An updated phylogeny of the Alphaproteobacteria reveals that the parasitic Rickettsiales and Holosporales have independent origins
- Dalhousie University, Canada
- University of Cologne, Germany
- Université de Montréal, Canada
Figures
Figure 1
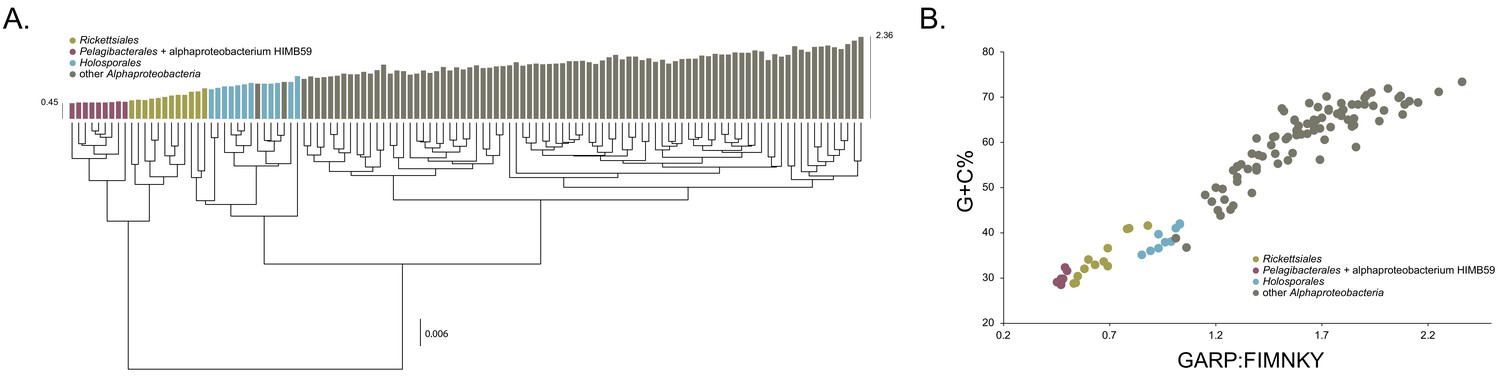
Compositional heterogeneity in the Alphaproteobacteria is a major factor that confounds phylogenetic inference.
There are great disparities in the genome G + C% content and amino acid compositions of the Rickettsiales, Pelagibacterales (including alphaproteobacterium HIMB59) and Holosporales with all other alphaproteobacteria. (A) A UPGMA (average-linkage) clustering of amino acid compositions (based on the 200 gene set for the Alphaproteobacteria) shows that the Rickettsiales (brown), Pelagibacterales (maroon), and Holosporales (light blue) all have very similar proteome amino acid compositions. At the tips of the tree, GARP:FIMNKY amino acid ratio values are shown as bars. (B) A scatterplot depicting the strong correlation between G + C% (nucleotide compositions) and GARP:FIMNKY ratios (amino acid composition) for the 120 taxa in the Alphaproteobacteria (and outgroup) shows a similar clustering of the Rickettsiales, Pelagibacterales (including alphaproteobacterium HIMB59) and Holosporales.
Figure 2 with 7 supplements
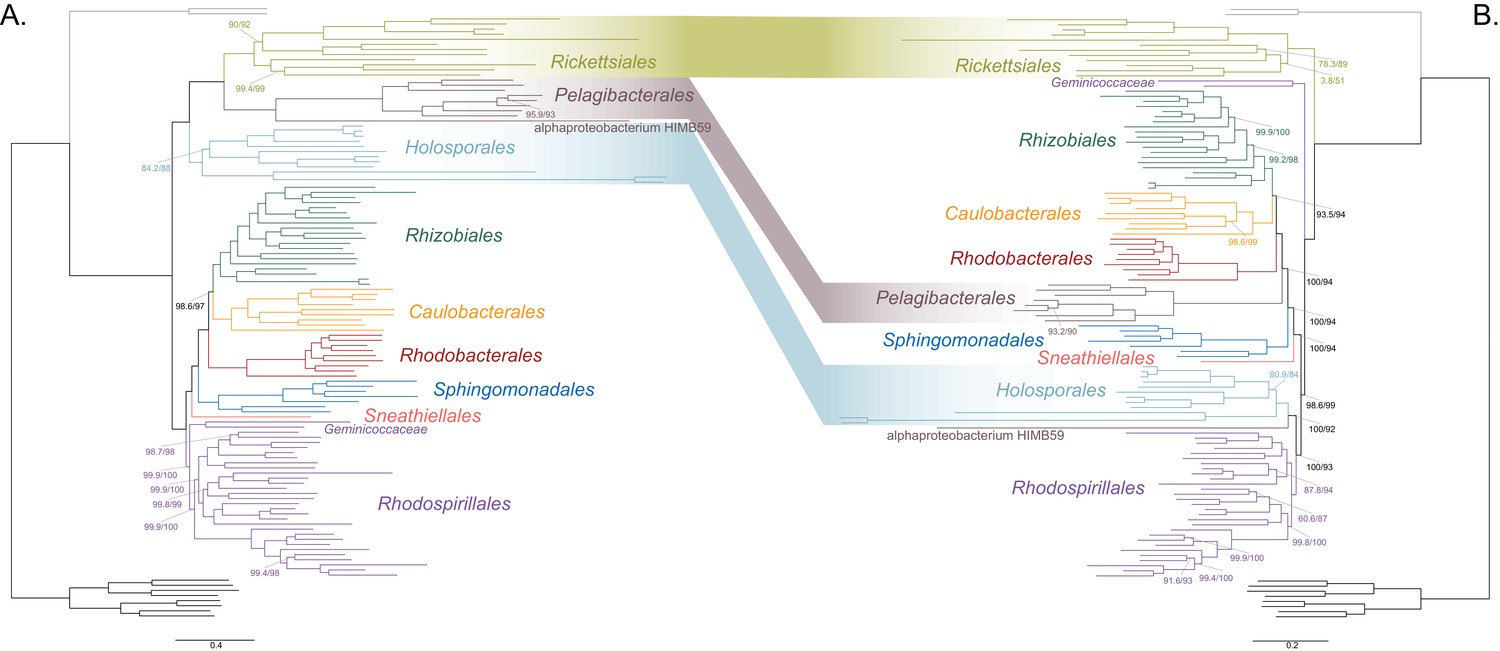
Decreasing compositional heterogeneity by removing compositionally biased sites disrupts the clustering of the Rickettsiales, Pelagibacterales (including alphaprotobacterium HIMB59) and Holosporales.
All branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A maximum-likelihood tree inferred under the LG + PMSF(ES60)+F + R6 model and from the untreated dataset which is highly compositionally heterogeneous. The three long-branched orders, the Rickettsiales, Pelagibacterales (including alphaprotobacterium HIMB59) and Holosporales, that have similar amino acid compositions form a clade. (B) A maximum-likelihood tree inferred under the LG + PMSF(ES60)+F + R6 model and from a dataset whose compositional heterogeneity has been decreased by removing 50% of the most biased sites according to ɀ. In this phylogeny, the clustering of the Rickettsiales, Pelagibacterales and Holosporales is disrupted. The Pelagibacterales is sister to the Rhodobacterales, Caulobacterales and Rhizobiales. The Holosporales, and alphaproteobacterium HIMB59, become sister to the Rhodospirillales. The Rickettsiales remains as the sister to the Caulobacteridae. See Figure 2—figure supplement 1 for taxon names. See Figure 2—figure supplement 3 for the Bayesian consensus trees inferred in PhyloBayes MPI v1.7 under the CAT-Poisson+Γ4 model. See also Figure 2—figure supplements 2 and 4–7.
Figure 2—figure supplement 1

A labeled version showing taxon names for Figure 2.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated.
