Proteome-wide signatures of function in highly diverged intrinsically disordered regions
- University of Toronto, Canada
- Harvard University, United States
- Max Planck Institute of Molecular Cell Biology and Genetics, Germany
- Technische Universität Dresden, Germany
- Hospital for Sick Children, Canada
Figures
Figure 1 with 2 supplements
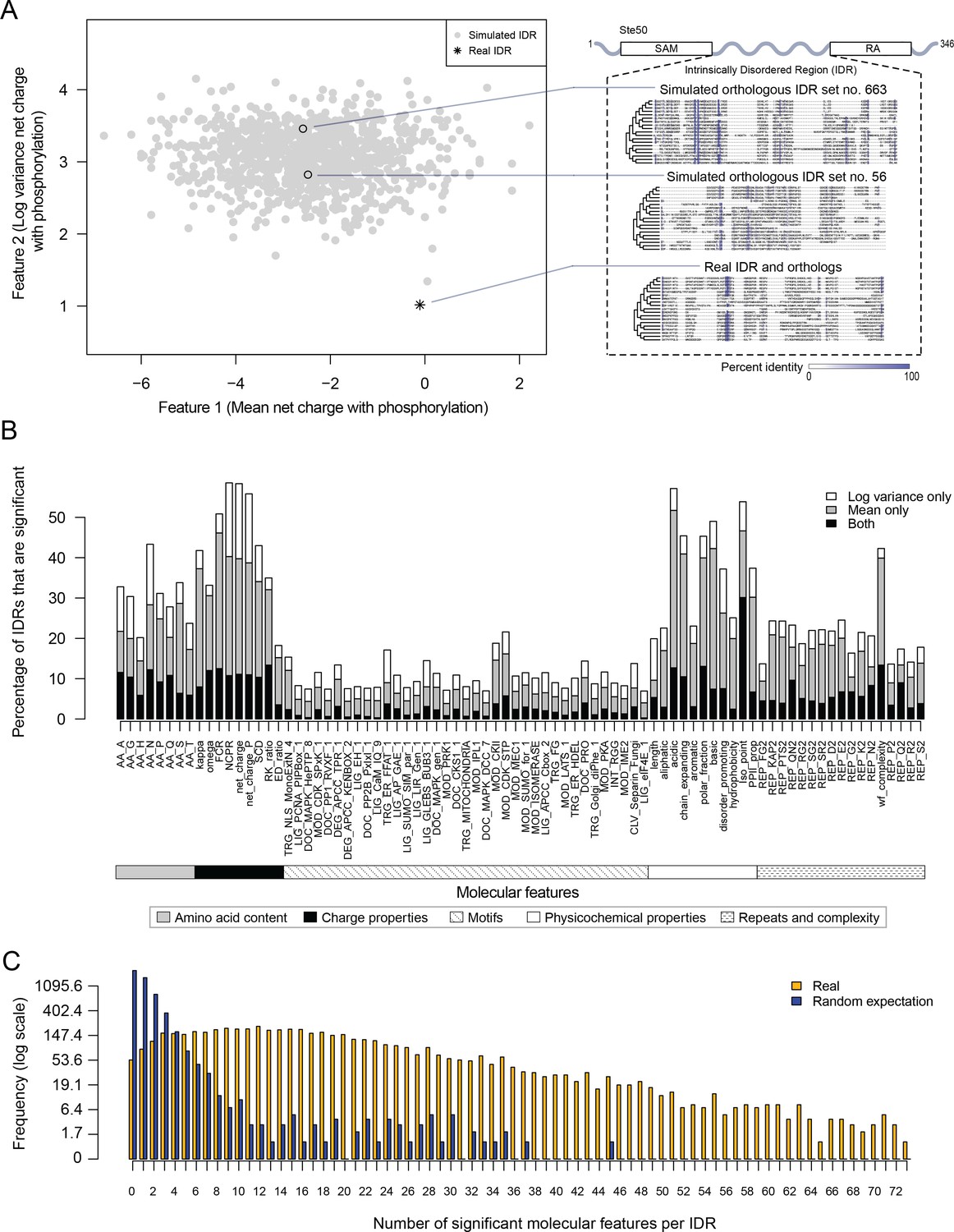
Proteome-wide evolutionary analysis reveals evolutionarily constrained sequence features are widespread in highly diverged intrinsically disordered regions.
(A) Left: Mean versus log variance of the ‘net charge with phosphorylation’ molecular feature for the real Ste50 IDR (a.a. 152–250) ortholog set and simulated Ste50 orthologous IDR sets (N = 1000). Right: Example simulated Ste50 orthologous IDR sets (no. 663 and no. 56 out of 1000) and the real Ste50 IDR and its orthologs, colored according to percent identity in the primary amino acid sequence. (B) Percentage of IDRs that are significantly deviating from simulations in mean, log variance, or both mean and log variance of each molecular feature. (C) Frequency [1 + log(frequency)] of number of significant molecular features per IDR for the real IDRs (yellow) versus the random expectation (blue) obtained from a set of simulated IDRs.
Figure 1—figure supplement 1
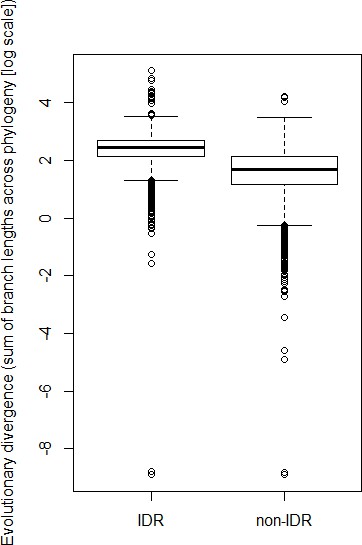
Predicted IDRs in the S. cerevisiae proteome (‘IDR’) are more highly diverged compared to regions that are not predicted to be disordered (‘non-IDR’) (p<2.2×10−16, Wilcoxon test).
Boxplot boxes represent the 25th-75th percentile of the data, the black line represents the median, and whiskers represent 1.5*the interquartile range. Outliers fall outside the 1.5*interquartile range, and are represented by unfilled circles.
Figure 1—figure supplement 2
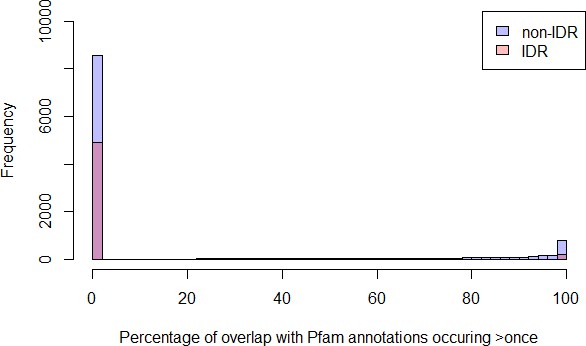
Percentage of overlap with Pfam domains for IDRs predicted to be disordered in the S. cerevisiae proteome that are >= 30 amino acids (‘IDR’) have less overlap with Pfam domains compared to all other regions that are >= 30 amino acids (‘non-IDR’) (p<2.2 × 10−16, Wilcoxon test).
