β-blockers augment L-type Ca2+ channel activity by targeting spatially restricted β2AR signaling in neurons
- Guangzhou Medical University, China
- University of California Davis, United States
- VA Northern California Health Care System, United States
Abstract
G protein-coupled receptors (GPCRs) transduce pleiotropic intracellular signals in mammalian cells. Here, we report neuronal excitability of β-blockers carvedilol and alprenolol at clinically relevant nanomolar concentrations. Carvedilol and alprenolol activate β2AR, which promote G protein signaling and cAMP/PKA activities without action of G protein receptor kinases (GRKs). The cAMP/PKA activities are restricted within the immediate vicinity of activated β2AR, leading to selectively enhance PKA-dependent phosphorylation and stimulation of endogenous L-type calcium channel (LTCC) but not AMPA receptor in rat hippocampal neurons. Moreover, we have engineered a mutant β2AR that lacks the catecholamine binding pocket. This mutant is preferentially activated by carvedilol but not the orthosteric agonist isoproterenol. Carvedilol activates the mutant β2AR in mouse hippocampal neurons augmenting LTCC activity through cAMP/PKA signaling. Together, our study identifies a mechanism by which β-blocker-dependent activation of GPCRs promotes spatially restricted cAMP/PKA signaling to selectively target membrane downstream effectors such as LTCC in neurons.
Introduction
GPCRs often signal not only through canonical G proteins but also through noncanonical G protein-independent signaling, frequently via G protein receptor kinases (GRKs) and β-arrestins (Lefkowitz, 2000; Xiang and Kobilka, 2003). One of the universal features of GPCRs is that they undergo ligand-induced phosphorylation at different sites by either GRKs or second messenger dependent protein kinases such as protein kinase A (PKA). The phosphorylated GPCRs thus may present distinct structural features that favor receptor binding to different signaling partners, engaging distinct downstream signaling cascades (Reiter and Lefkowitz, 2006; Lefkowitz, 2007; Nobles et al., 2011). Some ligands can differentially activate a GPCR via a phenomenon known as functional selectivity or biased signaling (Wisler et al., 2014; Zweemer et al., 2014). For example, stimulation of β2 adrenergic receptor (β2AR), a prototypical GPCR involved in memory and learning in the central nervous system (CNS) and regulation of metabolism and cardiovascular function, promotes phosphorylation by both GRKs and PKA (Najafi et al., 2016; Mammarella et al., 2016; Fu et al., 2017; Matera et al., 2018). We have recently identified spatially segregated subpopulations of β2AR undergoing exclusive phosphorylation by GRKs or PKA in a single cell. These findings indicate specific GPCR subpopulation-based signaling branches can co-exist in a single cell (Shen et al., 2018). GRK-mediated phosphorylation promotes pro-survival and cell growth signaling via β-arrestin-dependent mitogen-activated protein kinase (MAPK/ERK) pathways, prompting the search for biased ligands that preferentially activate β-arrestin pathways (Luttrell et al., 1999; Pierce et al., 2000; Kim et al., 2005; Ren et al., 2005; Zidar et al., 2009; Choi et al., 2018). On the other hand, our recent studies show that the cAMP/PKA-dependent phosphorylation of β2AR controls ion channel activity at the plasma membrane in primary hippocampal neurons (Shen et al., 2018).
β-blockers are thought to reduce cAMP signaling because they either reduce basal activity of βARs or block agonist-induced receptor activation. While β-blockers are successful in clinical therapies of a broad range of diseases, their utility is limited by side effects in both the CNS and peripheral tissues (Bakris, 2009; Gorre and Vandekerckhove, 2010). Indeed, studies have revealed that some β-blockers display partial agonism and can promote receptor-Gs coupling at high concentrations in vitro (Yao et al., 2009; DeVree et al., 2016; Gregorio et al., 2017). Accordingly, some β-blockers display intrinsic properties mimicking sympathetic activation (sympathomimetic β-blockers) (Maack et al., 2000; Brixius et al., 2001; Larochelle et al., 2014). The mechanism remains poorly understood because classic cAMP assay do not show even minimal cAMP signal induced by these β-blockers (Maack et al., 2000; Brixius et al., 2001).
In this study, we show that the β-blockers carvedilol and alprenolol can promote Gs protein coupling to β2AR and cAMP/PKA but not GRK activity at nanomolar concentrations. Thus, these β-blockers are emerging as partial agonists even at low concentrations rather than strict antagonists in mammalian cells. This cAMP/PKA signaling is spatially restricted, selectively promoting phosphorylation of β2AR and CaV1.2 by PKA which augments LTCC activity in primary hippocampal neurons. Furthermore, we have engineered a mutant β2AR that can be selectively activated by carvedilol but not by the orthosteric agonist isoproterenol (ISO) to stimulate PKA but not GRK. Together, these studies identify a unique mechanism by which β-blockers activate β2AR at low concentrations, which promotes Gs/cAMP/PKA signaling branch and selectively targets downstream LTCC channels in neurons. This observation may also explain sympathomimetic effects of β-blockers in the CNS.
Results
Carvedilol and alprenolol selectively promote β2AR-mediated PKA-phosphorylation of β2AR
In this study, we applied two sets of well-characterized phospho-specific antibodies, anti-pS261/262 and anti-pS355/356 to examine a series of β-blockers for their effects on the phosphorylation of β2AR at its PKA and GRKs sites, respectively (Shen et al., 2018; Tran et al., 2004; Tran et al., 2007). We found that various β-blockers including alprenolol (ALP), carvedilol (CAR), propranolol (PRO) and CGP12177 (177) were able to stimulate phosphorylation of β2AR at PKA sites expressed in HEK293 cells, similar to the βAR agonist isoproterenol (ISO) (Figure 1A and Figure 1—figure supplement 1A). In contrast, other β-blockers, that is ICI118551 (ICI), timolol (TIM) and metoprolol (MET), were not able to do so (Figure 1A). The ligand-induced phosphorylation of β2AR was blocked by β2AR-specific antagonist ICI but not β1AR-specific antagonist CGP20712A (CGP) (Figure 1B and C, and Figure 1—figure supplement 1B and C). We chose CAR and ALP for further study. We found that CAR and ALP promoted phosphorylation of β2AR by PKA even at nanomolar concentrations (Figure 2A and B, and Figure 2—figure supplement 1A and B), which was paralleled by concentration-dependent increases in phosphorylation of ERK (Figure 2—figure supplement 2). The roles of β2AR and PKA in this phenomenon were confirmed by inhibition of β2AR with ICI and inhibition of PKA with H89, respectively (Figure 2C and D, and Figure 2—figure supplement 1C and D). In contrast, those β-blockers induced at best minimal increases in phosphorylation of β2AR at GRK sites and only at high concentrations, consistent with a previous report (Wisler et al., 2007) (Figure 1A and Figure 2—figure supplement 3). As positive control, the βAR agonist ISO promoted robust increases in both PKA and GRK phosphorylation of the receptors at different concentrations ranging from nanomolar to micromolar (Figures 1 and 2, and Figure 2—figure supplements 2 and 3). In the CNS, β2AR emerges as a prevalent postsynaptic norepinephrine effector at glutamatergic synapses (Davare et al., 2001; Joiner et al., 2010; Wang et al., 2010; Qian et al., 2012). Consistent with the data from HEK293 cells, we found β-blockers CAR and ALP activated β2AR and promoted phosphorylation of the receptor by PKA in hippocampal neurons (Figure 2E). Together, these data suggest that certain β-blockers selectively promote PKA phosphorylation of β2AR in HEK293 and primary hippocampal neurons.
Figure 1 with 1 supplement see all

Carvedilol and alprenolol selectively promote phosphorylation of β2AR at PKA sites.
