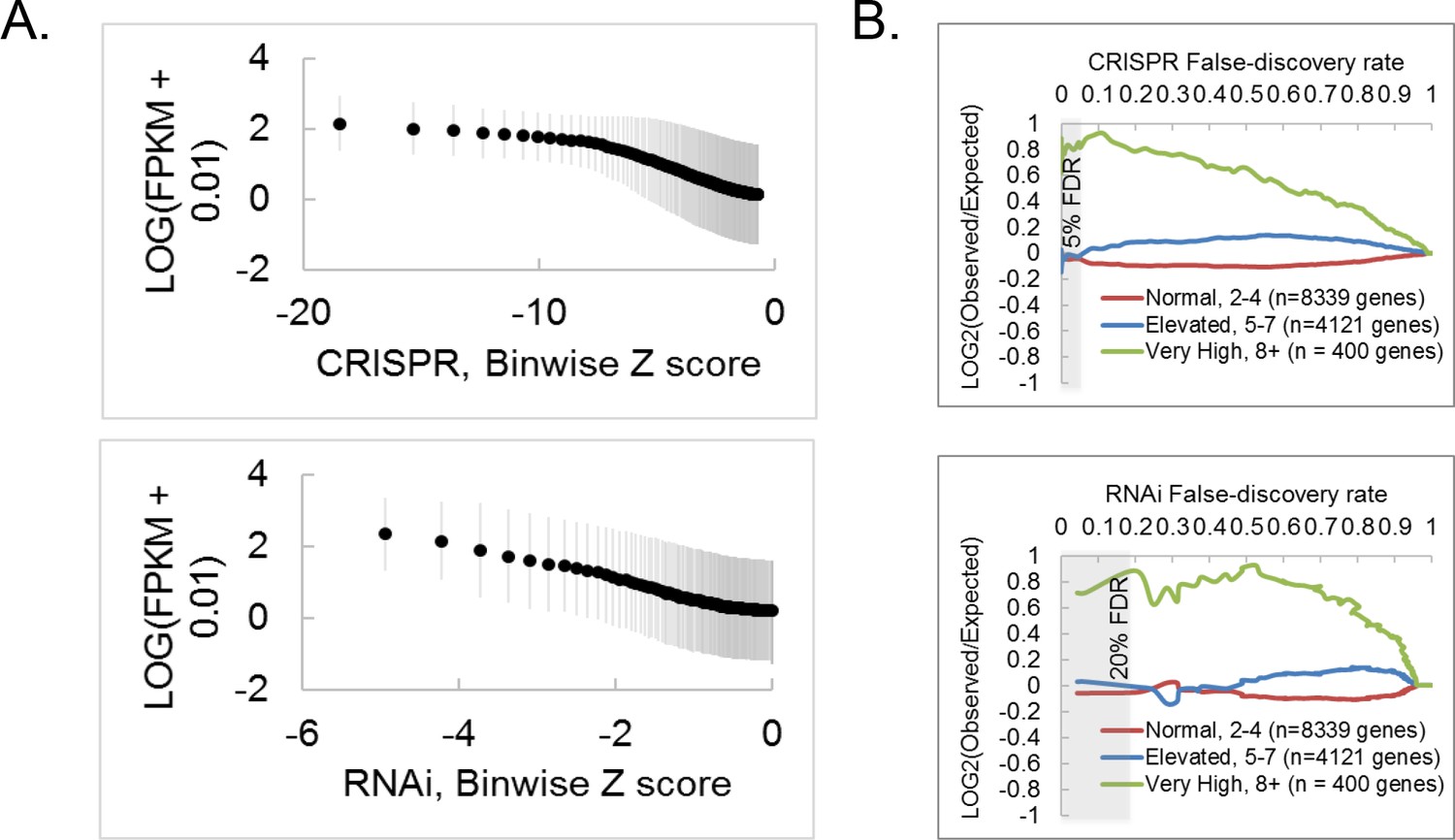
CRISPR and RNAi screen comparisons, continued.
(A) RNAi exhibits a stronger dependence on mRNA level than CRISPR. A rank-based binning function was applied to CRISPR or RNAi data, where every 100 genes was binned, and each biwize average Z-score plotted against each binwise RNAseq expression value (LOG10(RPKM + 0.010)). Error bars reflect standard deviation. (B) Effect of copy number amplification (CNA) on gene-level Z score assignment in CRISPR or RNAi screens. CNA data for S2R+ cells (Lee et al., 2014) was used to create bins according to copy number for each gene, and then log2(odds ratio) was determined for each bin. Notice that bins containing very high CNA (green) represent only ~3% of the genome, and genes in this bin were also hits in the RNAi screen, suggesting that CRISPR screening does not lead to spurious calls for genes with high CNA, but, rather, that genes with high CNA are enriched for fitness essentiality.