Percentage of regions with 0% Pfam overlap for IDRs is 91%, whereas for non-IDRs it is 74%.
Figure 2 with 1 supplement
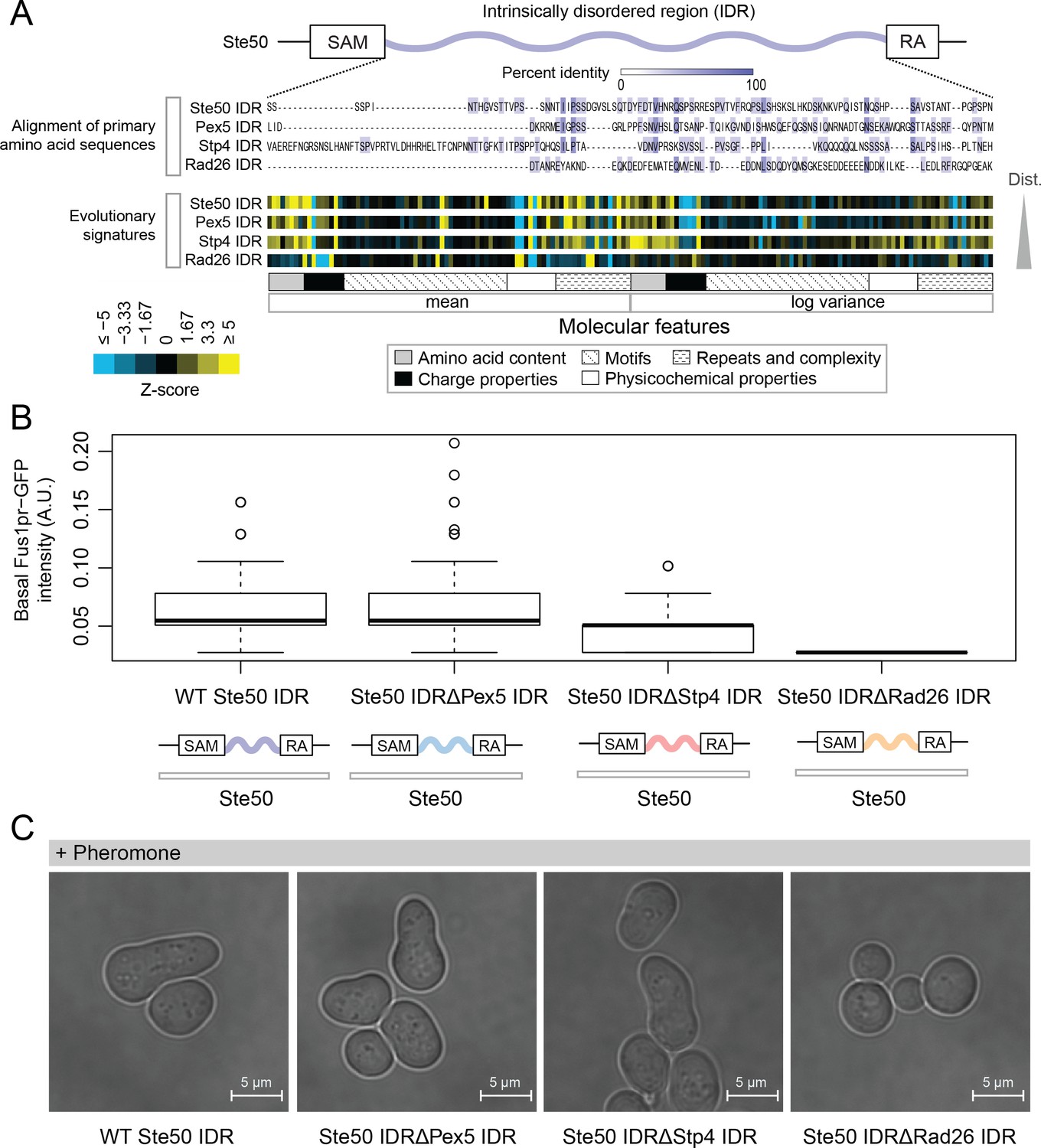
Intrinsically disordered regions with similar evolutionary signatures can rescue wildtype phenotypes, while those with different evolutionary signatures cannot.
(A) Multiple sequence alignment of Ste50 IDR (a.a. 152–250), Pex5 IDR (a.a. 77–161), Stp4 (a.a. 144–256), and Rad26 IDR (a.a. 163–239) shows negligible similarity when their primary amino acid sequences are aligned, while evolutionary signatures show that the Pex5 and Stp4 IDRs are more similar to the Ste50 IDR than the Rad26 IDR. While the Ste50 IDR has five consensus phosphorylation sites that are implicated in its function (Hao et al., 2008; Yamamoto et al., 2010; Zarin et al., 2017), the Pex5 IDR and Rad26 IDR have none, and the Stp4 IDR has 4. IDRs are presented in order of increasing Euclidian distance between their evolutionary signatures, though we do not recommend using this measure to quantitate similarity between evolutionary signatures independently (see Discussion). The Ste50 IDR is located between the Sterile Alpha Motif (SAM) and Ras Association (RA) domains in the Ste50 protein. (B) Boxplots show distribution of values corresponding to basal Fus1pr-GFP activity in an S. cerevisiae strain with the wildtype Ste50 IDR compared to strains with the Pex5, Stp4, or Rad26 IDR swapped to replace the Ste50 IDR in the genome. Boxplot boxes represent the 25th-75th percentile of the data, the black line represents the median, and whiskers represent 1.5*the interquartile range. Outliers fall outside the 1.5*interquartile range, and are represented by unfilled circles. Distribution of GFP activity is based on quantification of GFP intensity in single cells pooled from four colonies (which we define as biological replicates) for each strain; sample sizes for each distribution are as follows: WT n = 588 cells, Pex5 IDR n = 196 cells, Stp4 IDR n = 228 cells, Rad26 IDR n = 271 cells. (C) Brightfield micrographs showing each strain from part B following exposure to pheromone. Shmooing cells are those which have elongated cell shape, representing mating projections.
Figure 2—figure supplement 1

Full field-of-view micrographs of pheromone-exposed S. cerevisiae strains from Figure 2C.