HEK293 cells stably expressing FLAG-tagged β2AR were either directly stimulated for 5 min with the βAR agonist ISO or different β-blockers at indicated concentrations (A) n = 4), or pretreated for 15 min with 1 μM β1AR antagonist CGP20712A (B) n = 5) or 10 μM β2AR antagonist ICI118551 (C) n = 4) before the treatment. The phosphorylation of β2AR on its PKA and GRK sites were determined with phospho-specific antibodies, and signals were normalized to total β2AR detected with anti-FLAG antibody. NT, no treatment; ISO, isoproterenol; TIM, timolol; ALP, alprenolol; CAR, carvedilol; ICI, ICI118551; PRO, propranolol; MET, metoprolol; 177, CGP12177; CGP, CGP20712A. Error bars denote s.e.m., P values are computed by one-way ANOVA followed by Tukey’s test between NT and other groups.
-
Figure 1—source data 1
Excel spreadsheet containing the individual numeric values of phosphorylated β2AR / total β2AR relative density analyzed in Figure 1.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig1-data1-v3.xlsx
Figure 2 with 3 supplements see all
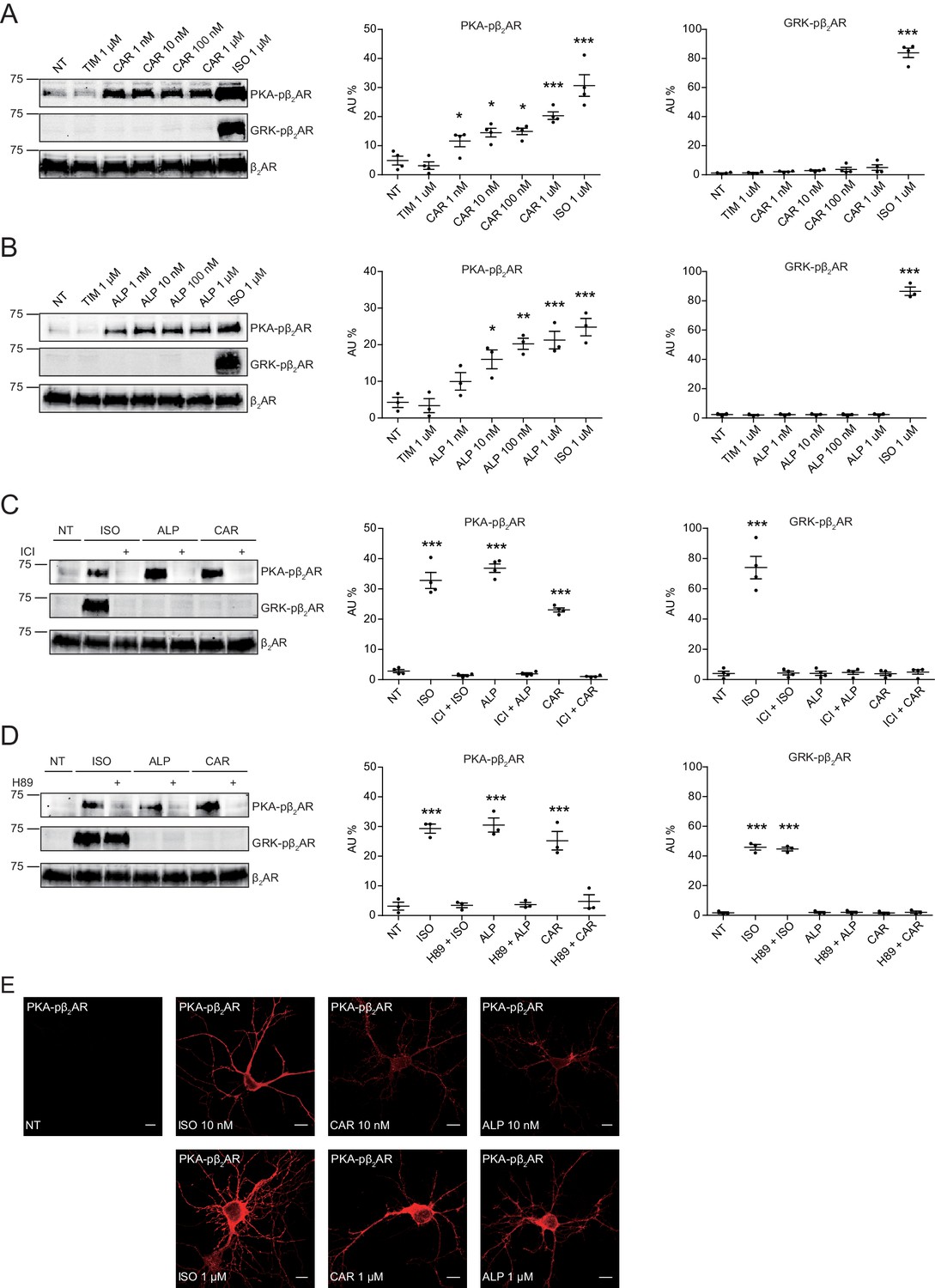
Carvedilol and alprenolol induce concentration-dependent PKA phosphorylation of β2AR in HEK293 and hippocampal neurons.
HEK293 cells stably expressing FLAG-tagged β2AR were treated with increasing concentrations of CAR (A) n = 4) and ALP (B) n = 3), or pretreated for 15 min with 10 μM β2AR antagonist ICI118551 (C) n = 4) and PKA inhibitor H89 (D) n = 3) before stimulated with 1 μM indicated drugs for 5 min. The phosphorylation of β2AR on its PKA and GRK sites were determined with phospho-specific antibodies, and signals were normalized to total β2AR detected with anti-FLAG antibody. Experiments were performed in the presence of 1 μM β1AR-selective antagonist CGP20712A to block endogenous β1AR signaling. NT, no treatment; ISO, isoproterenol; ALP, alprenolol; CAR, carvedilol; ICI, ICI118551. Error bars denote s.e.m., P values are computed by one-way ANOVA followed by Tukey’s test between NT and other groups. (E) Rat hippocampal neurons expressing β2AR were treated for 5 min with 10 nM or 1 μM indicated drugs on 12 days in vitro (DIV), and immuno-stained for PKA-phosphorylated β2AR. Confocal images show PKA-phosphorylated β2AR in agonist- or β-blocker-stimulated neurons have similar distribution. Scale bar, 10 μm. Representative of 6 images for each condition, three experiments.
-
Figure 2—source data 1
Excel spreadsheet containing the individual numeric values of phosphorylated β2AR / total β2AR relative density analyzed in Figure 2A-D.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig2-data1-v3.xlsx
Carvedilol and alprenolol promote Gsα recruitment to β2AR and increase spatially restricted cAMP signal
The western blot data on PKA phosphorylation of β2AR indicates a stimulation of the receptor-mediated Gs/AC/cAMP pathway by these β-blockers. We measured ligand-induced Gsα recruitment to β2AR with an in situ proximity ligation assay (PLA), which allows direct visualization and quantification of protein-protein interactions. We showed that ISO, CAR and ALP were able to increase the PLA signals between β2AR and Gsα, indicating recruitment of Gsα to β2AR (Figure 3A). As control, TIM had no effect on the recruitment of Gsα to β2ARs. The role of Gs/AC in CAR-induced PKA phosphorylation of β2AR was further validated by AC-specific inhibition with 2',5'-dideoxyadenosine (ddA, Figure 3—figure supplement 1). These data indicate that CAR and ALP are able to stimulate β2AR-Gs signal to increase PKA phosphorylation of the receptor.