Figure 3
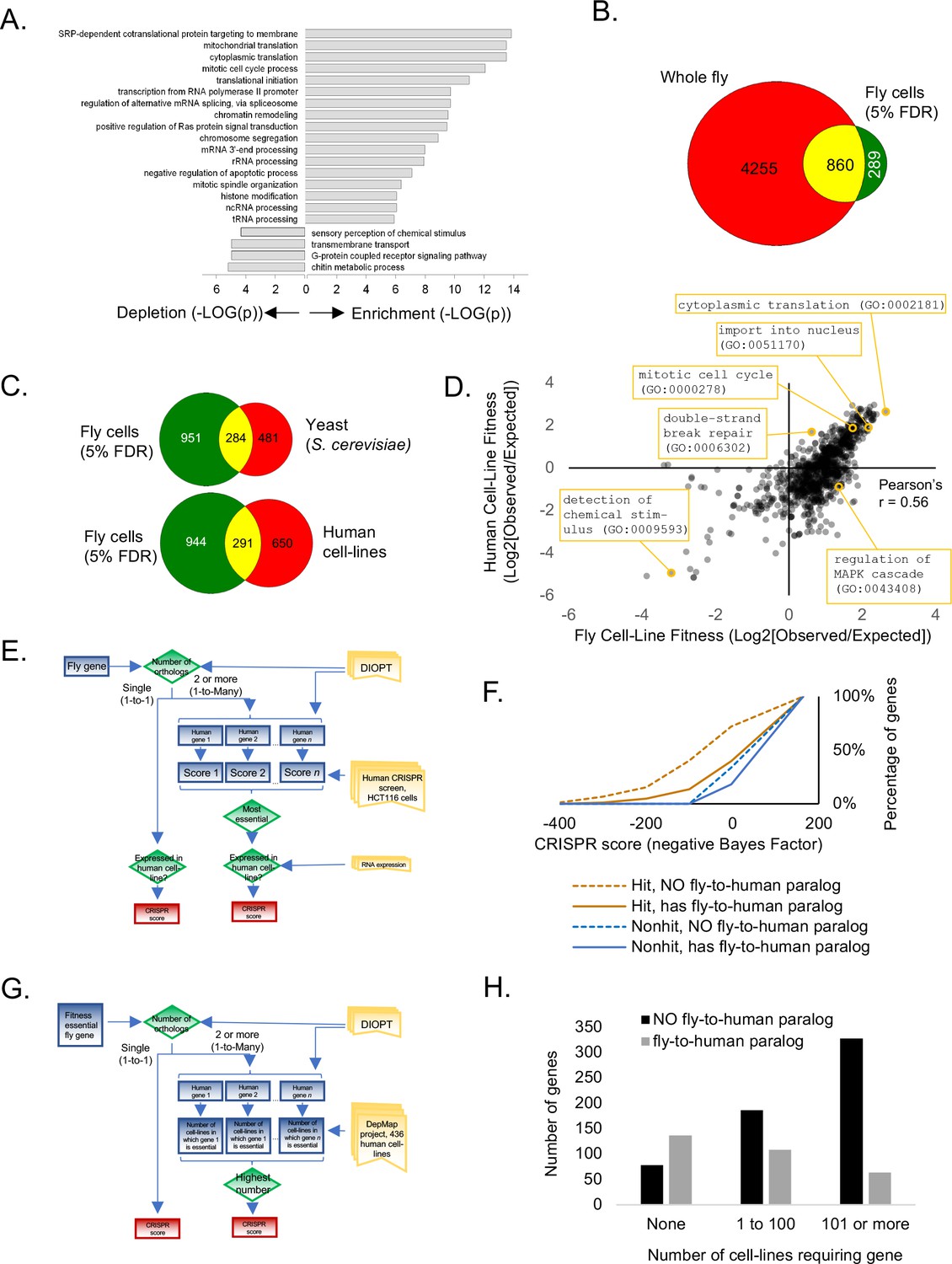
Analysis of Drosophila S2R+ fitness genes.