Figure 3
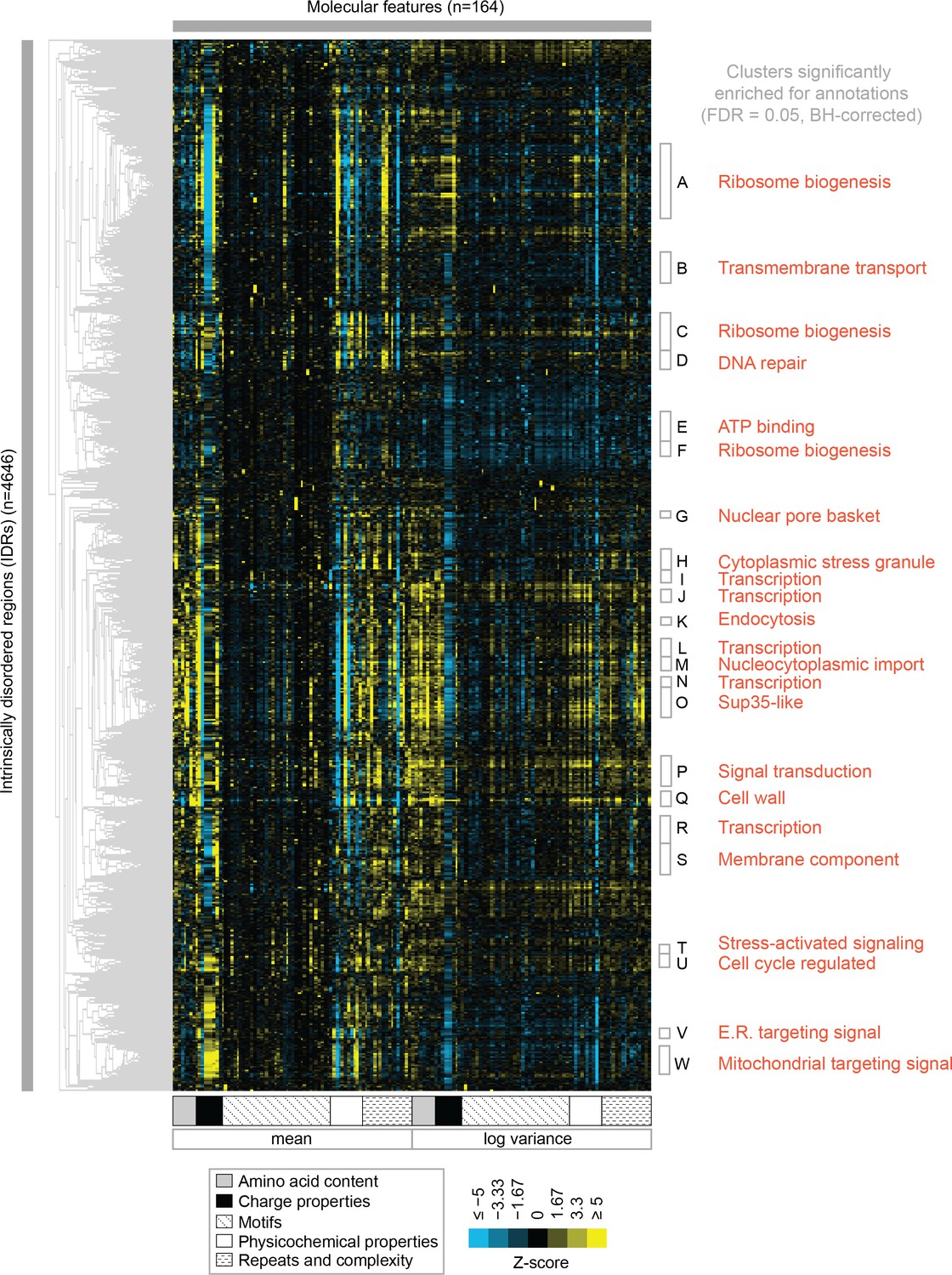
Clustering evolutionary signatures shows that IDRs in the proteome share evolutionary signatures, and that these clusters of IDRs are associated with specific biological functions.
A-W show clusters significantly enriched for annotations (see Table 1; full table of enrichments in supplementary data). Cluster names represent summary of enriched annotations.
-
Figure 3—source data 1
Clustered IDR data and mapping between IDRs and Cluster ID assigned in Figure 3.
- https://cdn.elifesciences.org/articles/46883/elife-46883-fig3-data1-v4.tar.gz
Figure 4
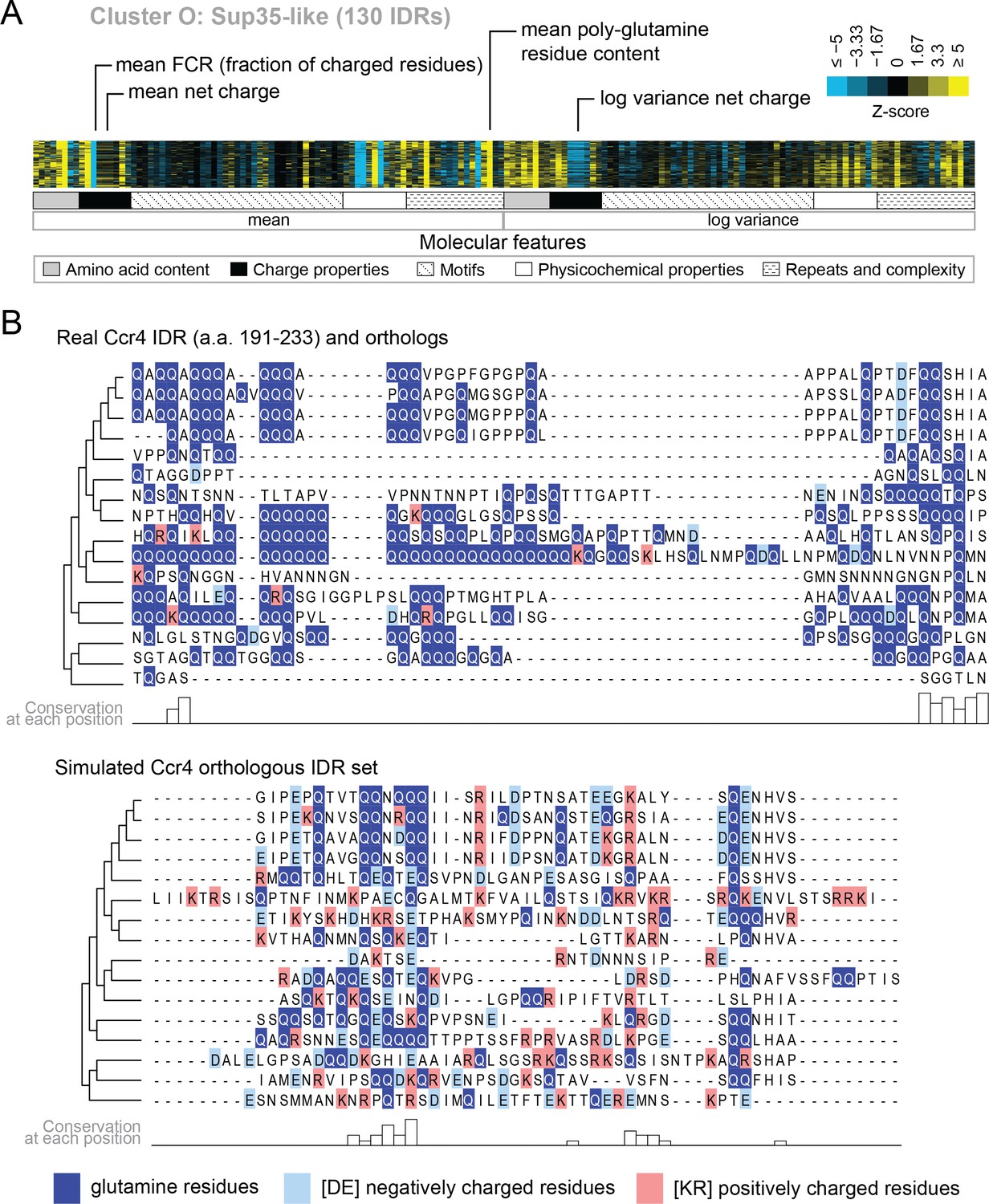
Evolutionary signatures in cluster O contain some molecular features that are typically associated with IDRs as well as some that are not.