Figure 3 with 3 supplements see all
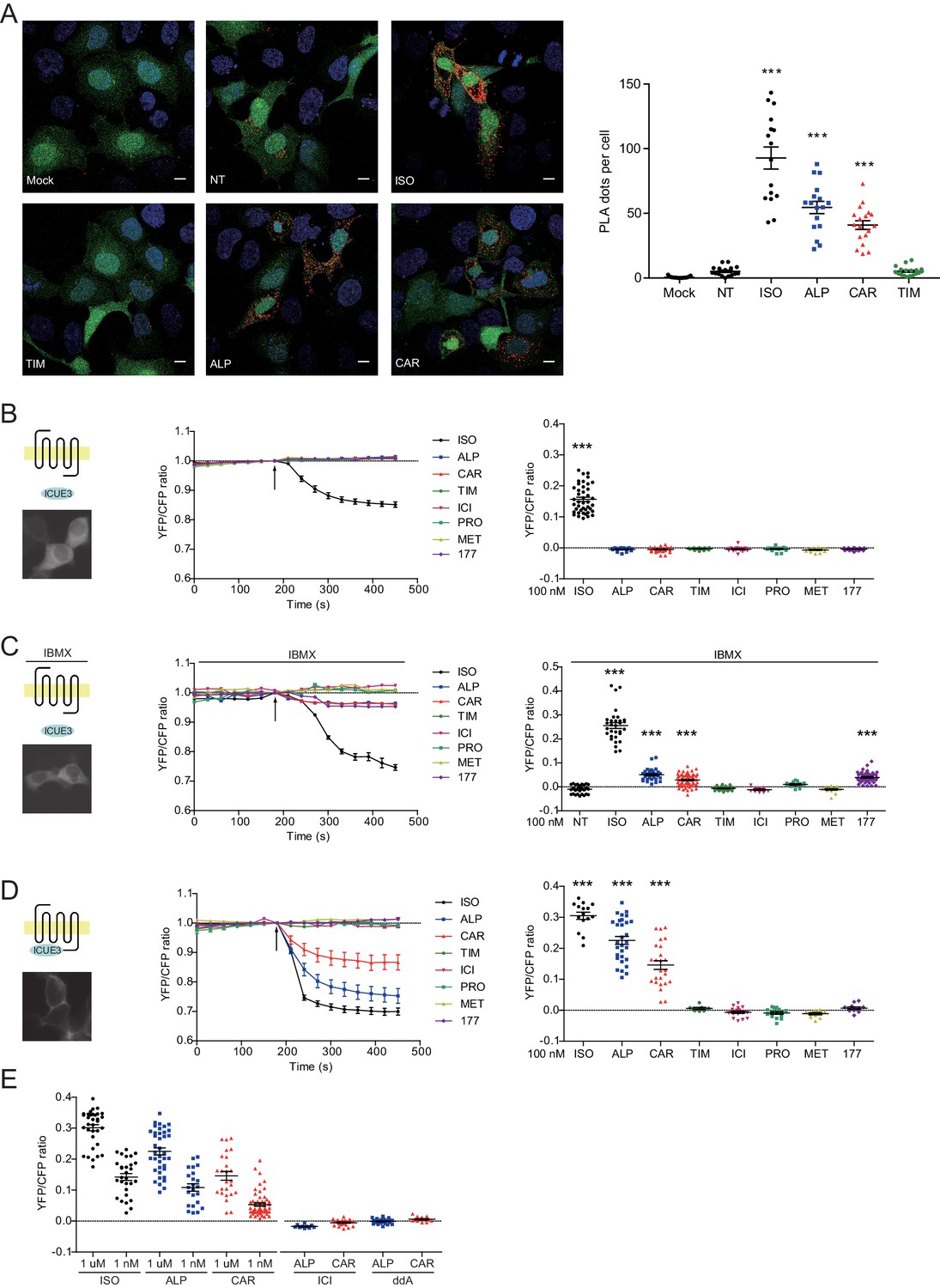
Carvedilol and alprenolol promote Gsα recruitment to β2AR and increase spatially restricted cAMP signal.
(A) HEK293 cells co-expressing FLAG-tagged β2AR, HA-tagged Gsα and EGFP were stimulated with 100 nM ISO or indicated β-blockers for 5 min. In proximity ligation assay (PLA), cells were immuno-stained with HA and β2AR antibody, nuclei were counterstained with DAPI. The green EGFP signal represents transfected cells, and red PLA signal represents Gsα and β2AR interactions. Carvedilol and alprenolol promoted Gsα recruitment to β2AR, but timolol could not. Scale bar, 10 μm. Representative of n = 15, 16, 16, 17, 18 and 18 images respectively, three experiments. (B–C) HEK293 cells expressing ICUE3 biosensor were treated with 1 μM ISO or indicated β-blockers (B), or together with 100 μM phosphodiesterase inhibitor IBMX (C). (D–E) HEK293 cells expressing β2AR-ICUE3 biosensor were treated with indicated concentration of ISO or β-blockers. In some cases, cells were pretreated for 30 min with the β2AR antagonist ICI (10 μM) or the adenylate cyclase inhibitor ddA (50 μM) before adding β-blockers. Changes in ICUE3 FRET ratio (an indication of cAMP activity) were measured. Experiments were performed in the presence of 1 μM β1AR-selective antagonist CGP20712A to block endogenous β1AR signaling. Mock, no primary antibody; NT, no treatment; ISO, isoproterenol; TIM, timolol; ALP, alprenolol; CAR, carvedilol; ICI, ICI118551; PRO, propranolol; MET, metoprolol; 177, CGP12177, IBMX, 3-isobutyl-1-methylxanthine; ddA, 2',5'-dideoxyadenosine. Each dot in the scatter dot plot in B–E represents a value from an individual tested cell. Error bars denote s.e.m., P values are computed by one-way ANOVA followed by Tukey’s test between NT (A) or TIM (B–E) and other groups.
-
Figure 3—source data 1
Excel spreadsheet containing the individual numeric values of PLA dots / cell number in each raw image analyzed in Figure 3A, and the individual numeric values for maximum FRET responses in Figure 3B-E.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig3-data1-v3.xlsx
β-blockers have been thought to generally block β2AR-induced cAMP signal. We hypothesized that the cAMP signal induced by β-blockers is restricted to local plasma membrane domains containing activated receptor, which is not detectable with traditional cAMP assays likely due to limited sensitivity. We applied the highly sensitive FRET-based biosensor ICUE3 to detect the dynamics of cAMP signal in living cells (DiPilato and Zhang, 2009; De Arcangelis et al., 2009). The full agonist ISO promoted cAMP signal in HEK293 cells while all β-blockers failed to do so (Figure 3B), in agreement with the classic definition of β-blockers. However, when cells were treated with non-selective phosphodiesterase (PDE) inhibitor IBMX, CAR, ALP and CGP12177 were able to induce small but significant cAMP signal in HEK293 cells (Figure 3C), indicating a role of PDE in suppressing and restricting the distribution of cAMP in the cells. When β2AR was exogenously expressed in HEK293 cells, CAR and ALP were able to induce cAMP signal in HEK293 cells even without PDE inhibition (Figure 3—figure supplement 2), probably due to insufficient cAMP-hydrolytic activity of endogenous PDEs to counter cAMP production induced from overexpressed β2AR. We then engineered a targeted cAMP biosensor by fusing the biosensor ICUE3 to the C-terminus of β2AR (β2AR-ICUE3), aiming to detect increases of cAMP within the local domain of the receptor. CAR and ALP promoted cAMP signals within the immediate vicinity of activated β2AR even at nanomolar concentrations (Figure 3D and E). The local increases of cAMP were abolished by inhibition of β2AR with ICI or inhibition of ACs with ddA (Figure 3E). We also used two generic plasma membrane (PM) targeted ICUE3 sensors to further characterize how the CAR and ALP generated cAMP signals are localized when compared to the full agonist ISO. Interestingly, neither CAAX-ICUE3 targeting to the non-rafts regions of PM nor LYN-ICUE3 targeting to the rafts regions of PM could sense cAMP induced by CAR and ALP, while ISO induced cAMP were readily detectable on PM (Figure 3—figure supplement 3), this further demonstrates that CAR and ALP only promote cAMP within the immediate vicinity of β2AR. These data confirm that CAR and ALP promote cAMP/PKA activity within the immediate vicinity of activated β2AR, in contrast to the broad distribution of cAMP/PKA activities induced by ISO in the cells.