Figure 2—figure supplement 2
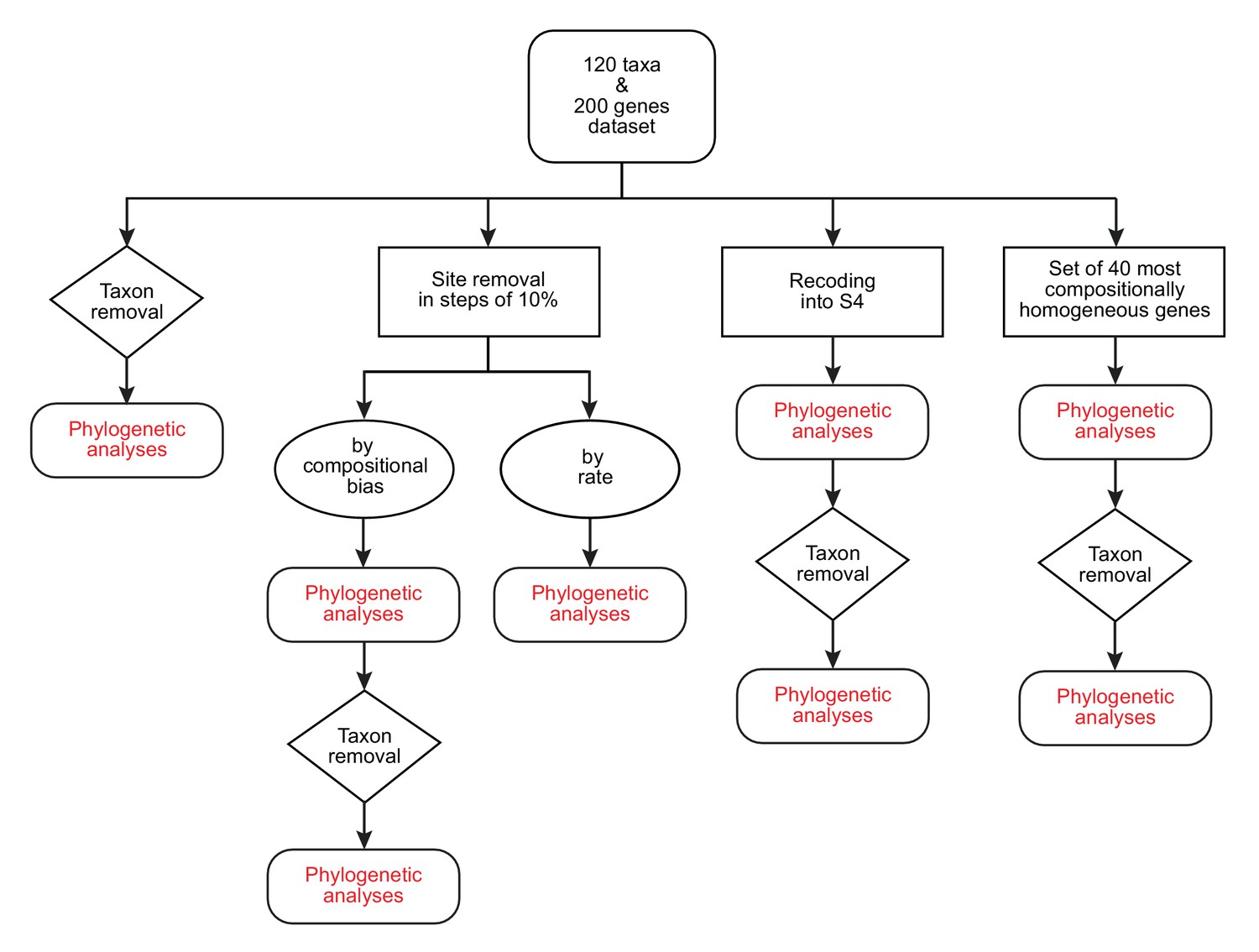
A diagram of the strategies and phylogenetic analyses employed in this study.
https://doi.org/10.7554/eLife.42535.007
Figure 2—figure supplement 3
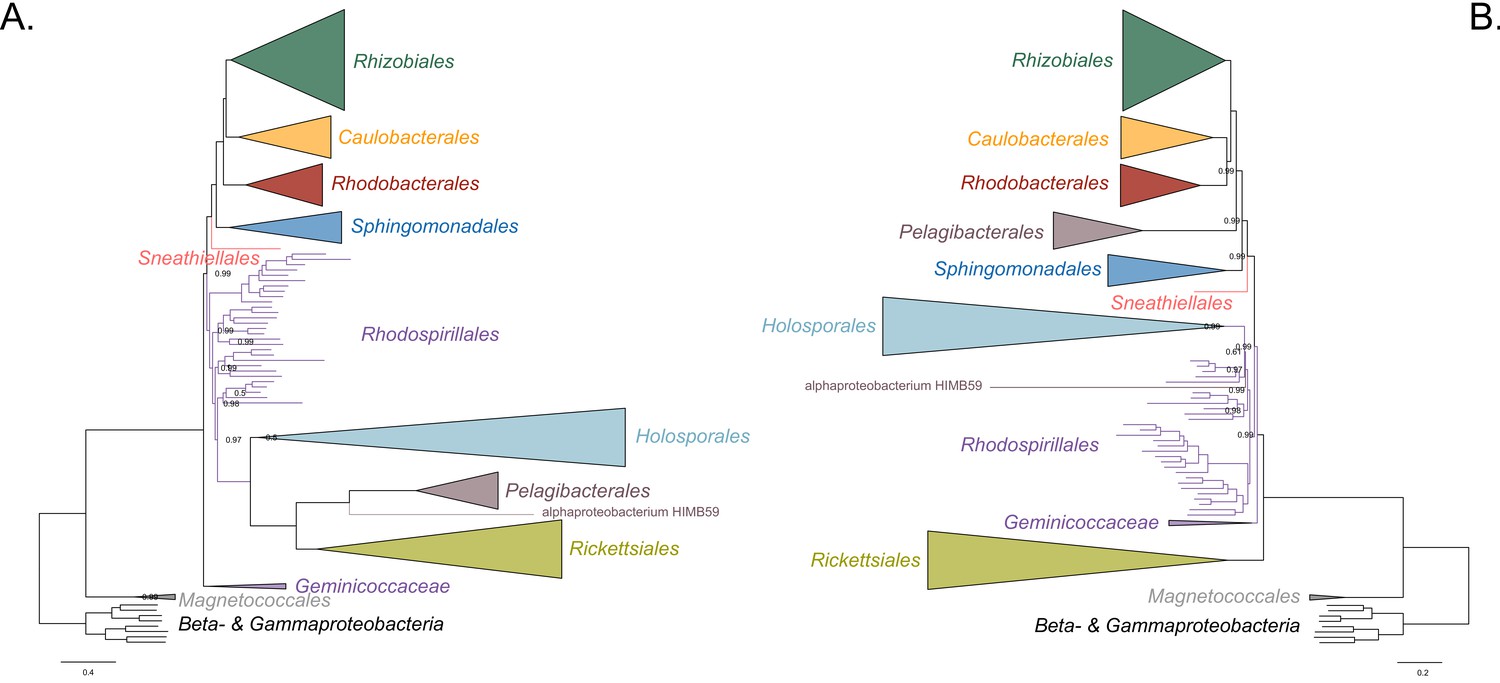
Bayesian consensus trees inferred with PhyloBayes MPI v1.7 and the CAT-Poisson+Γ4 model.
Branch support values are 1.0 posterior probabilities unless annotated. (A) Bayesian consensus tree inferred from the full dataset which is highly compositionally heterogeneous. (B) Bayesian consensus tree inferred from a dataset whose compositional heterogeneity has been decreased by removing 50% of the most biased sites according to ɀ. See Figure 2A and B for the most likely trees inferred in IQ-TREE v1.5.5 and the LG + PMSF(C60)+F + R6 model.
Figure 2—figure supplement 4
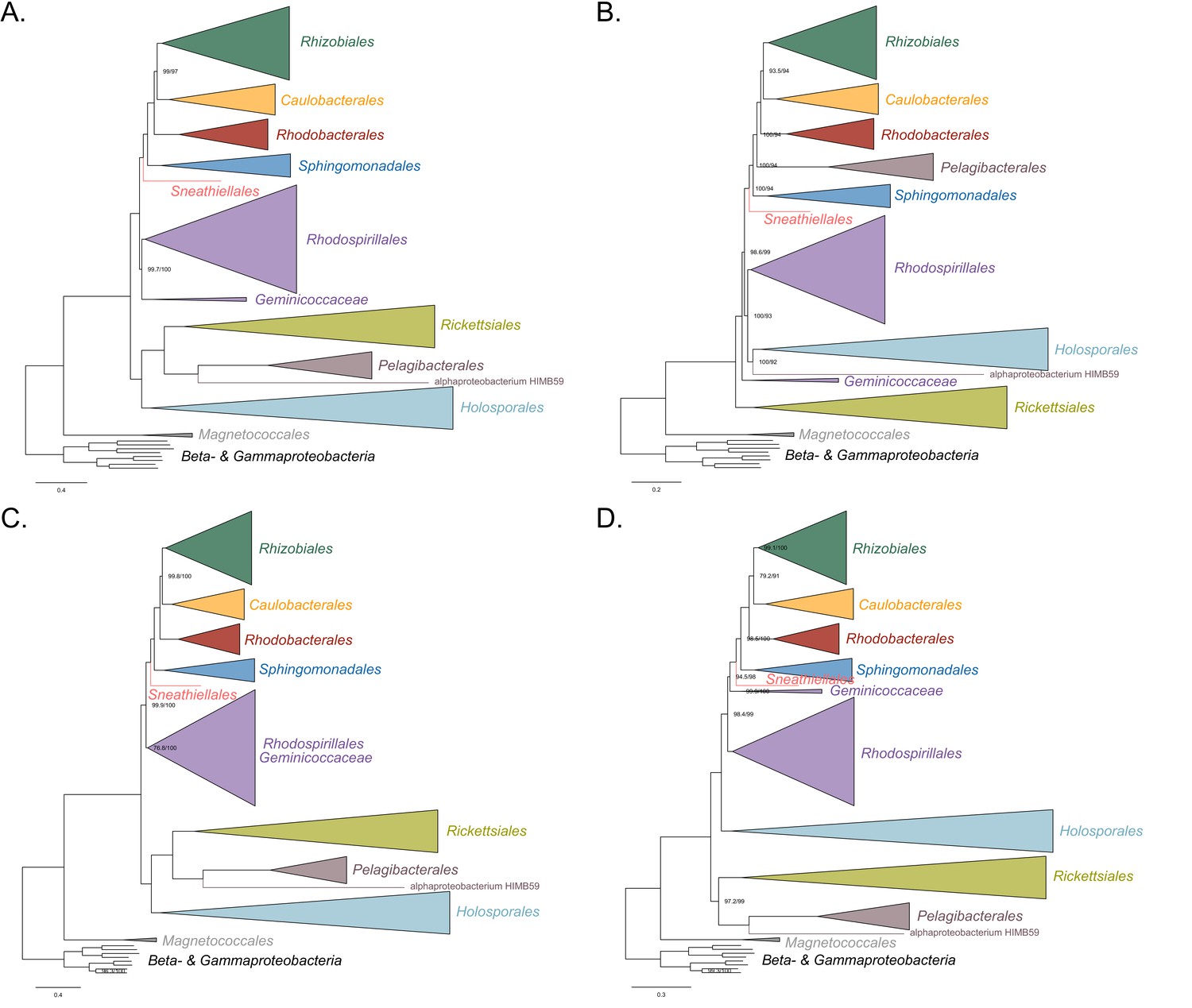
Maximum-likelihood trees to assess the placements of the Holosporales, Rickettsiales, Pelagibacterales and alphaproteobacterium HIMB59 when all four groups are included.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A tree that results from the analysis of the untreated dataset. (B) A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: RNCM EHIPTWV ADQLKS GFY). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes.