(A) Pattern of evolutionary signatures in cluster O. (B) Example disordered region from cluster O, Ccr4, with a subset of highlighted molecular features compared between its real set of orthologs and an example set of simulated orthologous IDRs. Species included in phylogeny in order from top to bottom are S. cerevisiae, Saccharomyces mikatae, Saccharomyces kudriavzevii, Saccharomyces uvarum, Candida glabrata, Kazachstania naganishii, Naumovozyma castellii, Naumovozyma dairenensis, Tetrapisispora blattae, Tetrapisispora phaffii, Vanderwaltozyma polyspora, Zygosaccharomyces rouxii, Torulaspora delbrueckii, Kluyveromyces lactis, Eremothecium (Ashbya) cymbalariae, Lachancea waltii.
Figure 5
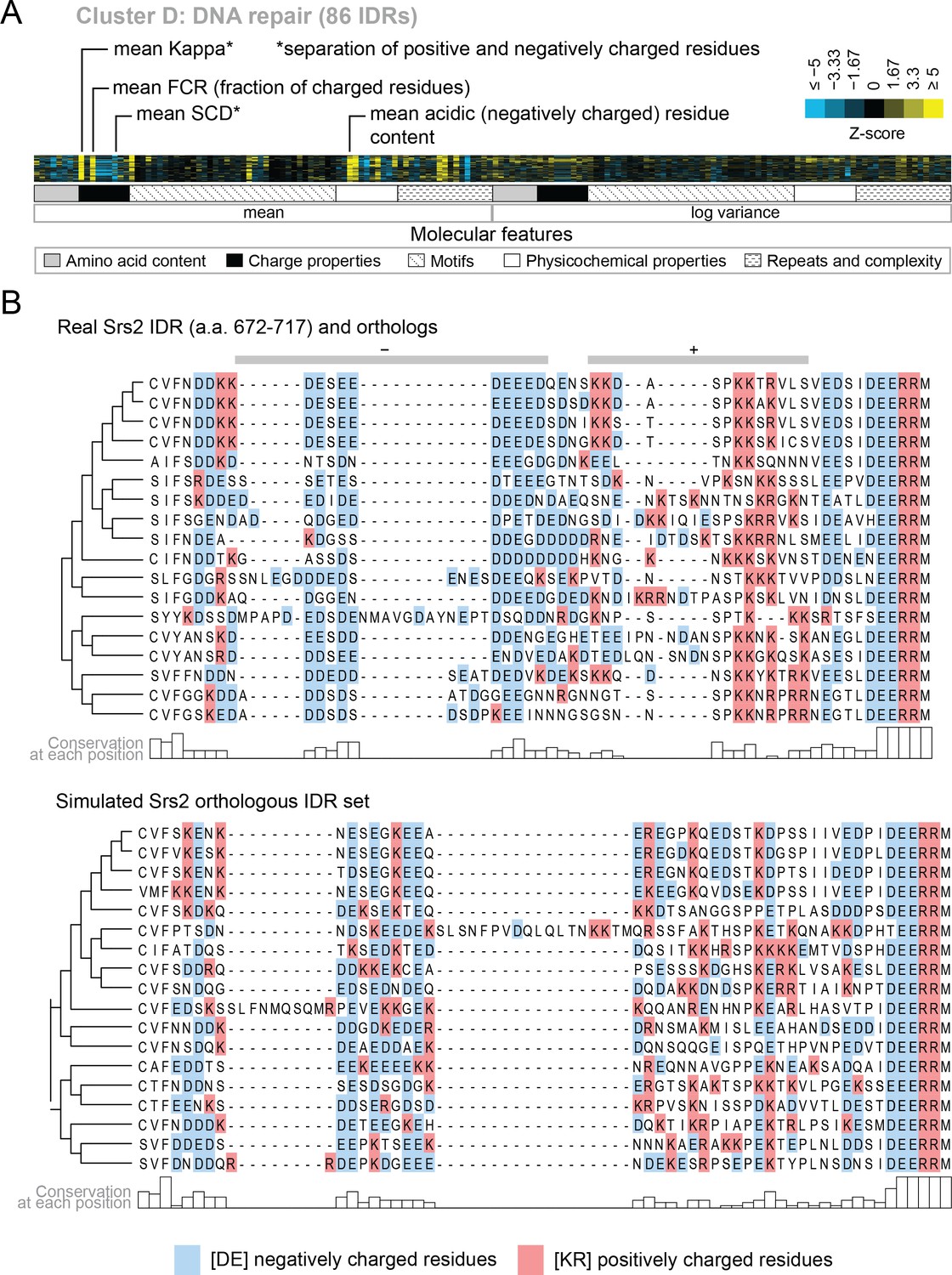
Cluster D contains disordered regions associated with DNA repair.
(A) Pattern of evolutionary signatures in cluster D. (B) Example disordered region from cluster D, Srs2, with a subset of highlighted molecular features compared between its real set of orthologs and an example set of simulated orthologous IDRs. Species included in phylogeny in order from top to bottom are S. cerevisiae, S. mikatae, S. kudriavzevii, S. uvarum, C. glabrata, Kazachstania africana, K. naganishii, N. castellii, N. dairenensis, T. phaffii, Z. rouxii, T. delbrueckii, K. lactis, Eremothecium (Ashbya) gossypii, E. cymbalariae, Lachancea kluyveri, Lachancea thermotolerans, L. waltii.