Carvedilol augments the endogenous β2AR-dependent PKA phosphorylation of CaV1.2 and its channel activity in hippocampal neurons
Local cAMP signals possess the potential to selectively regulate downstream effectors in receptor complexes or within the vicinity of activated receptors. In the CNS, β2AR emerges as a prevalent postsynaptic norepinephrine effector at glutamatergic synapses, where β2AR functionally interacts with AMPA receptor (AMPAR) and L-type Ca2+ channel (LTCC) CaV1.2, and regulates neuronal excitability and synaptic plasticity (Davare et al., 2001; Joiner et al., 2010; Wang et al., 2010; Qian et al., 2012). CAR and ALP, but not TIM significantly increased PKA phosphorylation of S1928 and S1700 of central α11.2 subunit of CaV1.2 in hippocampal neurons when both β2AR and LTCC were endogenously expressed (Figure 4A, and Figure 4—figure supplement 1A). However, CAR and ALP failed to promote phosphorylation of the AMPAR subunit GluA1 on its PKA site serine 845 (Figure 4B, and Figure 4—figure supplement 1B). Like CaV1.2, AMPARs are associated with β2AR, Gs, AC and PKA (Davare et al., 2001; Joiner et al., 2010; Wang et al., 2010; Qian et al., 2012). These results indicate high selectivity in targeting downstream substrates by this β-blocker-induced signaling in hippocampal neurons. Meanwhile, the CAR and ALP-induced PKA phosphorylation of LTCC were blocked by β2AR inhibitor ICI, AC inhibitor ddA, and PKA inhibitor H89, but not CaMKII inhibitor KN93, validating the activation of β2AR-cAMP-PKA pathway (Figure 4C, and Figure 4—figure supplement 1C). We then examined the effects of CAR on PKA-dependent activation of LTCC CaV1.2 channels using cell-attached single channel recordings in hippocampal neurons. As shown before, ISO stimulates LTCC activity (Figure 5) (Shen et al., 2018; Davare et al., 2001). Consistent with the phosphorylation data, CAR but not TIM significantly increased the open probability, channel availability and mean ensemble average of endogenous LTCC in rat hippocampal neurons (Figure 5 and Figure 5—figure supplement 1). CAR stimulated channel activity when present in the patch pipette solution but not when applied outside the patch via bath perfusion (Figure 5 and Figure 5—figure supplement 1D). Moreover, backfilling experiments with CAR found that L-type channels activity was relatively low at the beginning of the recording but then it significantly increased as the drug diffused to the pipette tip (Figure 5F–G and Figure 5—figure supplement 1C). Consistent with our hypothesis and prior studies (Davare et al., 2001), ISO applied outside the patch via bath perfusion was still able to stimulate LTCC activity (Figure 5—figure supplement 1B). These data indicate that CAR promotes spatially restricted cAMP/PKA activities for selective augmentation of LTCC activities in neurons. We further found that the activation of LTCC by CAR promoted cell death in cortical neuron cultures, and inhibition of β2AR or LTCC counteracted carvedilol-induced neuronal toxicities (Figure 5—figure supplement 2).
Figure 4 with 1 supplement see all
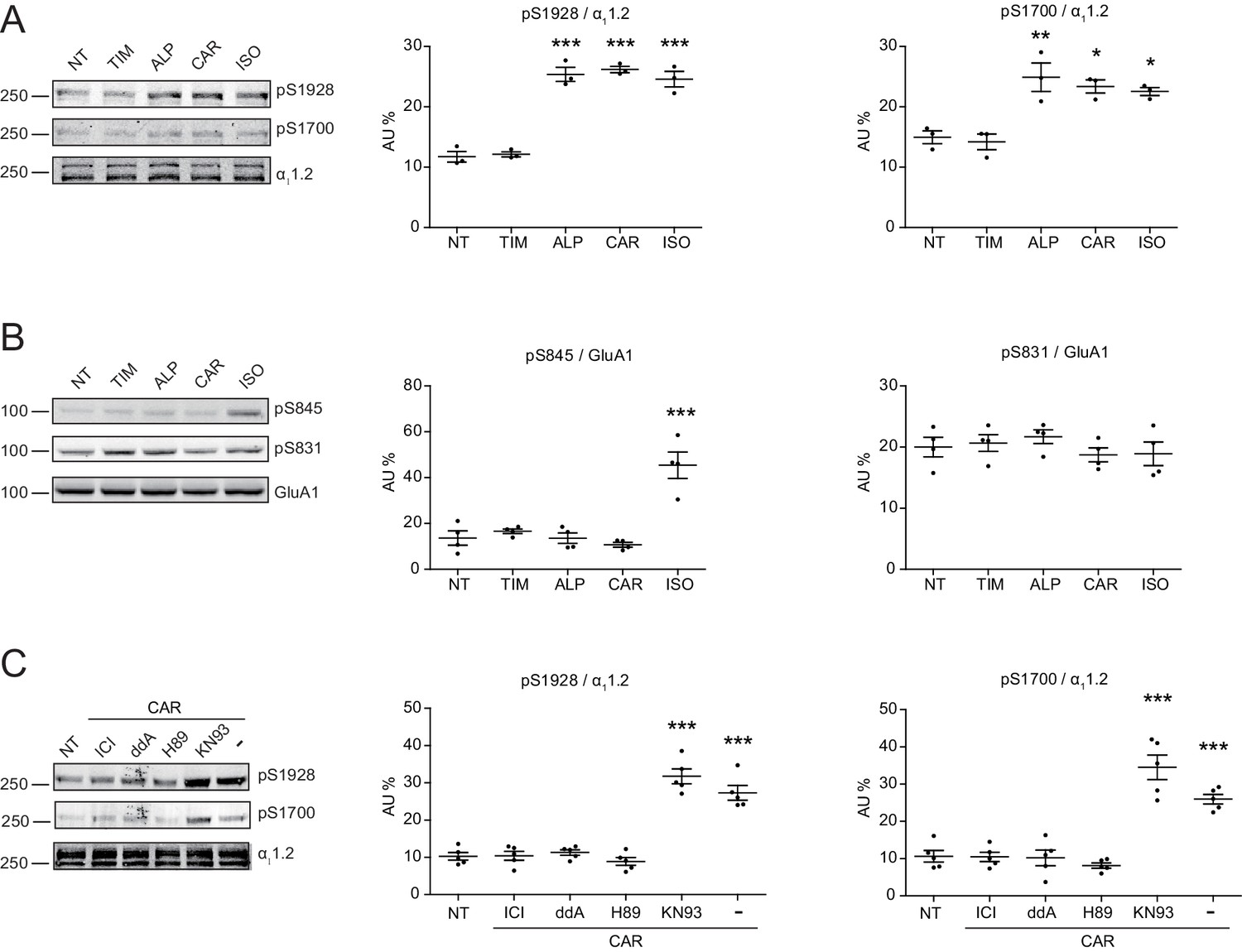
Carvedilol promotes endogenous β2AR-dependent phosphorylation of LTCC α11.2 by PKA in neurons.
(A) Rat neurons on 10–14 days in vitro (DIV) were treated for 5 min with 1 μM indicated drugs. The phosphorylation of endogenous LTCC α11.2 subunit was determined with phospho-specific antibodies, and normalized to total α11.2, n = 3. (B) Rat neurons on 10–14 DIV were treated for 5 min with 1 μM indicated drugs. The phosphorylation of endogenous AMPAR GluA1 subunit was determined with phospho-specific antibodies, and signals were normalized to total GluA1, n = 4. (C) Neurons were pretreated for 30 min with 10 μM β2AR inhibitor ICI, 50 μM AC inhibitor ddA, 10 μM PKA inhibitor H89 or 10 μM CaMKII inhibitor KN93 and then stimulated with 1 μM CAR for 5 min. Carvedilol-induced LTCC phosphorylation depends on endogenous β2AR, AC and PKA, but not CaMKII, n = 5. NT, no treatment; ISO, isoproterenol; TIM, timolol; ALP, alprenolol; CAR, carvedilol. Error bars denote s.e.m., P values are computed by one-way ANOVA followed by Tukey’s test between NT and other groups.