Figure 2—figure supplement 5
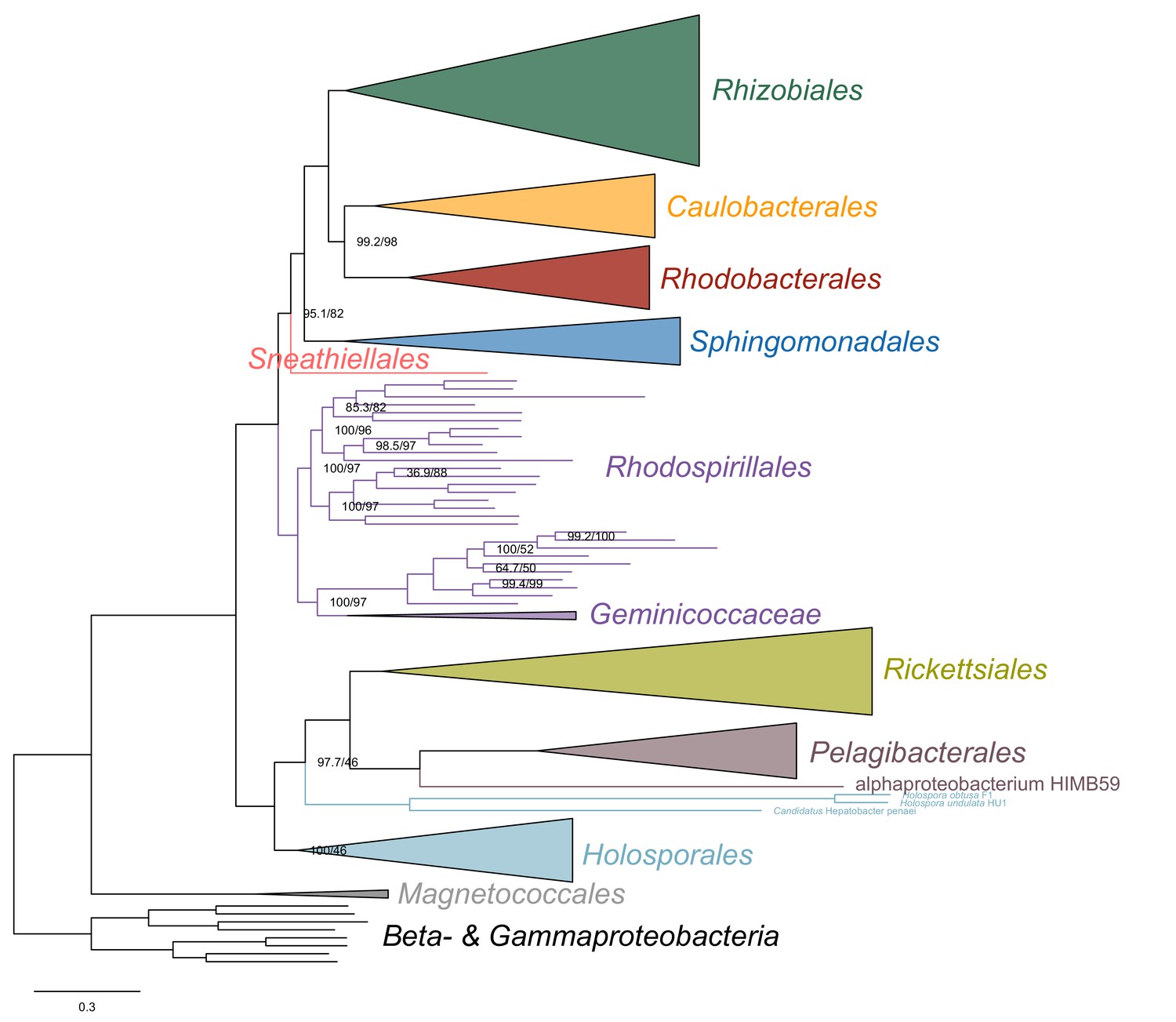
Maximum-likelihood tree from the untreated dataset from which no taxon has been removed and analyzed under simpler LG4X model.
In this tree, derived from an analysis using a model that does not account for compositional heterogeneity across sites, the Geminicoccaceae has a more derived placements within the Rhodospirillales as sister to the Acetobacteraceae.
Figure 2—figure supplement 6
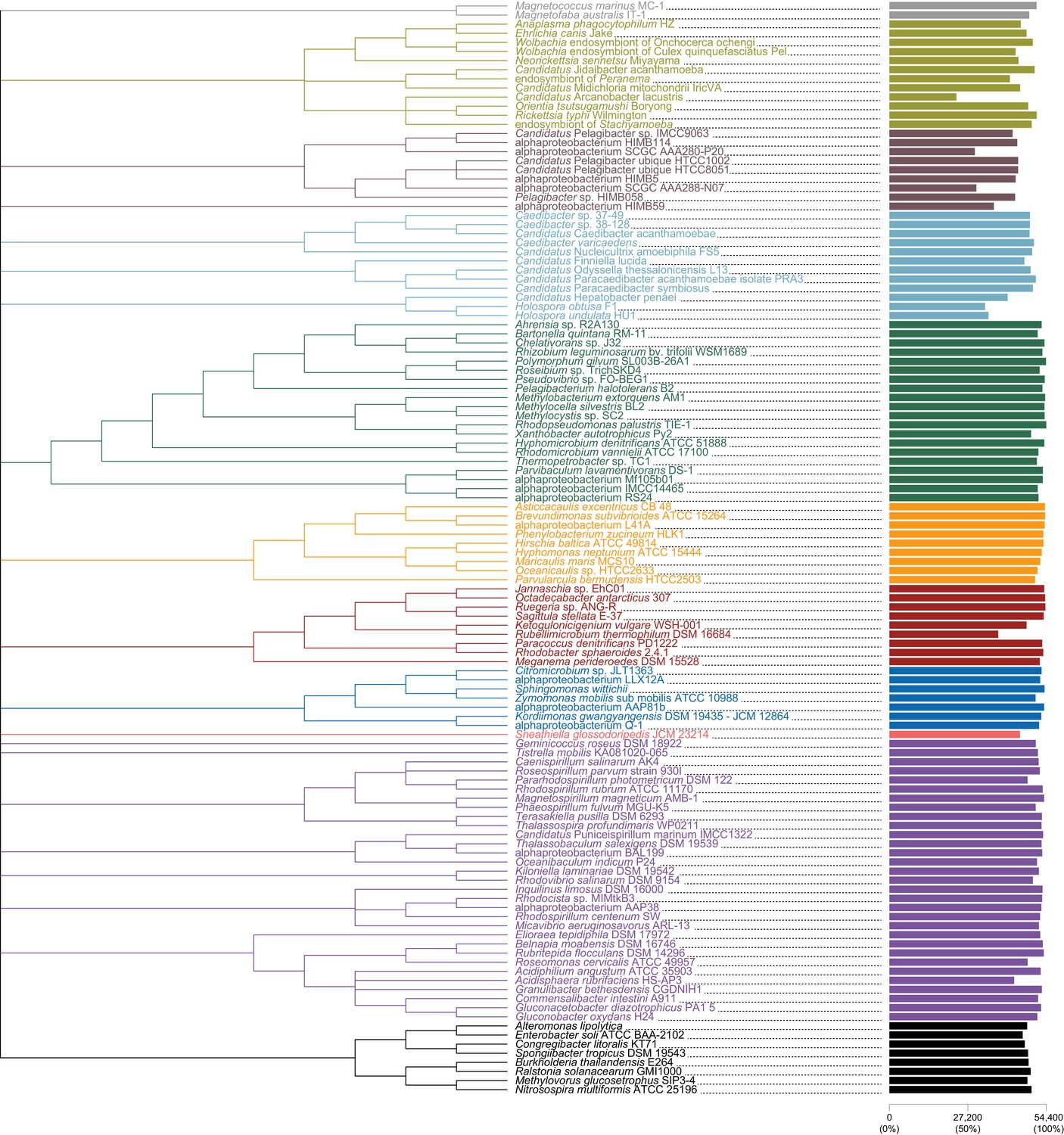
Constraint tree, used for IQ-TREE analyses, labeled with taxon names and also degree of missing data per taxon.
Magnetococcales in gray; Rickettsiales in brown; Pelagibacterales in maroon; Holosporales in light blue; Rhizobiales in green; Caulobacterales in orange; Rhodobacterales in red; Sneathiellales in pink; Rhodospirillales in purple; Beta- and Gammaproteobacteria in black.