Figure 6 with 4 supplements
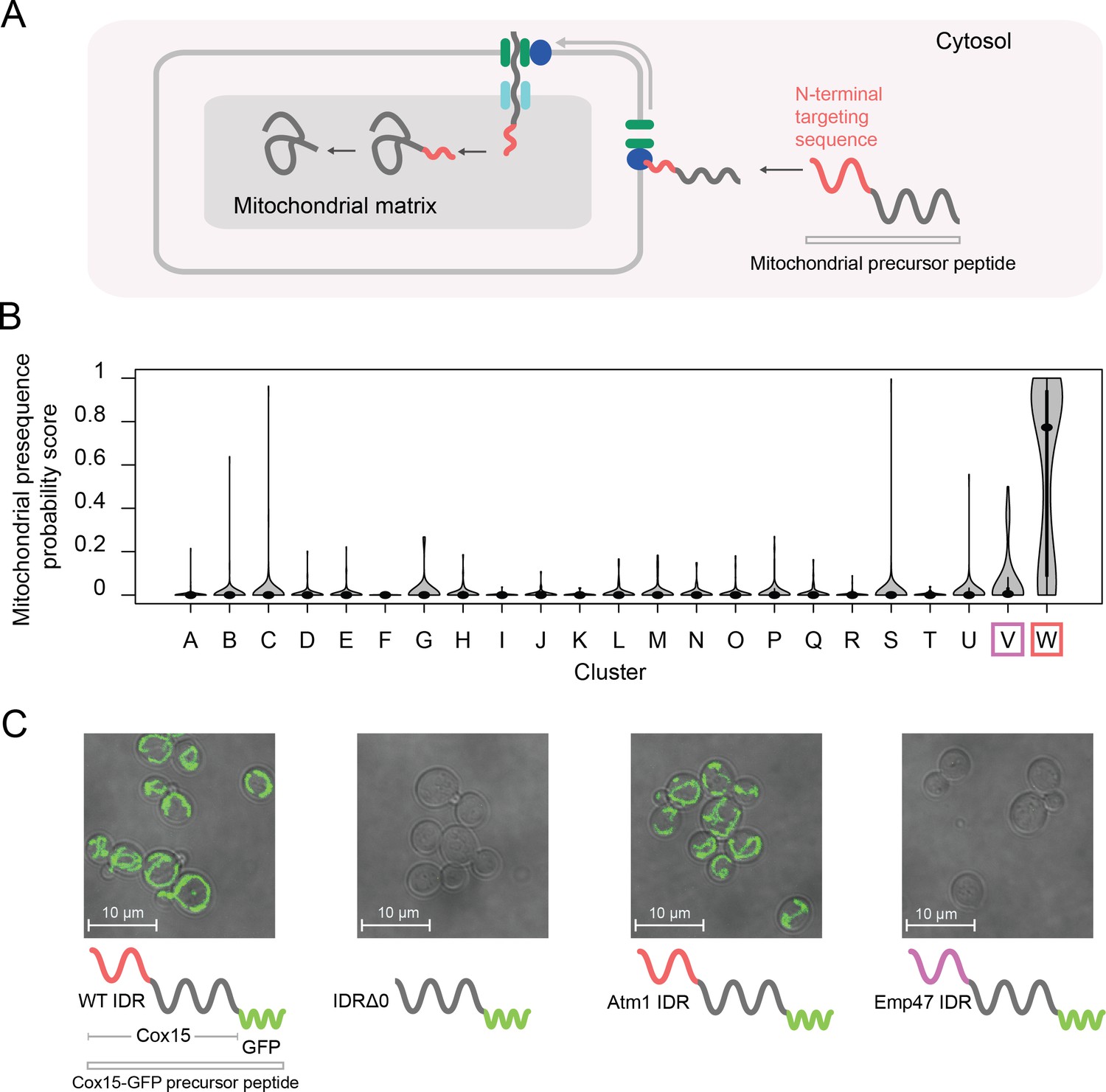
Cluster W is associated with mitochondrial N-terminal targeting signals.
(A) Schematic (not to scale) showing the path of a mitochondrial precursor peptide (with N-terminal targeting sequence in red) from the cytosol, where it is translated, to the mitochondrial matrix, where the peptide folds and targeting sequence is cleaved. (B) Violin plots (median indicated by black dot, thick black line showing 25th-75th percentile, and whiskers showing outliers) show distributions of mitochondrial presequence probability scores for all IDRs in each cluster. The cluster that we predict to contain mitochondrial N-terminal targeting signals is outlined in red, while the cluster that we predict to contain endoplasmic reticulum targeting signals is outlined in purple. (C) Micrographs of S. cerevisiae strains in which Cox15 is tagged with GFP, with either the wildtype Cox15 IDR, deletion of the Cox15 IDR, replacement of the Cox15 IDR with the Atm1 IDR (also in the mitochondrial targeting signal cluster), or replacement of the Cox15 IDR with the Emp47 IDR (from the endoplasmic reticulum targeting signal cluster).
Figure 6—figure supplement 1
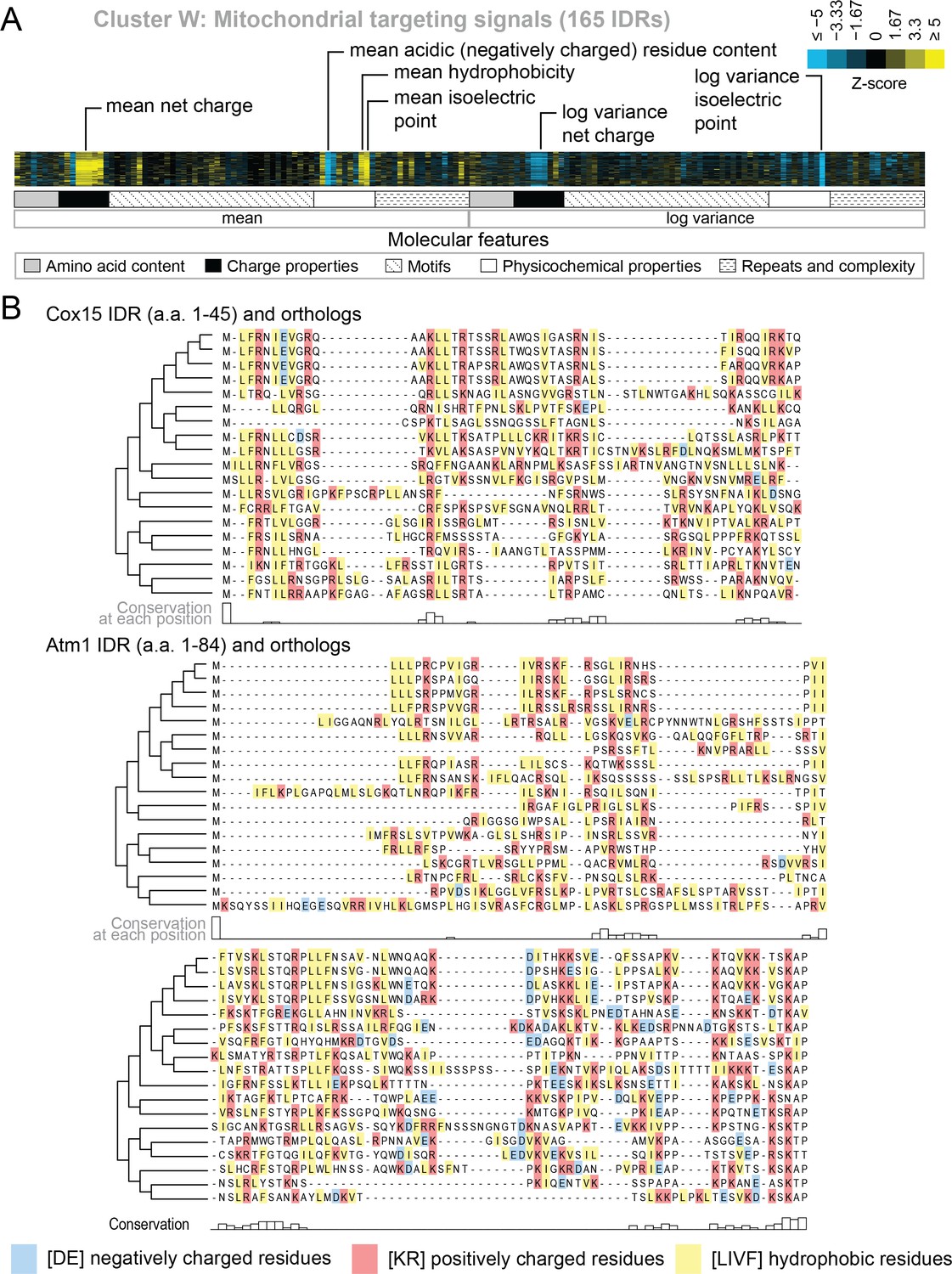
Evolutionary signatures in cluster W contain molecular features that have been previously reported for mitochondrial N-terminal targeting signals.