-
Figure 4—source data 1
Excel spreadsheet containing the individual numeric values of phosphorylated a11.2 or GluA1 / total a11.2 or GluA1 relative density analyzed in Figure 4.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig4-data1-v3.xlsx
Figure 5 with 2 supplements see all
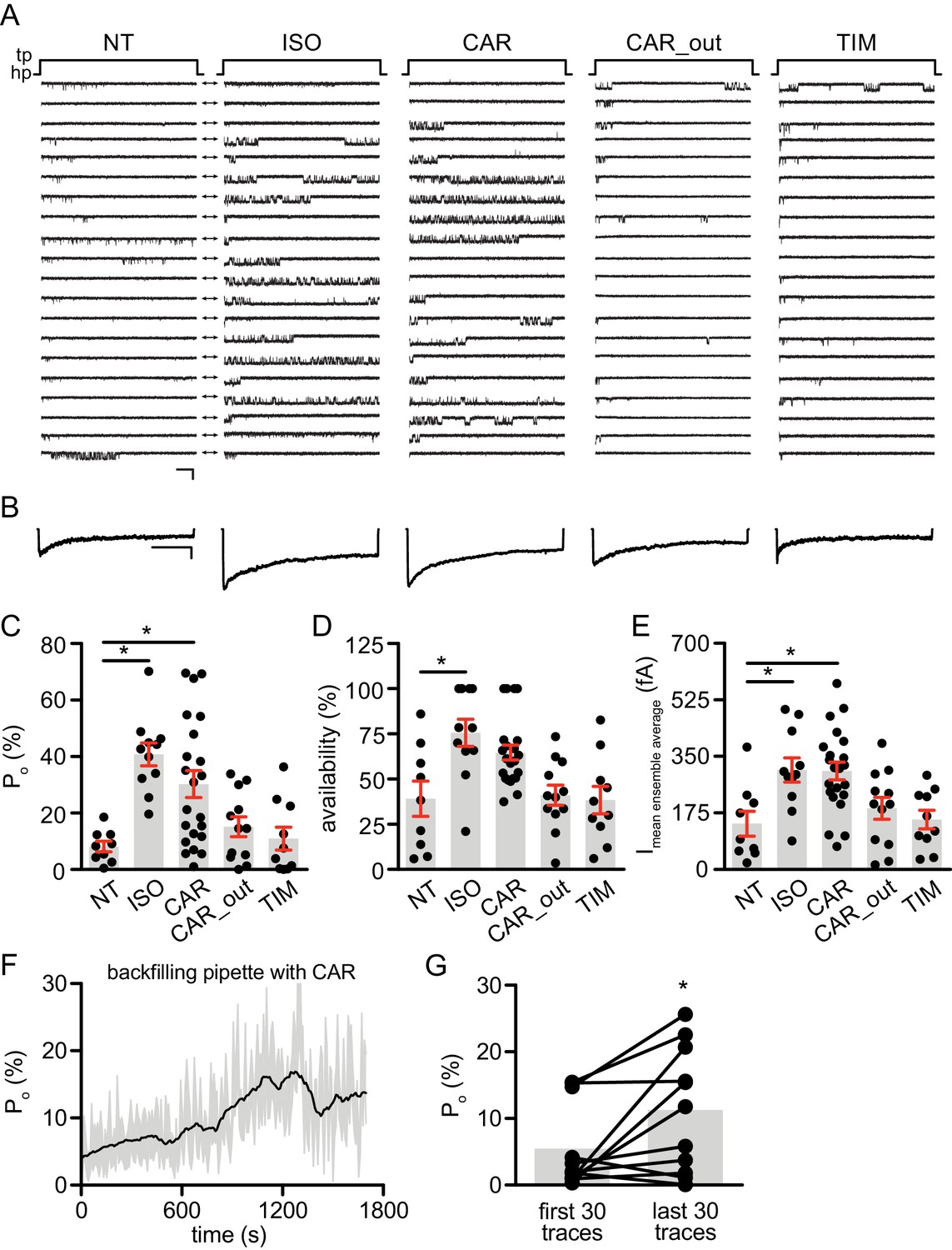
Carvedilol augments LTCC CaV1.2 channel activity in neurons.
(A) Representative single channel recordings of LTCC CaV1.2 currents using 110 mM Ba2+ as charge carrier in rat hippocampal neurons on 7–10 days in vitro (DIV) after depolarization from −80 (hp) to 0 mV (tp) in control patches (NT), patches containing 1 μM isoproterenol (ISO), 1 μM carvedilol (CAR) or 1 μM timolol (TIM) in the patch pipette or after addition of 1 μM CAR to the bath while the patch pipette contained a control pipette solution (CAR_out). Shown are 20 consecutive sweeps from representative experiments. Arrows throughout the figure indicate the 0-current level (closed channel). Scale bar denotes 2 pA and 200 ms. (B) Ensemble average currents as determined from all sweeps recorded for all the experimental conditions. Scale bar denotes 50 fA and 400 ms. (C–E) Mean ± s.e.m. for (C) Po (%), (D) availability (i.e. likelihood that a sweep had at least one event) (%) and (E) the mean ensemble average current (fA) for each experimental condition. *p<0.05 with Kruskal Wallis – Dunn’s multiple comparison test. Sweep and n numbers as well as summary statistics are in Supplementary file 1. (F) Ensemble Po versus time measurements obtained with a pipette backfilled with 1 μM CAR. The solid dark line represents the mean Po over time and the gray area is the s.e.m. at each time point. The mean line was smoothed to 15 neighbors on each size with a second order polynomial smoothing in PRISM for representation purposes only. (G) Mean Po of the first 30 traces versus the last 30 traces obtained with a pipette backfilled with 1 μM CAR. The gray boxes highlight the mean on each group. n = 11 patches. *p<0.05 with Mann-Whitney test.
-
Figure 5—source data 1
Excel spreadsheet containing the individual numeric values of Po, availability and current analyzed in Figure 5C-G.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig5-data1-v3.xlsx
Carvedilol but not isoproterenol selectively activates a mutant β2AR to augment LTCC activity in neurons
Structure-functional analyses of β2AR have previously revealed distinct residues important for binding to catecholamines and β-blockers (Strader et al., 1989; Liapakis et al., 2000; Warne et al., 2012; Ring et al., 2013). We hypothesized that mutation of Ser204 and Ser207 sites within β2AR binding pocket would abolish receptor hydrogen bonds with the catecholamine phenoxy moieties, thus reducing binding affinity to agonist ISO while having no effect on β-blocker binding (Figure 6A). Such a mutant β2AR could thus be selectively activated by CAR. We co-expressed the cAMP biosensor ICUE3 together with either wild-type (WT) β2AR or mutant S204A/S207A β2AR in MEF cells lacking endogenous β1AR and β2AR (DKO) to detect receptor signaling induced by different ligands. The mutant S204A/S207A β2AR induced a moderate cAMP signal at high but not low concentrations of ISO (Figure 6B). In contrast, after stimulation with CAR, the β2AR mutant S204A/S207A promoted significant cAMP signals at nanomolar concentrations; the overall concentration response curve was similar to those induced by WT β2AR (Figure 6B). Accordingly, the ISO-induced PKA phosphorylation of β2AR S204A/S207A mutant was selectively abolished at nanomolar concentrations. At higher concentrations, ISO was able to induce reduced PKA phosphorylation of the β2AR S204A/S207A mutant when compared to WT β2AR, consistent with the data of cAMP signals (Figure 6C, and Figure 6—figure supplement 1A). Meanwhile, ISO failed to induce GRK phosphorylation of β2AR S204A/S207A mutant at different concentrations (Figure 6C). In comparison, CAR induced equivalent PKA phosphorylation of β2AR WT and S204A/S207A mutant at different concentrations (Figure 6D, and Figure 6—figure supplement 1B). These data suggest that CAR, but not ISO selectively activates the S204A/S207A mutant β2AR at nanomolar concentrations. We then tested the effects of β2AR S204A/S207A mutant on LTCC channel activity after treatment with CAR in hippocampal neurons. In DKO neurons expressing the mutant S204A/S207A β2AR, CAR, but not ISO (30 nM) promoted PKA phosphorylation of LTCC α11.2 (Figure 7A and B and Figure 7—figure supplement 1). In agreement, CAR, but not ISO significantly increased the open probability, channel availability and mean ensemble average of LTCC (Figure 7C–7G). Together, CAR but not ISO selectively activates the S204A/S207A mutant β2AR at low concentrations and increases channel opening probabilities.