Figure 2—figure supplement 7
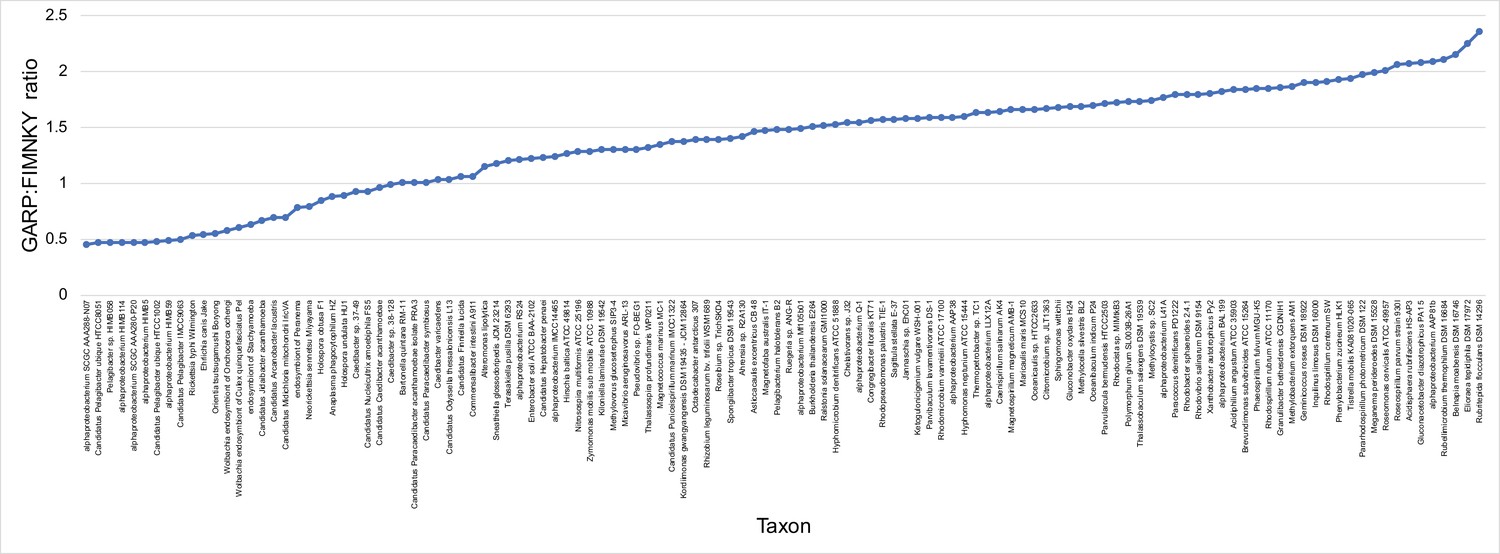
GARP:FIMNKY ratios across the proteomes of the 120 alphaproteobacteria and outgroup used in this study.
https://doi.org/10.7554/eLife.42535.012
Figure 3 with 8 supplements
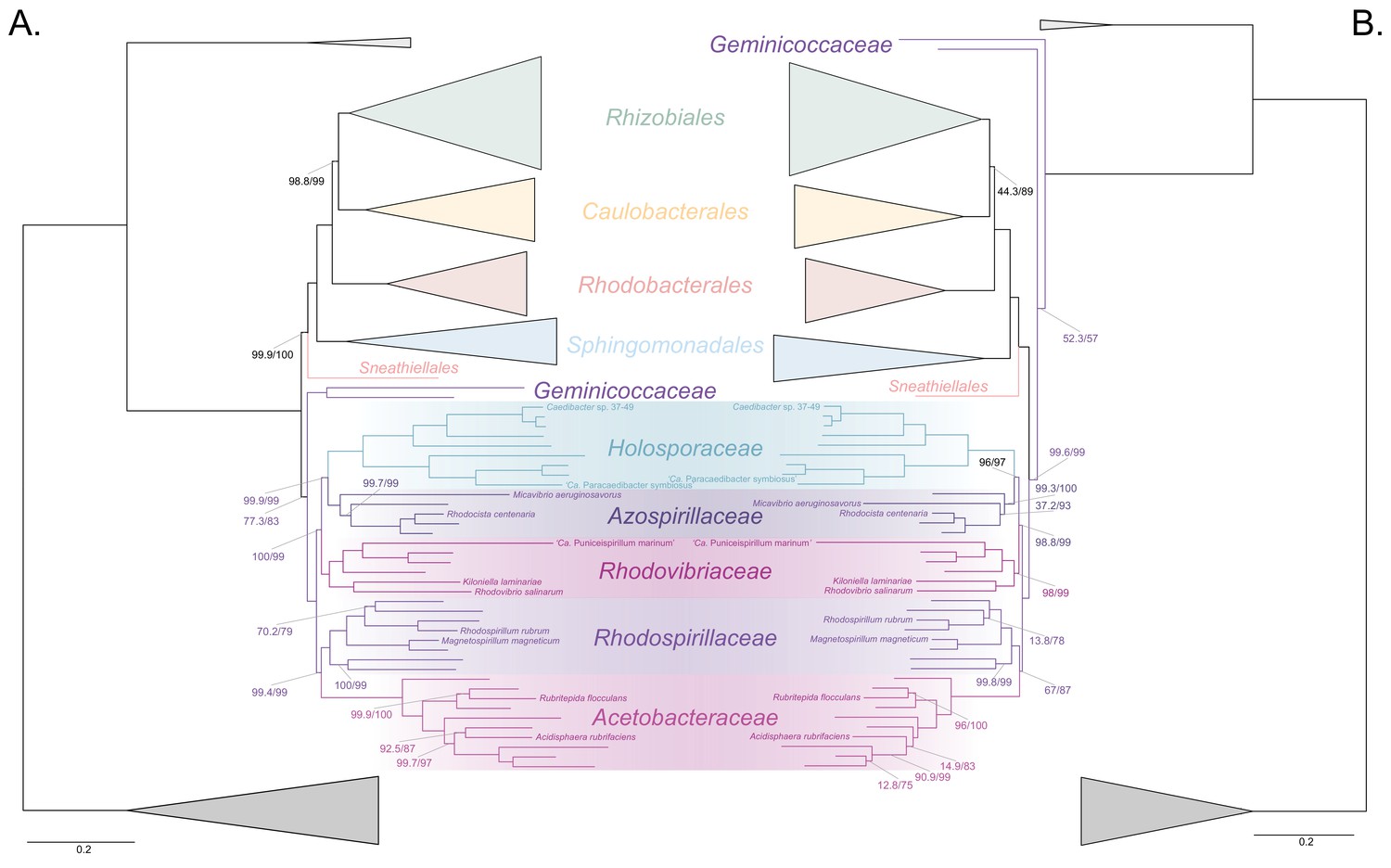
The Holosporales (renamed and lowered in rank to the Holosporaceae family here) branches in a derived position within the Rhodospirillales when compositional heterogeneity is reduced and the long-branched and compositionally biased Rickettsiales, Pelagibacterales, and alphaproteobacterium HIMB59 are removed.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A maximum-likelihood tree, inferred under the LG + PMSF(ES60)+F + R6 model, to place the Holosporaceae in the absence of the Rickettsiales, Pelagibacterales, and alphaproteobacterium HIMB59 and when compositional heterogeneity has been decreased by removing 50% of the most biased sites. The Holosporaceae is sister to the Azospirillaceae fam. nov. within the Rhodospirillales. (B) A maximum-likelihood tree, inferred under the GTR + ES60 S4+F + R6 model, to place the Holosporaceae in the absence of the Rickettsiales, Pelagibacterales, and alphaproteobacterium HIMB59, and when the data have been recoded into a four-character state alphabet (the dataset-specific recoding scheme S4: ARNDQEILKSTV GHY CMFP W) to reduce compositional heterogeneity. This phylogeny shows a pattern that matches that inferred when compositional heterogeneity has been alleviated through site removal. See Figure 3—figure supplement 6 for the Bayesian consensus trees inferred in PhyloBayes MPI v1.7 and under the and the CAT-Poisson+Γ4 model. See also Figure 3—figure supplements 1–5 and 7–8.