(A) Pattern of evolutionary signatures in cluster W. (B) Multiple sequence alignments of example disordered regions from Cox15 (top) and Atm1 (bottom) from cluster W, showing a subset of highlighted molecular features. Species included in phylogeny in order from top to bottom are S. cerevisiae, S. mikatae, S. kudriavzevii, S. uvarum, C. glabrata, K. africana, K. naganishii, N. castellii, N. dairenensis, T. phaffii, V. polyspora, Z. rouxii, T. delbrueckii, K. lactis, E. gossypii, E. cymbalariae, L. kluyveri, L. thermotolerans, L. waltii.
Figure 6—figure supplement 2
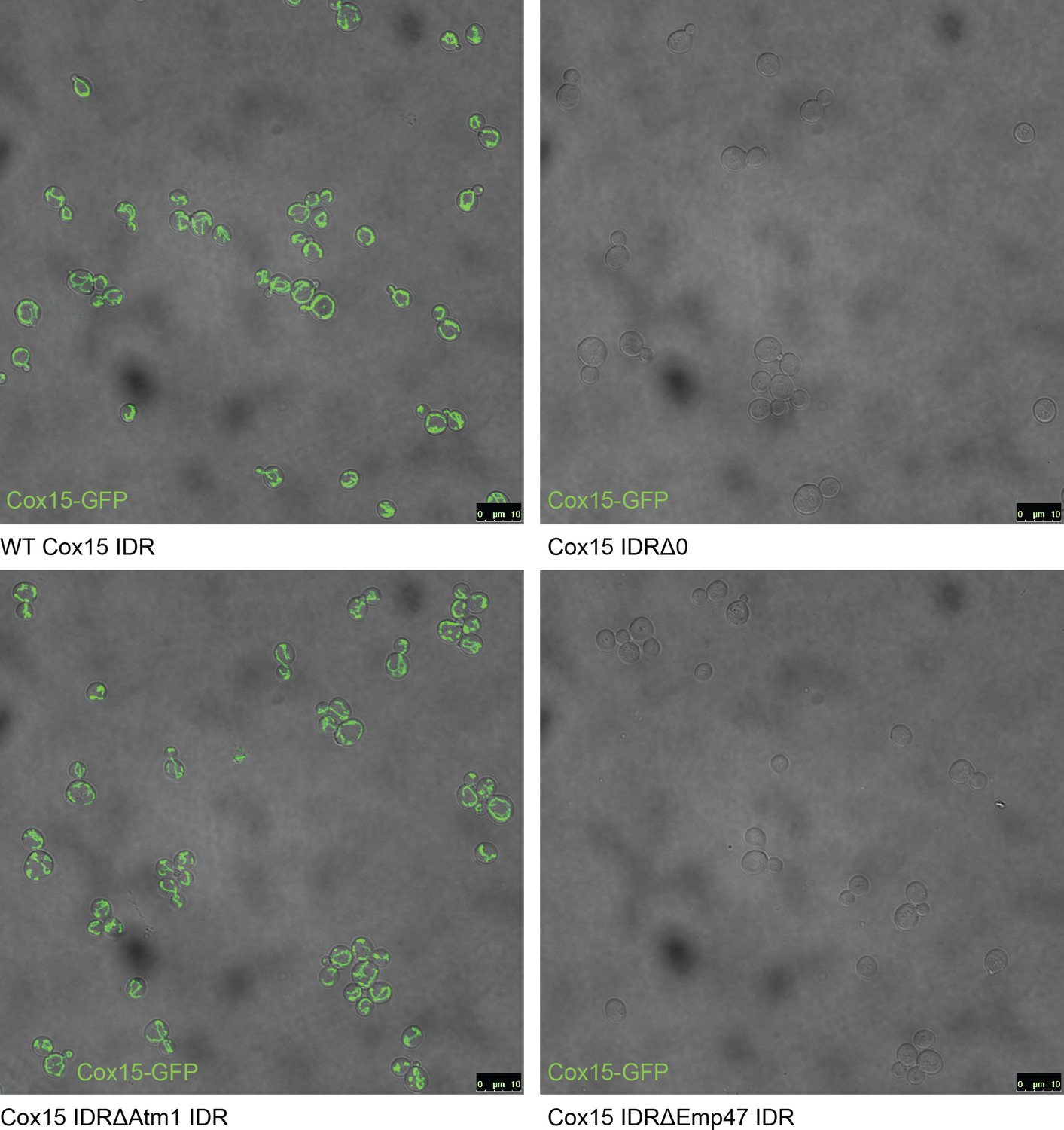
Full field-of-view micrographs of S. cerevisiae strains from Figure 6C.