Figure 6 with 1 supplement see all
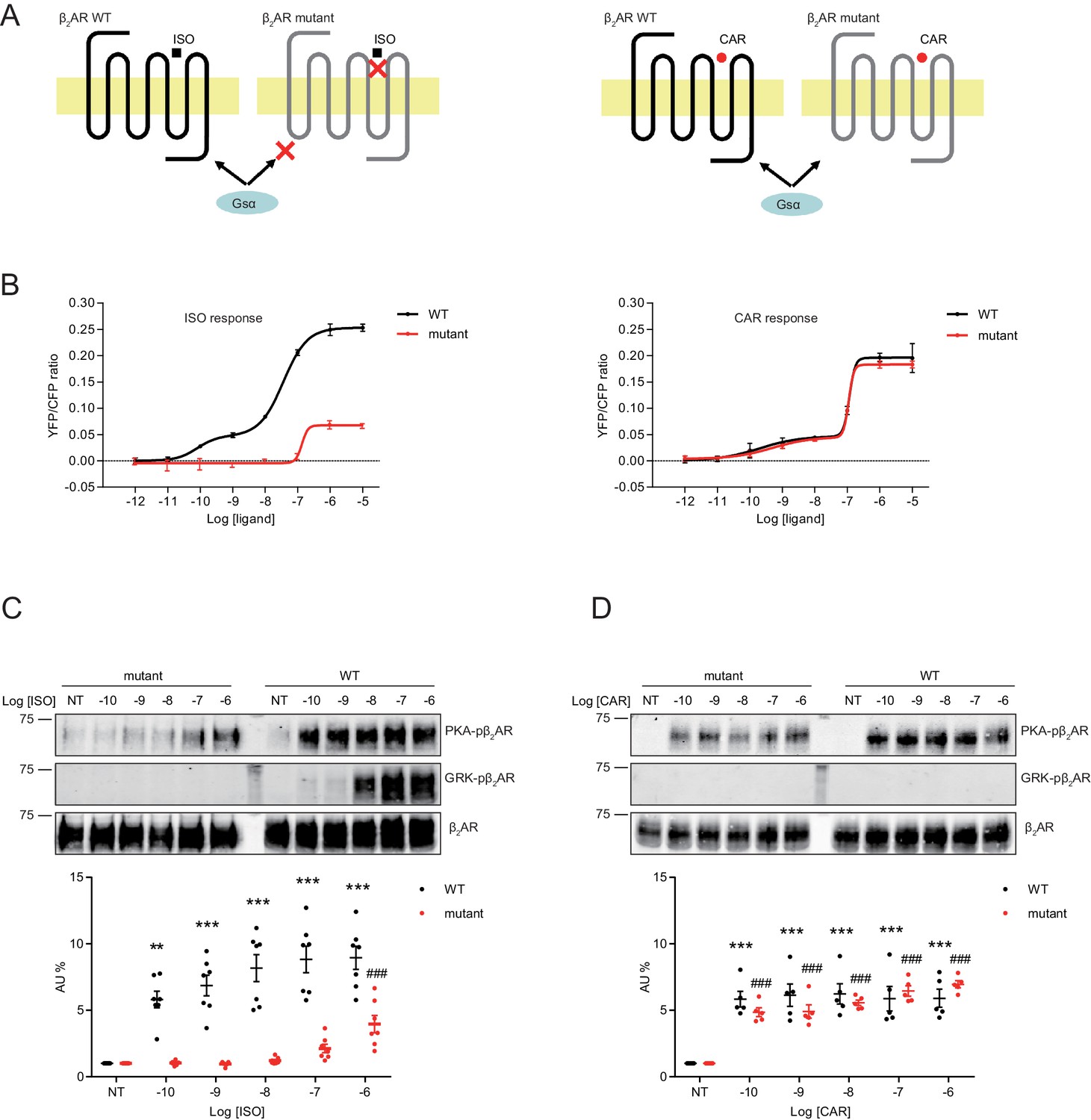
A mutant β2AR is selectively activated by carvedilol but not isoproterenol.
(A) Schematic of an engineered β2AR with S204/207A double serine mutations that loses high affinity binding to ISO but not CAR at nanomolar range. (B) cAMP biosensor ICUE3 and β2AR wild-type (WT) or mutant were co-expressed in MEF cells lacking both β1AR and β2AR. Changes of cAMP FRET ratio by increasing concentrations of ISO or CAR were measured. n = 5–29 cells. (C–D) HEK293 cells stably expressing FLAG-tagged β2AR WT or mutant were stimulated for 5 min with increasing concentrations of ISO (C), n = 7) or CAR (D), n = 5). The phosphorylation of β2AR on its PKA and GRK sites were determined with phospho-specific antibodies, and signals were normalized to total β2AR detected with anti-FLAG antibody. Experiments were performed in the presence of 1 μM β1AR-selective antagonist CGP20712A to block endogenous β1AR signaling. NT, no treatment; ISO, isoproterenol; CAR, carvedilol. Error bars denote s.e.m., P values are computed by one-way ANOVA followed by Tukey’s test between NT and other concentrations.