Figure 3—figure supplement 1
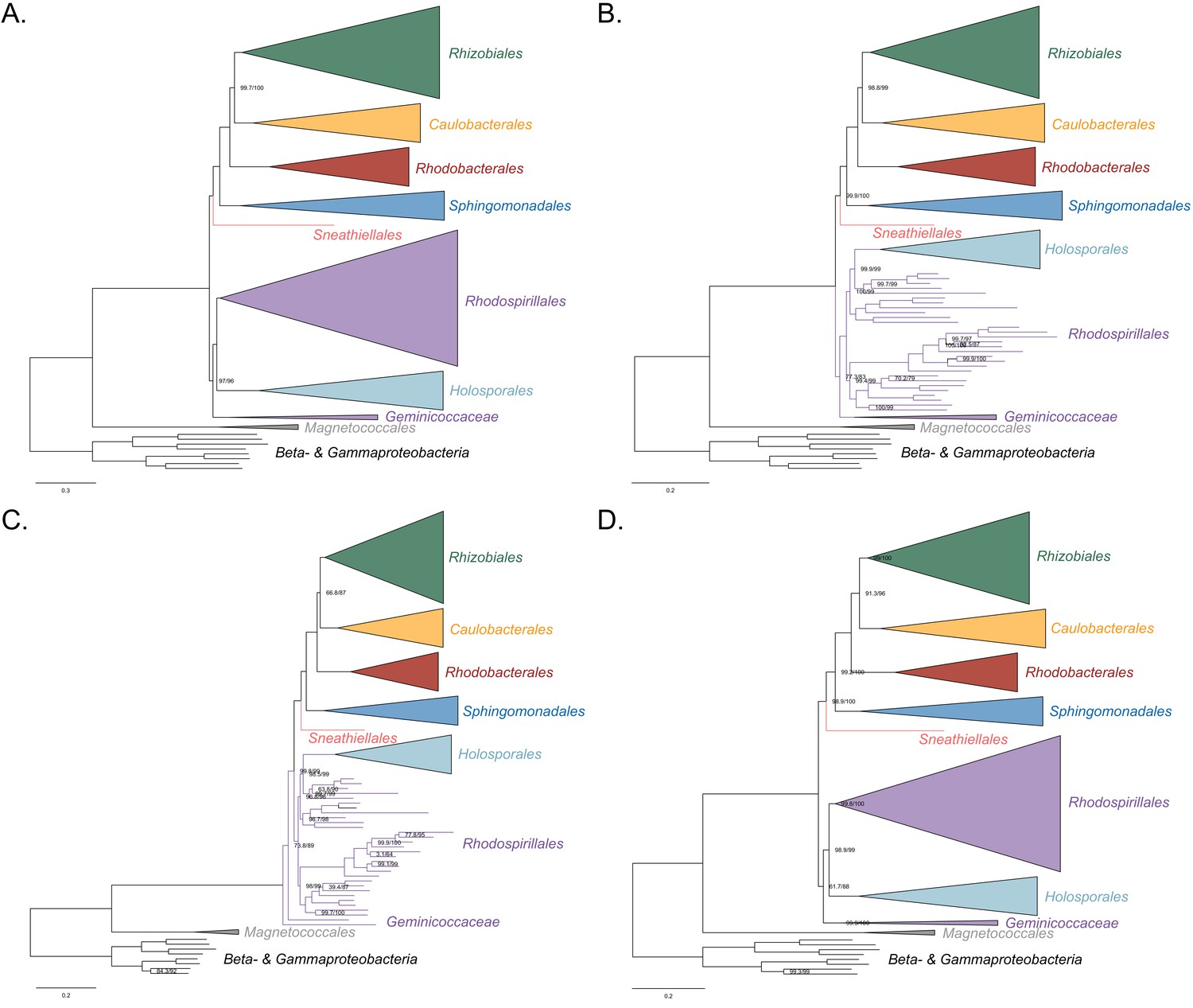
Maximum-likelihood trees to assess the placement of the Holosporales in the absence of the Rickettsiales, Pelagibacterales and alphaproteobacterium HIMB59.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A tree that results from the analysis of the untreated dataset. () A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: ARNDQEILKSTV GHY CMFP W). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes.
Figure 3—figure supplement 2
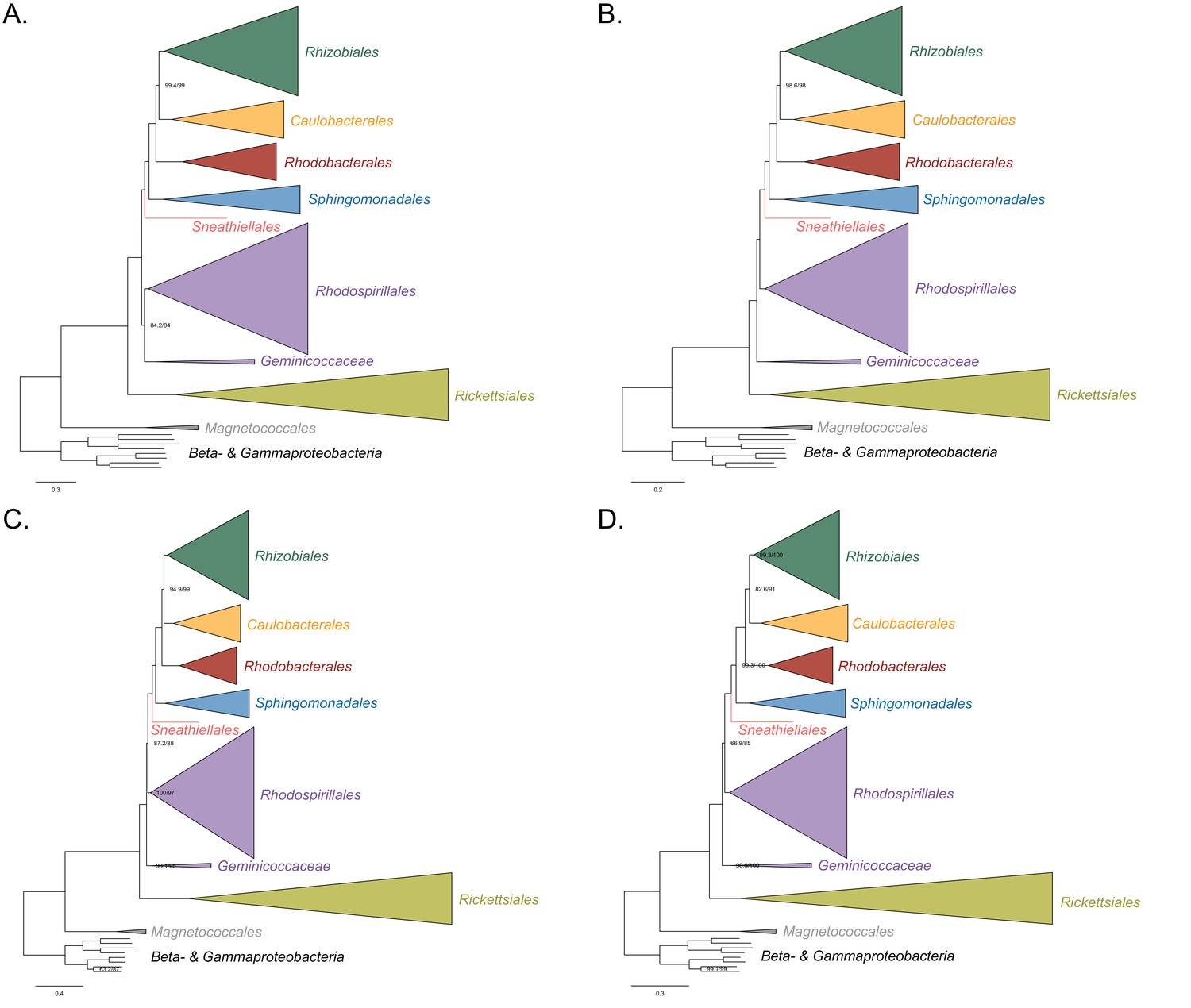
Maximum-likelihood trees to assess the placement of the Rickettsiales in the absence of the Holosporales, Pelagibacterales, and alphaproteobacterium HIMB59.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A tree that results from the analysis of the untreated dataset. (B) A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: PY RNMF GHLKTW ADCQEISV). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes.
Figure 3—figure supplement 3
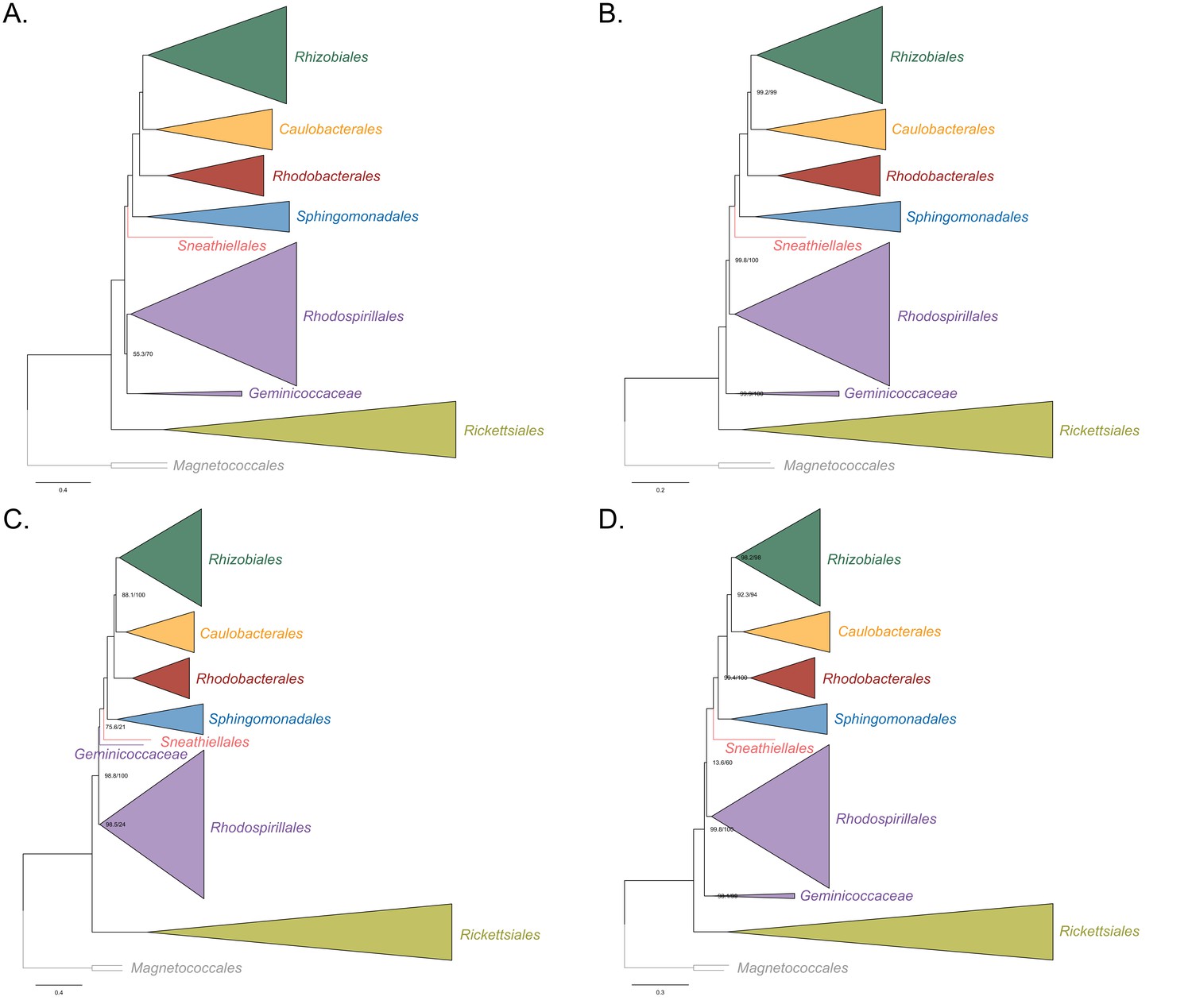
Maximum-likelihood trees to assess the placement of the Rickettsiales in the absence of the Holosporales, Pelagibacterales, alphaproteobacterium HIMB59 and the Beta-, and Gammaproteobacteria outgroup.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A tree that results from the analysis of the untreated dataset. (B) A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: RNMF GHLKTW ADCQEISV PY). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes. Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated.