Figure 6—figure supplement 3
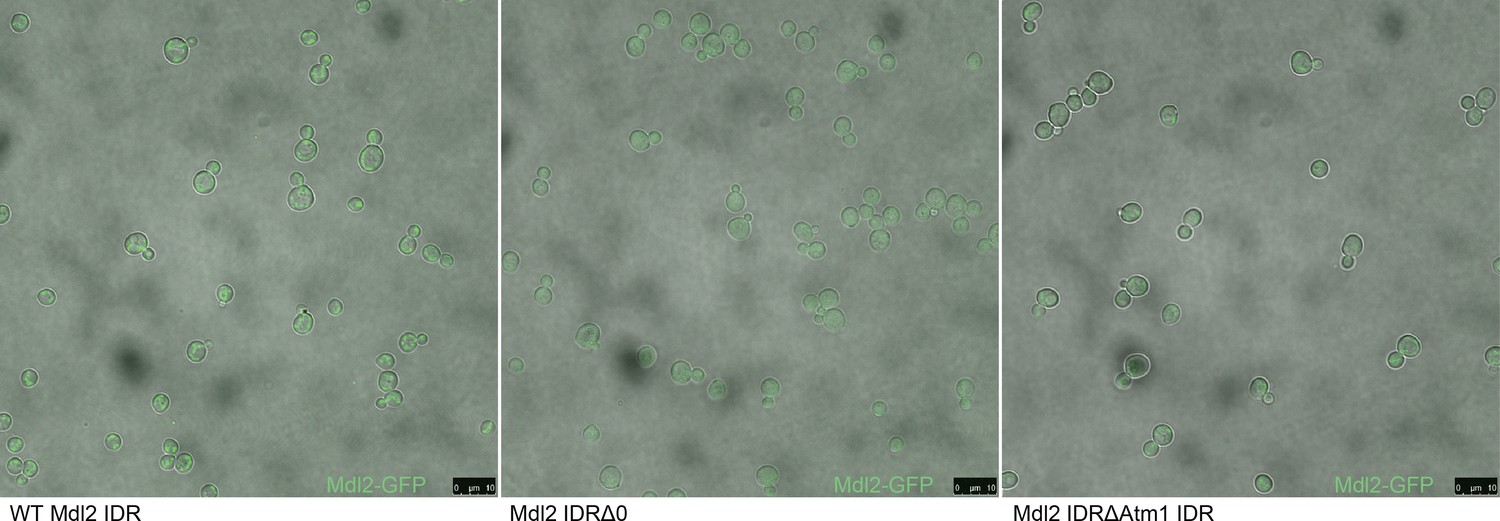
Micrographs of S. cerevisiae strains with three different genotypes.
From left to right: Mdl2-GFP has a mitochondrial localization in the wildtype (WT) strain, knocking out the Mdl2 IDR abolishes wildtype localization, and replacing the Mdl2 IDR with that of Atm1 rescues mitochondrial localization.
Figure 6—figure supplement 4
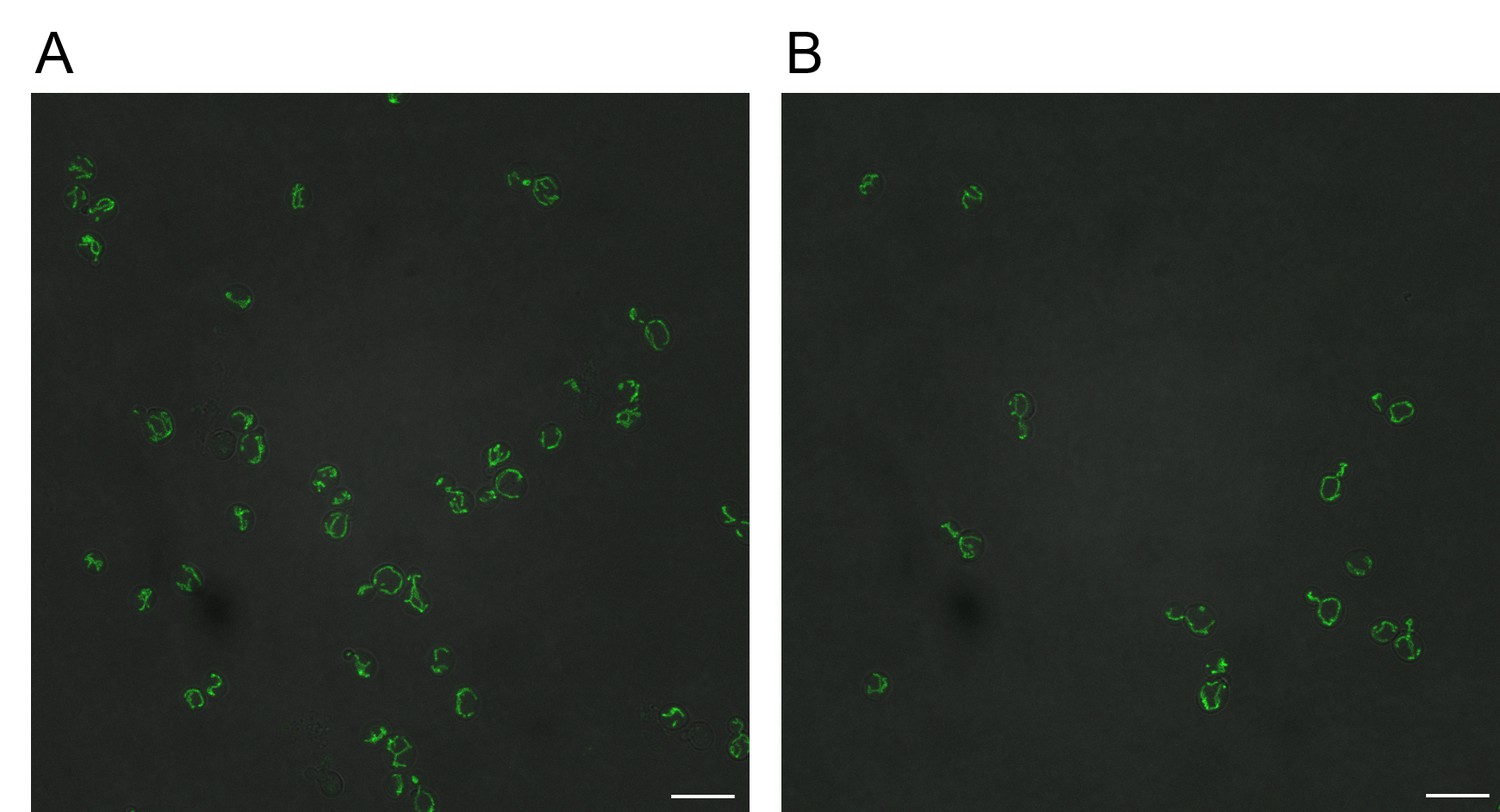
Reverse transformation of GFP-tagged Cox15 IDR∆0 and Cox15∆Emp47 strains to wildtype Cox15 IDR rescues mitochondrial localization of Cox15-GFP.
Scale bars represent 10 micrometers. (A) GFP-tagged Cox15 IDR∆Emp47 reverted to wildtype Cox15-GFP. (B) GFP-tagged Cox15 IDR∆0 reverted to wildtype Cox15-GFP.
Tables
Table 1
Top five enriched GO term annotations and top three enriched phenotype annotations for each cluster (in order of decreasing corrected p-values).