-
Figure 6—source data 1
Excel spreadsheet containing the individual numeric values for maximum FRET responses in Figure 6B, and the individual numeric values of phosphorylated β2AR / total β2AR relative density analyzed in Figure 6C-D.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig6-data1-v3.xlsx
Figure 7 with 1 supplement see all
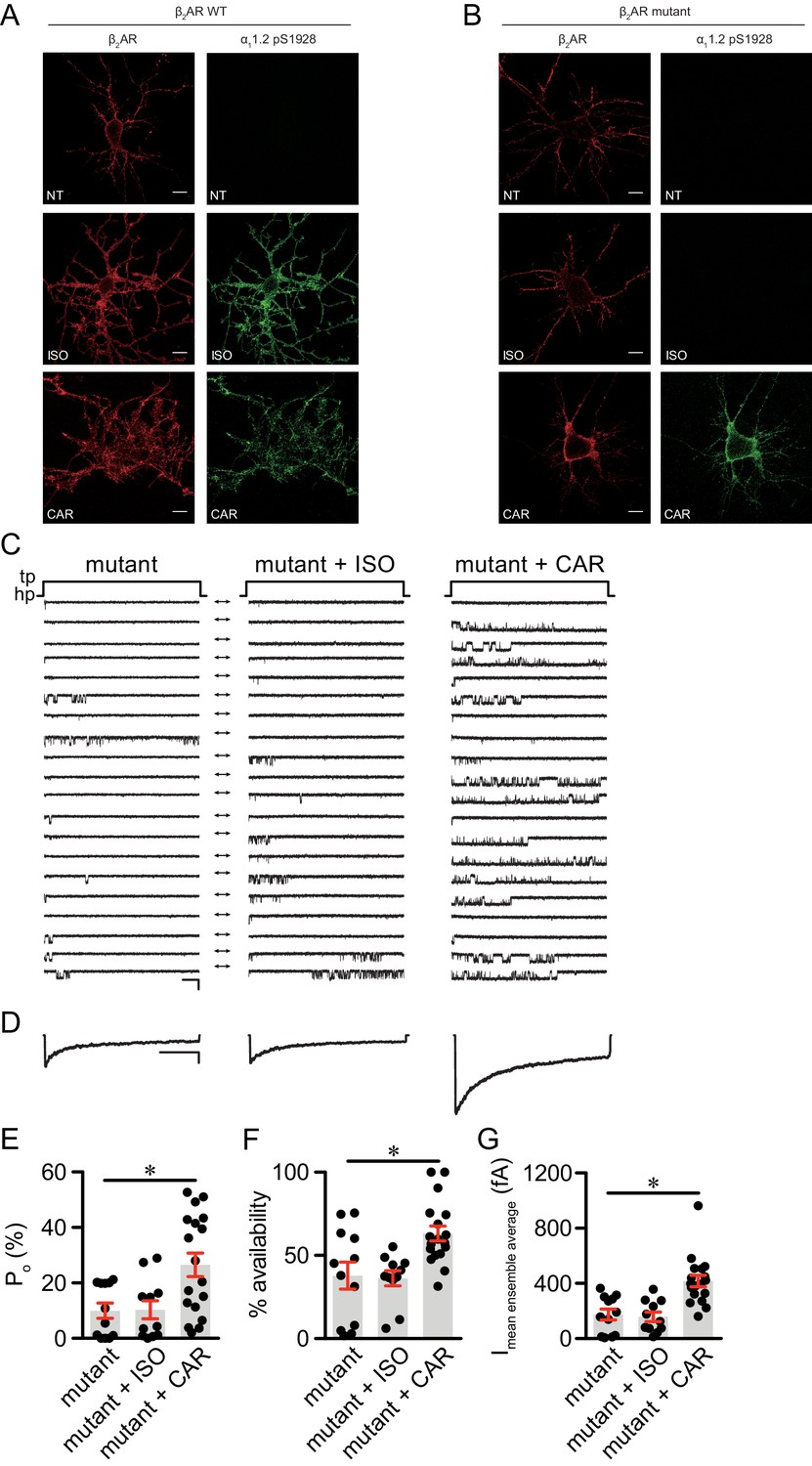
The β2AR mutant selectively supports carvedilol-induced augmentation of LTCC activity in neurons.
(A–B) β1AR/β2AR double knockout (DKO) mouse hippocampal neurons on 7–10 days in vitro (DIV) were cotransfected with FLAG-tagged β2AR WT (A) or mutant (B) and HA-tagged LTCC α11.2 subunit, 24 hr later cells were either mock treated (NT), or treated for 5 min with 10 nM isoproterenol (ISO) or carvedilol (CAR), fixed and labeled with anti-FLAG and a phospho-specific antibody for S1928 phosphorylated α11.2. Confocal images show mutant β2AR losts the ability of promoting LTCC phosphorylation upon ISO stimulation but remained the ability upon CAR stimulation in neurons. Scale bar, 10 μm. Representative of 6 images for each condition, three experiments. (C) Representative single channel recordings of LTCC CaV1.2 currents using 110 mM Ba2+ as charge carrier in DKO neurons on 7–10.days DIV expressing mutant β2AR after depolarization from −80 to 0 mV in in control patches (mutant) and patches containing 1 μM isoproterenol (ISO) or 1 μM carvedilol (CAR) in the patch pipette. Shown are 20 consecutive sweeps from representative experiments. Arrows throughout the figure indicate the 0-current level (closed channel). Scale bar denotes 2 pA and 200 ms. (D) Ensemble average currents as determined from all sweeps recorded for all the experimental conditions. Scale bar denotes 50 fA and 400 ms. (E–G) Mean ± s.e.m. for (E) Po (%), (F) availability (i.e. likelihood that a sweep had at least one event) (%) and (G) the mean ensemble average current (fA) for each experimental condition. *p<0.05 with Kruskal Wallis – Dunn’s multiple comparison test. Sweep and n numbers as well as summary statistics are in Supplementary file 2.
-
Figure 7—source data 1
Excel spreadsheet containing the individual numeric values of Po, availability and current analyzed in Figure 7E-G.
- https://cdn.elifesciences.org/articles/49464/elife-49464-fig7-data1-v3.xlsx
Discussion
In a classic view, agonist stimulation promotes both PKA and GRK phosphorylation of activated GPCRs. Although previous studies have reported that some β-blockers promote βAR-Gs coupling and thus might display partial agonism, this phenomenon is only observed at high concentrations and in vitro with reconstituted systems (Yao et al., 2009; DeVree et al., 2016; Gregorio et al., 2017). In this study, using a combination of highly sensitive tools such as engineered FRET-based cAMP sensors and single channel recording together with detection by phospho-specific antibodies, we show for the first time that β-blockers such as CAR and ALP can promote receptor-Gs coupling at nanomolar concentrations in living cells, which is clinically relevant in contrast to superphysiological concentrations in previous studies. In detail, as low as 1 nM alprenolol as well as 1 nM carvedilol induce 20–40% of maximal effects (as obtained with 1 μM isoproterenol) with respect to phosphorylation of β2AR by PKA and to cAMP production detected by the ICUE3 sensor coupled to β2AR. Unlike agonists, activation of β2AR by β-blockers selectively transduce G protein/cAMP/PKA signaling but not GRK signaling. More importantly, the β2AR-induced cAMP signal is highly spatially restricted to the local domain of activated β2AR, which selectively promotes activation of receptor-associated LTCC but not receptor-associated AMPAR, two downstream ion channels essential for adrenergic regulation of neuronal excitability in hippocampal neurons. The differential signaling by carvedilol with respect to LTCC and AMPAR is especially remarkable because both channels form complexes with β2AR that are localized within dendritic spines. Moreover, we have engineered a mutant β2AR that is selectively activated by β-blockers but not by catecholamines at low concentration. Our study defines CAR and ALP as Gs-biased partial agonists of βAR for highly spatially restricted cAMP/PKA signaling to CaV1.2 in neurons. The study exemplifies a unique mechanism by which β-blockers shape the compartmentalization of βAR signaling and a highly restrictive distribution of ligand-induced activation of GPCR targeting a specific downstream effector.
PKA-mediated phosphorylation is thought to play critical roles in heterologous desensitization of GPCRs and in receptor switching from Gs to Gi coupling (Daaka et al., 1997; Zamah et al., 2002), whereas GRK-mediated phosphorylation is implicated in β-arrestin recruitment and β-arrestin-dependent ERK activation (Luttrell et al., 1999; Pierce et al., 2000; Kim et al., 2005; Ren et al., 2005; Zidar et al., 2009; Choi et al., 2018). We have recently characterized that PKA and GRKs phosphorylate distinct subpopulations of β2AR in a single fibroblast or neuron (Shen et al., 2018). While GRK phosphorylation of β2AR is only observed at high concentrations of agonists, PKA phosphorylation can be induced with minimal doses of agonist (Shen et al., 2018; Tran et al., 2004; Tran et al., 2007; Liu et al., 2009). Here, our data show CAR does not promote GRK phosphorylation at low concentrations and induces a slow and minimal GRK effect at high concentrations when compared to those induced by ISO. The CAR-induced GRK effects are minimally related to the PKA effects. Previously, CAR has been recognized as a biased β-blocker that preferentially activates β-arrestin/ERK pathways (Wisler et al., 2007; Kim et al., 2008). Despite the prominent role of GRK phosphorylation in full agonist ISO-induced β2AR-β-arrestin/ERK signaling, our data clearly indicate that GRK phosphorylation of β2AR is not necessary for CAR-induced activation of ERK, consistent with a recent study showing a distinct general mechanism of β-arrestin activation that does not require the GRK-phosphorylated tail of different GPCRs (Eichel et al., 2018). Meanwhile, other studies show that in the absence of all G proteins, GPCRs fail to transduce β-arrestin/ERK signaling (Grundmann et al., 2018). These data indicate the necessity of G proteins in GPCR-induced arrestin activation. In our study, we observed a concentration-dependent correlation between PKA phosphorylation of β2AR with ERK activity induced by β-blockers, suggesting the potential role of Gs and PKA in CAR-induced β2AR-β-arrestin/ERK signaling are overlooked. In comparison, Gi is not required for CAR-induced β2AR/β-arrestin signaling even though CAR induces Gi recruitment to β1AR for transducing β1AR/β-arrestin signaling (Wang et al., 2017). Moreover, our results are also in line with a recent report that activation of β2AR with as low as femtomolar concentrations of ligands causes sustained ERK signaling (Civciristov et al., 2018), further support a PKA but GRK-dependent mechanism in GPCR-induced ERK activation. Future studies will help us understand how ligand-induced GPCRs utilize distinct mechanisms in activating β-arrestin/ERK pathway.