Figure 3—figure supplement 4
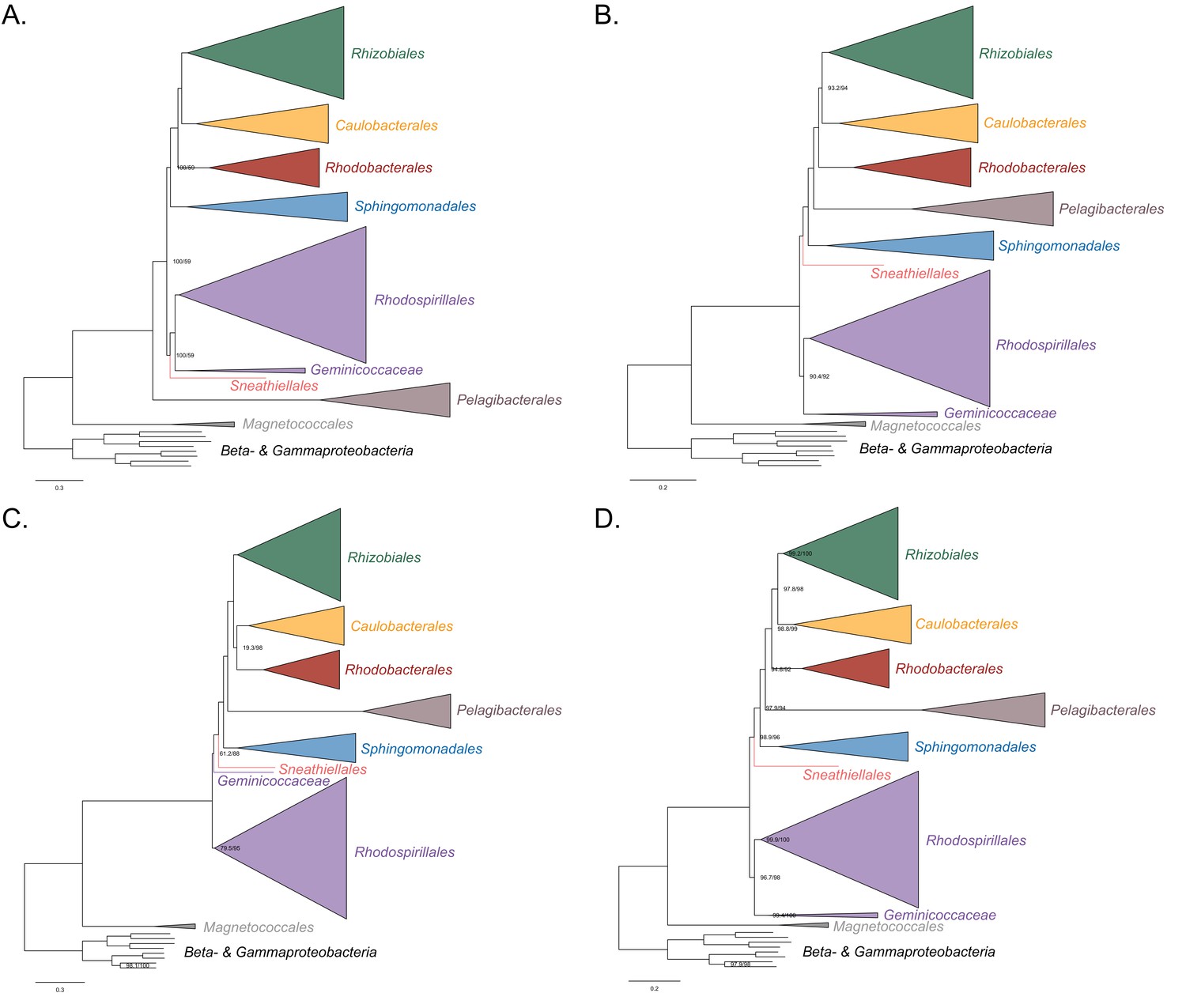
Maximum-likelihood trees to assess the placement of the Pelagibacterales in the absence of the Holosporales, Rickettsiales and alphaproteobacterium HIMB59.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated. (A) A tree that results from the analysis of the untreated dataset. (B) A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: EGIV ARNDQHKMPSY LFT CW). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes.
Figure 3—figure supplement 5
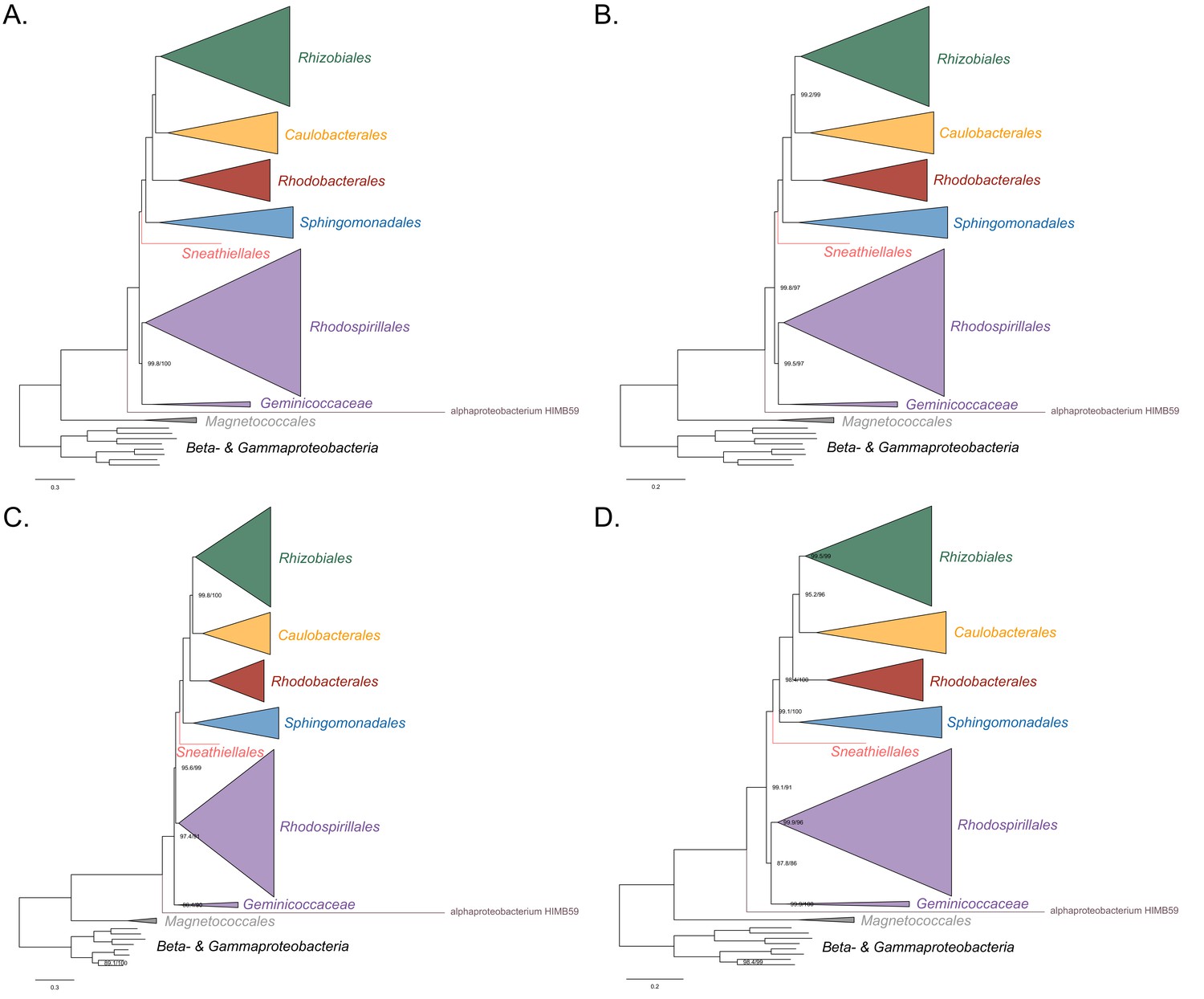
Maximum-likelihood trees to assess the placement of alphaproteobacterium HIMB59 in the absence of the Holosporales, Rickettsiales and Pelagibacterales.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated.( A) A tree that results from the analysis of the untreated dataset. (B) A tree that results from the analysis of a dataset from which the 50% most compositionally biased sites have been removed. (C) A tree that results from the analysis of a dataset that has been recoded into the four-character state recoding scheme S4 (recoding scheme: RLKMT ANDQEIPSV CW GHFY). (D) A tree that results from the analysis of a dataset that only comprises the 40 most compositionally homogeneous genes.