Full table of >1000 significant GO term, phenotype, and literature enrichments in supplementary data.
| ID | Annotations (Positive proteins in cluster/Total proteins in cluster) | Corrected P <= |
|---|---|---|
| A | nucleus (201/295), rRNA processing (40/295), ribosome biogenesis (39/295), nucleolus (50/295), maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) (14/295), inviable (110/295), RNA accumulation decreased (46/295), RNA accumulation increased (39/295) | 1.46e-03 |
| B | amino acid transmembrane transport (8/140), amino acid transmembrane transporter activity (8/140), transmembrane transport (21/140), amino acid transport (9/140) | 1.11e-02 |
| C | nucleolus (42/159), rRNA processing (27/159), ribosome biogenesis (26/159), nucleus (107/159), preribosome, large subunit precursor (13/159), RNA accumulation increased (28/159), inviable (60/159), RNA accumulation decreased (27/159) | 4.88e-03 |
| D | nucleus (72/86), DNA repair (20/86), cellular response to DNA damage stimulus (18/86), DNA binding (28/86), damaged DNA binding (7/86), mutation frequency increased (14/86), chromosome plasmid maintenance decreased (29/86), cell cycle progression in S phase increased duration (4/86) | 4.21e-02 |
| E | motor activity (4/89), ATP binding (25/89), ASTRA complex (3/89) | 4.23e-02 |
| F | 90S preribosome (11/73), rRNA processing (14/73), ribosome biogenesis (14/73), endonucleolytic cleavage in ITS1 to separate SSU-rRNA from 5.8S rRNA and LSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) (6/73), nucleolus (15/73) | 2.49e-02 |
| G | nuclear pore nuclear basket (4/35), nucleocytoplasmic transporter activity (4/35) | 4.54e-02 |
| H | nucleic acid binding (16/66), translational initiation (7/66), cytoplasmic stress granule (9/66), mRNA binding (13/66), translation initiation factor activity (6/66) | 3.60e-03 |
| I | regulation of transcription, DNA-templated (23/52), transcription, DNA-templated (22/52), positive regulation of transcription from RNA polymerase II promoter (12/52) | 6.58e-03 |
| J | RNA polymerase II transcription factor activity, sequence-specific DNA binding (10/52), positive regulation of transcription from RNA polymerase II promoter (14/52), regulation of transcription, DNA-templated (21/52), RNA polymerase II core promoter proximal region sequence-specific DNA binding (9/52), transcription, DNA-templated (19/52) | 1.22e-02 |
| K | trehalose biosynthetic process (2/19), Golgi to endosome transport (3/19), ubiquitin binding (4/19) | 3.81e-02 |
| L | sequence-specific DNA binding (21/70), RNA polymerase II core promoter proximal region sequence-specific DNA binding (13/70), DNA binding (27/70), positive regulation of transcription from RNA polymerase II promoter (17/70), regulation of transcription, DNA-templated (27/70) | 6.75e-05 |
| M | structural constituent of nuclear pore (8/54), protein targeting to nuclear inner membrane (5/54), nuclear pore central transport channel (6/54), mRNA transport (9/54), nuclear pore (8/54) | 5.87e-05 |
| N | sequence-specific DNA binding (18/39), DNA binding (19/39), zinc ion binding (11/39), regulation of transcription, DNA-templated (19/39), RNA polymerase II transcription factor activity, sequence-specific DNA binding (8/39) | 6.21e-04 |
| O | regulation of transcription, DNA-templated (53/130), transcription, DNA-templated (50/130), sequence-specific DNA binding (25/130), positive regulation of transcription from RNA polymerase II promoter (26/130), nuclear-transcribed mRNA catabolic process, deadenylation-dependent decay (8/130), endocytosis decreased (26/130), invasive growth increased (37/130), cell shape abnormal (15/130) | 1.29e-02 |
| P | intracellular signal transduction (19/129), protein kinase activity (22/129), protein serine/threonine kinase activity (22/129), kinase activity (24/129), phosphorylation (24/129) | 3.34e-06 |
| Q | extracellular region (33/67), fungal-type cell wall (30/67), cell wall (25/67), anchored component of membrane (20/67), cell wall organization (23/67) | 1.01e-20 |
| R | positive regulation of transcription from RNA polymerase II promoter (21/119), DNA binding (32/119), RNA polymerase II core promoter proximal region sequence-specific DNA binding (12/119), transcription factor activity, sequence-specific DNA binding (10/119), transcription, DNA-templated (33/119) | 1.55e-02 |
| S | integral component of membrane (59/133), membrane (68/133), fungal-type vacuole membrane (18/133), vacuole (18/133), L-tyrosine transmembrane transporter activity (4/133) | 5.48e-03 |
| T | stress-activated protein kinase signaling cascade (4/33), regulation of apoptotic process (4/33) | 3.57e-02 |
| U | cytoskeleton (15/80), spindle (6/80), kinetochore microtubule (3/80) | 1.47e-02 |
| V | fungal-type vacuole (15/43), mannosylation (7/43), integral component of membrane (28/43), cell wall mannoprotein biosynthetic process (6/43), alpha-1,6-mannosyltransferase activity (4/43) | 1.45e-05 |
| W | mitochondrion (144/165), mitochondrial inner membrane (57/165), mitochondrial matrix (34/165), oxidation-reduction process (31/165), mitochondrial translation (22/165), respiratory growth decreased rate (81/165), respiratory growth absent (71/165), mitochondrial genome maintenance absent (25/165) | 3.15e-15 |
-
Table 1—source data 1
Full table of enrichments for Table 1.
- https://cdn.elifesciences.org/articles/46883/elife-46883-table1-data1-v4.csv
Table 2
Evolutionary signatures of function can be used for functional annotation of previously uncharacterized proteins and IDRs.
| ID | Name | Description | % Disorder | Cluster ID |
|---|---|---|---|---|
| YCL028W | RNQ1 | Protein whose biological role is unknown; localizes to the cytosol | 96 | M: Nucleocytoplasmic transport |
| YKL105C | SEG2 | Protein whose biological role is unknown; localizes to the cell periphery | 92 | P: Signal transduction |
| YGR196C | FYV8 | Protein whose biological role is unknown; localizes to the cytoplasm in a large-scale study | 89 | A: Ribosome biogenesis R: Transcription |
| YGL023C | PIB2 | Protein whose biological role is unknown; localizes to the mitochondrion in a large-scale study | 86 | R: Transcription |
| YOL036W | Protein whose biological role and cellular location are unknown | 84 | P: Signal transduction R: Transcription | |
| YNL176C | TDA7 | Protein whose biological role is unknown; localizes to the vacuole | 83 | Q: Cell wall organization |
| YFR016C | Protein whose biological role is unknown; localizes to both the cytoplasm and bud in a large-scale study | 83 | A: Ribosome biogenesis | |
| YBL081W | Protein whose biological role and cellular location are unknown | 82 | M: Nucleocytoplasmic transport | |
| YBR016W | Protein whose biological role is unknown; localizes to the bud membrane and the mating projection membrane | 82 | O: Sup35-like | |
| YOL070C | NBA1 | Protein whose biological role is unknown; localizes to the bud neck and cytoplasm and colocalizes with ribosomes in multiple large-scale studies | 81 | Does not fall into annotated cluster; close to ribosome biogenesis cluster |
Additional files
-
Supplementary file 1
Includes Tables S1-S4.
- https://cdn.elifesciences.org/articles/46883/elife-46883-supp1-v4.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/46883/elife-46883-transrepform-v4.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Proteome-wide signatures of function in highly diverged intrinsically disordered regions
eLife 8:e46883.
https://doi.org/10.7554/eLife.46883




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}