(A) Top enriched gene ontology (GO) terms for screen hits at 5% FDR compared with top enriched GO terms for non-hits. (B) Overlap between cell-line CRISPR hits at 5% FDR and all ‘lethal’ Flybase entries after subtracting entries with no allele information. (C) Overlap between Drosophila CRISPR hits at 5% FDR and orthologs in yeast (S. cerevisiae) or human cell-lines. (D) Gene ontology terms enriched in human CRISPR fitness screens (Hart et al., 2015) compared with fly CRISPR fitness screens. Selective listing of co-enriched terms, a co-depleted term, and outliers. (E) Schematic for ‘fly-to-human paralog’ assignment and testing using high-resolution human CRISPR screen data (Hart et al., 2015). For each fly gene with a unique human ortholog, no selection was performed. For fly genes with multiple human orthologs, the most essential human ortholog was chosen. Genes were included in the analysis only if expressed in the human cell-line. Ortholog assignment used DIOPT ‘high’ and ‘moderate’ confidence mapping calls (Hu et al., 2011). (F) Effect of fly-to-human paralogs on hit-calling in human CRISPR screens. Cumulative average of gene fitness essentiality (negative Bayes Factor) for high-resolution human cell-line CRISPR screen (Lenoir et al., 2018; Hart et al., 2015) examining indicated genesets: those with paralogs are dashed; orthologs of fly fitness genes are brown; orthologs of non-hits are blue. (G) Schematic for ‘fly-to-human paralog’ assignment and testing using cancer Dependency Map data (Tsherniak et al., 2017). A CERES score of <0.8 was used for fitness calls. (H) Effect of fly-to-human paralogs on number of cell-lines requiring a particular gene for fitness.
Figure 4
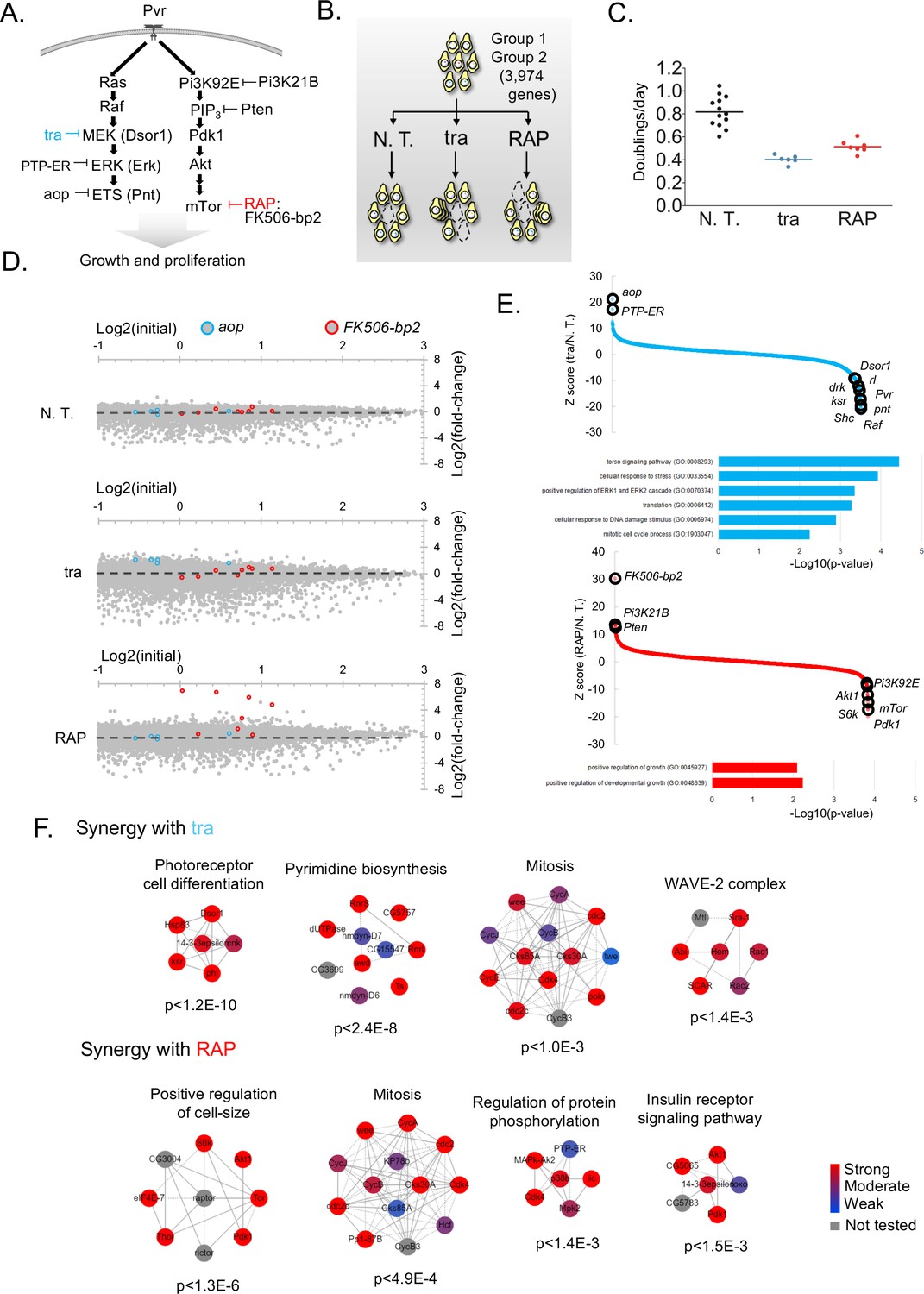
Screens to identify genes regulating cell growth and proliferation.