Figure 3—figure supplement 6
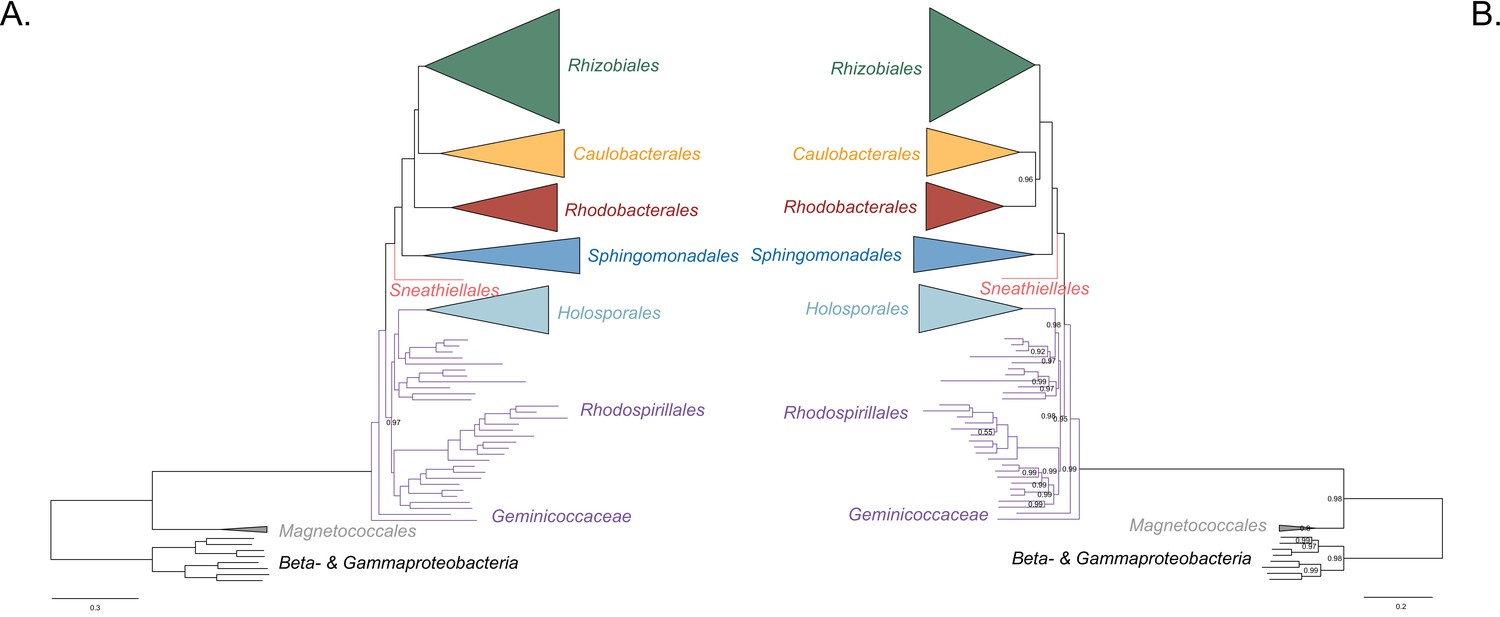
Bayesian consensus trees inferred with PhyloBayes MPI v1.7 and the CAT-Poisson+Γ4 model.
Branch support values are 1.0 posterior probabilities unless annotated. (A) Bayesian consensus tree inferred to place the Holosporales in the absence of the Rickettsiales and the Pelagibacterales and when compositional heterogeneity has been decreased by removing 50% of the most biased sites according to ɀ. (B) Bayesian consensus tree inferred to place the Holosporales in the absence of the Rickettsiales and the Pelagibacterales and when the data have been recoded into a four-character state alphabet (the dataset-specific recoding scheme S4: ARNDQEILKSTV GHY CMFP W) to reduce compositional heterogeneity. See Figure 2A and B for the most likely trees inferred in IQ-TREE v1.5.5 and the LG + PMSF(C60)+F + R6 and GTR + ES60 S4+F + R6 models, respectively.
Figure 3—figure supplement 7
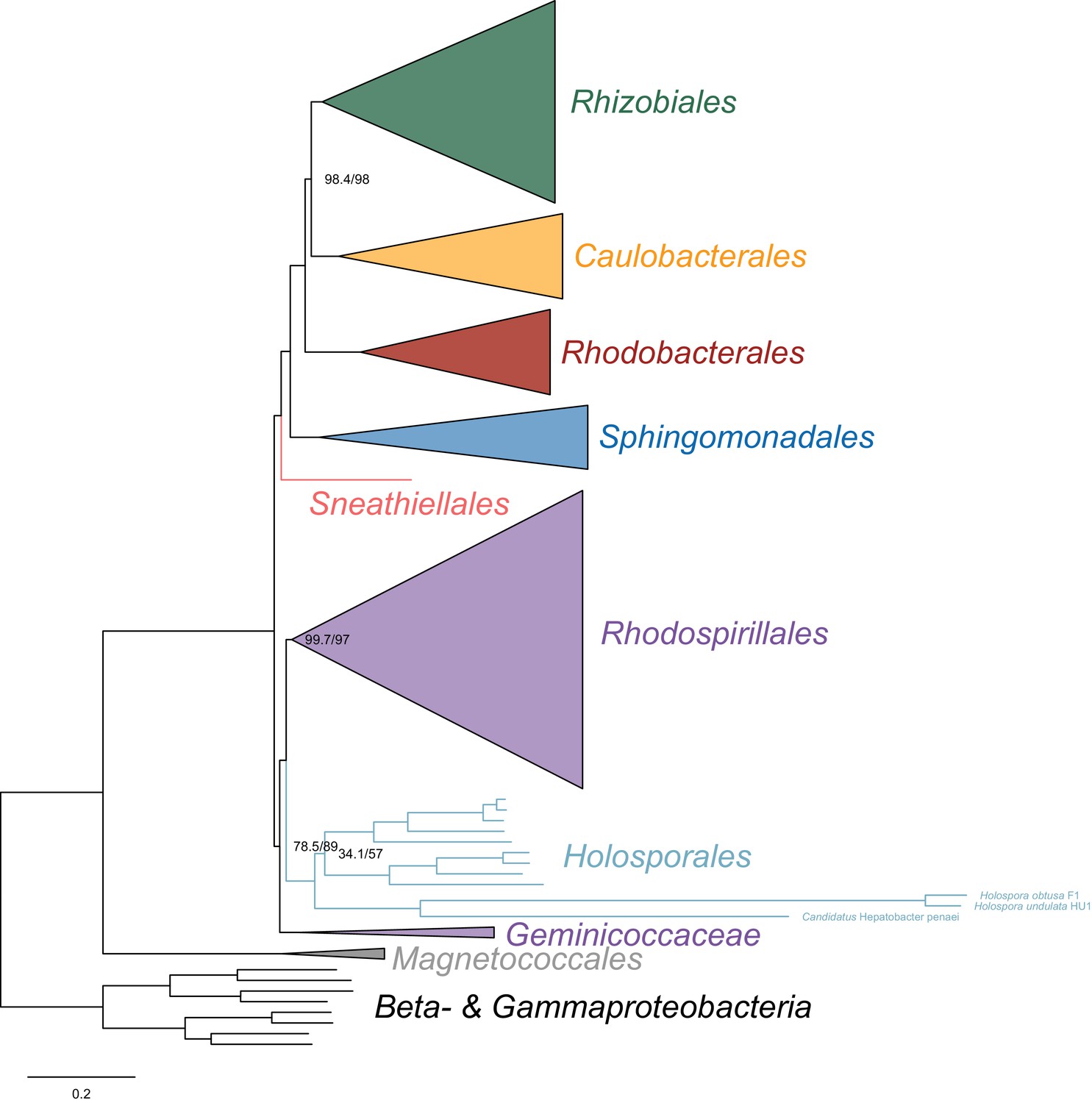
Maximum-likelihood trees to assess the placement of the Holosporales when the fast-evolving Holospora and ‘Candidatus Hepatobacter’ are also included in the absence of the Rickettsiales, Pelagibacterales and alphaproteobacterium HIMB59.
Branch support values are 100% SH-aLRT and 100% UFBoot unless annotated.
Figure 3—figure supplement 8
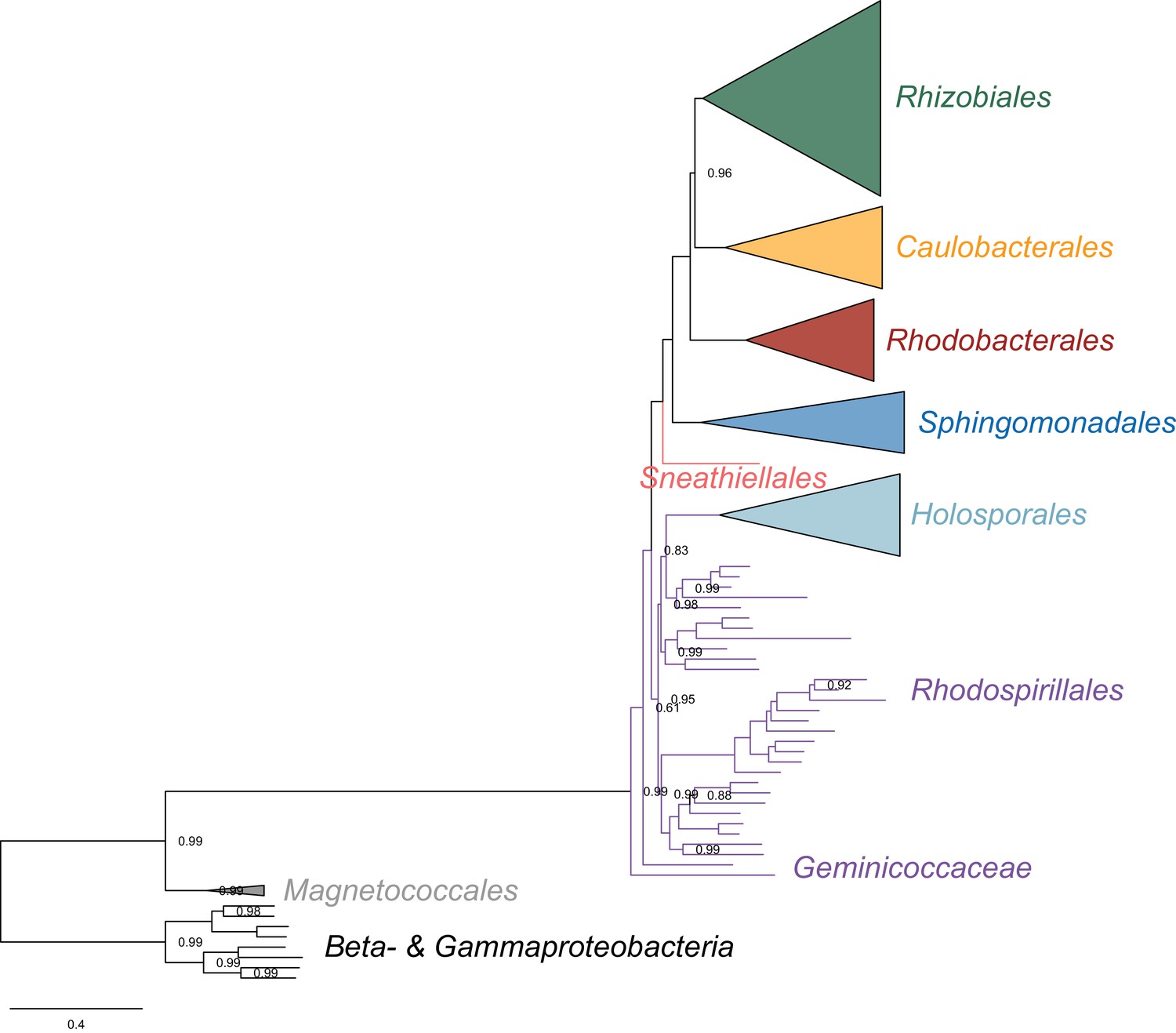
Bayesian consensus tree inferred to place the Holosporales in the absence of the Pelagibacterales, alphaproteobacterium HIMB59, and Rickettsiales, and when the data have been recoded into a six-character state alphabet (the dataset-specific recoding scheme S6: AQEHISV RKMT PY DCLF NG W) to reduce compositional heterogeneity.
Branch support values are 1.0 posterior probabilities unless annotated.
Tables
Table 1
Genome features for the three novel rickettsialeans sequenced in this study.
See Supplementary file 1 as well.
| Species | ‘Candidatus Finniella inopinata’ | Stachyamoeba-associated rickettsialean | Peranema-associated rickettsialean |
|---|---|---|---|
| Genome size | 1,792,168 bp | 1,738,386 bp | 1,375,759 bp |
| N50 | 174,737 bp | 1,738,386 bp | 28,559 bp |
| Contig number | 28 | 1 | 125 |
| Gene number† | 1741 | 1588 | 1223 |
| A + T% content | 56.58% | 67.01% | 59.13% |
| Family | 'Candidatus Paracaedibacteraeae' | Rickettsiaceae | ‘Candidatus Midichloriaceae’ |
| Order | Holosporales | Rickettsiales | Rickettsiales |
| Completeness‡ | 94.96% | 97.12% (=100%) | 92.08% |
| Redundancy‡ | 0.0% | 0.0% | 2.1% |
-
†as predicted by Prokka v.1.13 (rRNA genes were searched with BLAST).
‡as estimated by Anvi’o v.2.4.0 using the Campbell et al., 2013 marker gene set.
Table 2
A higher-level classification scheme for the Alphaproteobacteria and the Magnetococcia classes within the Proteobacteria, and the Rickettsiales and Rhodospirillales orders within the Alphaproteobacteria.
https://doi.org/10.7554/eLife.42535.022| Class 1. Alphaproteobacteria Garrity et al., 2005 |
| Subclass 1. Rickettsidae Ferla et al., 2013 emend. Muñoz-Gómez et al. 2019 (this work) |
| Order 1. Rickettsiales Gieszczkiewicz, 1939 emend. Dumler et al., 2001 |
| Family 1. Anaplasmataceae Philip, 1957 Family 2. 'Candidatus Midichloriaceae' Montagna et al., 2013 Family 3. Rickettsiaceae Pinkerton, 1936 |
| Subclass 2. Caulobacteridae Ferla et al., 2013 emend. Muñoz-Gómez et al. 2019 |
| Order 1. Rhodospirillales Pfennig and Trüper, 1971 emend. Muñoz-Gómez et al. 2019 |
| Family 1. Acetobacteraceae (ex Henrici 1939) Gillis and De Ley, 1980 Family 2. Rhodospirillaceae Pfennig and Trüper, 1971 emend. Muñoz-Gómez et al. 2019 Family 3. Azospirillaceae fam. nov. Muñoz-Gómez et al. 2019 Family 4. Holosporaceae Szokoli et al., 2016 Family 5. Rhodovibriaceae fam. nov. Muñoz-Gómez et al. 2019 Family 6. Geminicoccaceae Proença et al., 2018 |
| Order 2. Sneathiellales Kurahashi et al., 2008 |
| Order 3. Sphingomonadales Yabuuchi and Kosako, 2005 |
| Order 4. Pelagibacterales Grote et al., 2012 |
| Order 5. Rhodobacterales Garrity et al., 2005 |
| Order 6. Caulobacterales Henrici and Johnson, 1935 |
| Order 7. Rhizobiales Kuykendall, 2005 |
| Class 2. Magnetococcia Parks et al., 2018 |
| Order 1. Magnetococcales Bazylinski et al., 2013 |
Additional files
-
Supplementary file 1
A 16S rRNA gene maximum-likelihood tree of the Rickettsiales and Holosporales that phylogenetically places the three endosymbionts whose genomes were sequenced in this study.
(1) ‘Candidatus Finniella inopinata’ endosymbiont of Viridiraptor invadens strain Virl02, (2) an alphaproteobacterium associated with Peranema trichophorum strain CCAP 1260/1B, and (3) an alphaproteobacterium associated with Stachyamoeba lipophora strain ATCC 50324. Branch support values are SH-aLRT and UFBoot.
- https://doi.org/10.7554/eLife.42535.023
-
Supplementary file 2
Supplementary tables.
(A) Ultrafast bootstrap (UFBoot) variation for several clades discussed in this study as compositionally biased sites, according to ɀ, are progressively removed in steps of 10%. (B) Ultrafast bootstrap (UFBoot) variation for several clades discussed in this study as the fastest sites are progressively removed in steps of 10%. (C) GenBank assembly accession numbers for the 120 alphaproteobacterial and outgroup genomes used in this study. (D) A list of the least compositionally heterogeneous genes out of the 200 single-copy and vertically inherited genes used in this study. (E) Model fit of amino acid replacement matrices as components of simple models that do not account for compositional heterogeneity across sites. Models are ordered from lowest to highest BIC. -LnL: log-likelihood; df: degrees of freedom or number of free parameters; AIC: Akaike information criterion; AICc: corrected Akaike information criterion; BIC: Bayesian information criterion. (F) Model fit of amino acid replacement matrices as components of complex models that account for compositional heterogeneity across sites. Models are ordered from lowest to highest BIC. -LnL: log-likelihood; df: degrees of freedom or number of free parameters; AIC: Akaike information criterion; AICc: corrected Akaike information criterion; BIC: Bayesian information criterion. (G) Model fit of LG + ES60+F for which the model component that accounts for rate heterogeneity across sites varies. Models are ordered from lowest to highest BIC. -LnL: log-likelihood; df: degrees of freedom or number of free parameters; AIC: Akaike information criterion; AICc: corrected Akaike information criterion; BIC: Bayesian information criterion. (H) Several summary statistics for the PhyloBayes MCMC chains run for each analysis under the CAT-Poisson+Γ4.
- https://doi.org/10.7554/eLife.42535.024
-
Transparent reporting form
- https://doi.org/10.7554/eLife.42535.025
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
An updated phylogeny of the Alphaproteobacteria reveals that the parasitic Rickettsiales and Holosporales have independent origins
eLife 8:e42535.
https://doi.org/10.7554/eLife.42535




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}