(A) Schematic of selected components of the Ras/ERK/ETS and PI3K/mTor pathways and of inhibition by trametinib (‘tra’) or rapamycin (‘RAP’). (B) Experimental schematic: pathway-specific perturbations to identify context-specific gene essentiality using Drosophila CRISPR screens. Dropout screens conducted with no additional treatment (N.T.) serves as a control. (C) Estimates of doubling per day obtained during periodic counting of cell pools to verify that tra and RAP partially inhibit cell growth. Each observation and mean doubling time plotted. (D) Plot of log2(initial distribution) versus log2(fold-change) for indicated sgRNAs in each screen. Pathway-specific resistance for sgRNAs targeting aop, a known suppressor of the Ras/ERK/ETS pathway in Drosophila (Lai and Rubin, 1992) or FK506-bp2, the putative cellular co-factor for rapamycin (Thomson and Johnson, 2010). (E) Computed maximum likelihood estimate (MLE) Z score based on sgRNA fold-change data comparing drug treatment condition with no treatment control. sgRNA fold-changes are mean of two independent replicates. Expected intra-pathway negative or positive regulators are noted (see Supplementary file 3 for complete hit list and raw data). GO terms for synergistic interactions are listed along with hypergeometric p-values for term assignment (PatherDB). (F) Physical protein-protein interaction (PPI) networks enriched using differential CRISPR screens in tra or RAP. PPI network prediction and reported p-values use COMPLEAT, and requires complexes to have >6 members per complex (Vinayagam et al., 2013).
Additional files
-
Supplementary file 1
Fitness essential gene data.
Worksheet 2 is a list of all genome-wide sgRNAs. Worksheet 1 contains computed MAGeCK (Li et al., 2014) MLE result for each independent replicate of the negative selection screen and the sgRNA-level average. Worksheet 3 contains primer sequences for cloning oligo pools. Worksheet 4 contains primers for primers for amplifying sgRNAs from cell pools (see protocol illustration in Figure 1—figure supplement 3).
- https://doi.org/10.7554/eLife.36333.012
-
Supplementary file 2
Uncharacterized (‘CG’) genes and insect-specific fitness essential genes.
- https://doi.org/10.7554/eLife.36333.013
-
Supplementary file 3
Context-specific fitness gene essentiality data.
Worksheet 1 contains computed MLE result of CRISPR screen conducted in each drug (tra or RAP) versus control. Worksheet 2 contains raw readcount file used to generate data.
- https://doi.org/10.7554/eLife.36333.014
-
Supplementary file 4
List file for group 1 Drosophila sgRNA library.
File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.015
-
Supplementary file 5
List file for group 2 Drosophila sgRNA library.
File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.016
-
Supplementary file 6
List file for group 3 Drosophila sgRNA library.
File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.017
-
Supplementary file 7
Readcount file for group 1, replicate 1 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.018
-
Supplementary file 8
Readcount file for group 2, replicate 1 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.019
-
Supplementary file 9
Readcount file for group 3, replicate 1 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.020
-
Supplementary file 10
Readcount file for group 1, replicate 2 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.021
-
Supplementary file 11
Readcount file for group 2, replicate 1 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.022
-
Supplementary file 12
Readcount file for group 3, replicate 1 of Drosophila sgRNA library following transfection and outgrowth.
Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.023
-
Supplementary file 13
Readcount file for average replicates 1 and 2, group 1.
Readcounts were internally normalized to a median value of 10000 prior to computing the average. Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.024
-
Supplementary file 14
Readcount file for average replicates 1 and 2, group 2.
Readcounts were internally normalized to a median value of 10000 prior to computing the average. Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.025
-
Supplementary file 15
Readcount file for average replicates 1 and 2, group 3.
Readcounts were internally normalized to a median value of 10000 prior to computing the average. Column 1 provides internal ID number compatible with Supplementary file 1. Column 2 provides targeted gene ID. Column 3, ‘REF’, provides readcount file from plasmid pool. Column 4 provides readcount following transfection and outgrowth. File is compatible with MAGeCK (Li et al., 2014).
- https://doi.org/10.7554/eLife.36333.026
-
Transparent reporting form
- https://doi.org/10.7554/eLife.36333.027
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells
eLife 7:e36333.
https://doi.org/10.7554/eLife.36333




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}