Engineered GPCRs have been widely applied in investigating structural and biological processes and behaviors by precisely controlling specific GPCR signaling branches (Lee et al., 2014). Previous mutagenesis studies have shown that β2AR with S204/207A mutation loses binding to adrenaline but still binds with several β-blockers including ALP (Liapakis et al., 2000). Based on this and recent advances in βAR structures with agonists and β-blockers (Warne et al., 2012; Ring et al., 2013), we have generated a S204/207A mutant that bestow β2AR with the ability to be selectively activated by β-blockers such as CAR and to transduce cAMP/PKA signaling. At nanomolar concentrations, while ISO fails to stimulate PKA phosphorylation of the S204/207A mutant β2AR, the mutant receptor still retains CAR-induced stimulation of PKA-phosphorylation of the receptor. The CAR-induced activation of mutant β2AR triggers the β2AR/Gs/cAMP/PKA signaling pathway and selectively targets downstream effectors in primary hippocampal neurons. Interestingly, the S204/207A β2AR mutant is not only refractory to its agonists but also completely lost both ISO- and CAR-induced GRK-phosphorylation of β2AR. Further studies comparing this mutant with previous reported β2AR-TYY and Y219A mutants that lack Gs and GRKs coupling, respectively (Choi et al., 2018; Shenoy et al., 2006), will facilitate the analysis of the physiological relevance of Gs/cAMP/PKA-dependent and GRK-dependent signaling pathways and enable researchers to explore β-arrestin/ERK pathway devoid of individual signaling branches.
β-blockers are a standard clinical treatment in a broad range of diseases. Many β-blockers possess intrinsic sympathomimetic activities (Bakris, 2009; Gorre and Vandekerckhove, 2010), which are problematic due to the side effects through stimulation of βARs (Bakris, 2009; Gorre and Vandekerckhove, 2010), a feature that limits the clinical utility of the drugs. Here, we show that β-blockers promote activation of β2AR by recruiting Gs that selectively transduces cAMP/PKA signal but not GRK signal. Meanwhile, binding of β-blockers to β1AR has been shown to enhance cAMP levels locally by dissociating a β1AR-PDE4 complex, thereby reducing the local cAMP-hydrolytic activity (Richter et al., 2013), β1AR and β2AR thus could utilize different mechanisms for β-blocker-induced signaling. Another interesting observation is that the β-blocker-induced β2AR-cAMP signal is sufficient to promote PKA phosphorylation of both β2AR and the receptor-associated CaV1.2 of LTCC, but not another substrate, the AMPAR GluA1 subunit. Both LTCC and AMPAR are shown to associate with the β2AR in hippocampal neurons (Davare et al., 2001; Joiner et al., 2010; Wang et al., 2010; Qian et al., 2012). Therefore, the preference of one local membrane target over another local target indicates a highly restricted nature of the cAMP-PKA activities, potentially dependent on the recently identified distinct subpopulations of β2AR and associated signaling molecules in the neurons (Shen et al., 2018). Nevertheless, the PKA phosphorylation leads to augmentation of LTCC activity, potentially contributing to the neuronal toxicities. Therefore, activation of GPCR at low ligand concentrations should be taken into consideration when designing and screening new therapeutic drugs.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain (Mus musculus) | β1AR/β2AR double knockout | Jackson Laboratories | Stock # 003810 | |
| Strain (Rattus norvegicus) | Sprague Dawley | Charles River Laboratories | ||
| Cell line (Homo sapiens) | HEK293/β2AR-WT | De Arcangelis et al., 2009 | HEK293 cells stably expressing FLAG-β2AR | |
| Cell line (Homo sapiens) | HEK293/β2AR-S204/207A | This paper | HEK293 cells stably expressing FLAG-β2AR-S204/207A | |
| Antibody | Phospho-β2AR (Ser261/262) (mouse monoclonal) | Dr. Richard Clark (UT Huston) | Clone 2G3 | IF (1 μg/ml), WB (1:1000) |
| Antibody | Phospho-β2AR (Ser355/356) (mouse monoclonal) | Dr. Richard Clark (UT Huston) | Clone 10A5 | WB (1:1000) |
| Antibody | β2AR (rabbit polyclonal) | Santa Cruz Biotechnology | sc-570 RRID:AB_2225412 | PLA (1:100), WB (1:1000) |
| Antibody | Phospho-β2AR (Ser355/356) (rabbit polyclonal) | Santa Cruz Biotechnology | sc-16719R RRID:AB_781609 | WB (1:1000) |
| Antibody | α11.2 (rabbit polyclonal) | Patriarchi et al., 2016 | FP1 | WB (1:1000) |
| Antibody | Phospho-α11.2 (Ser1928) (rabbit polyclonal) | Patriarchi et al., 2016 | CH3P | IF (1 μg/ml), WB (1:1000) |
| Antibody | Phospho-α11.2 (Ser1700) (rabbit polyclonal) | Patriarchi et al., 2016, Originally from Dr. William Catterall (U of Washington) | WB (1:1000) | |
| Antibody | GluA1 (rabbit polyclonal) | Patriarchi et al., 2016 | WB (1:1000) | |
| Antibody | Phospho-GluA1 (Ser831) (rabbit polyclonal) | Patriarchi et al., 2016 | WB (1:1000) | |
| Antibody | Phospho-GluA1 (Ser845) (rabbit polyclonal) | Patriarchi et al., 2016 | WB (1:1000) | |
| Antibody | FLAG-M1 | Sigma-Aldrich | F3040 RRID:AB_439712 | IF (1 μg/ml), WB (1:1000) |
| Antibody | HA | Covance | MMS-101R RRID:AB_291262 | PLA (1:1000) |
| Recombinant DNA reagent | β2AR-mutant | This paper | FLAG-tagged human β2AR with S204/207A double mutations | |
| Recombinant DNA reagent | β2AR-ICUE3 | This paper | ICUE3 fused to the C-terminal of human β2AR | |
| Commercial assay or kit | Duolink in situ detection reagents | Sigma-Aldrich | DUO92007 | PLA |
| Software, algorithm | pCLAMP10 | Molecular Devices | electrophysiology | |
| Software, algorithm | MetaFluor | Molecular Devices | FRET |
Animals
β1AR/β2AR double knockout (DKO) mouse were obtained from Jackson Laboratories to produce P0-P1 postnatal DKO pups, SD pregnant rats were obtained from Charles River Laboratories to provide E17-E19 embryonic rats. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#20234 and #20673) of the University of California at Davis and in accordance with the NIH guidelines.
Plasmids
DNA constructs expressing FLAG-tagged human β2AR (FLAG-β2AR) and HA-tagged rat L-type calcium channel (LTCC) α11.2 were described before (Shen et al., 2018). FLAG-tagged human β2AR with S204/207A double mutations (FLAG-mutant) was generated by Gibson assembly method (Thermo Fisher) using FLAG-β2AR and synthetic gBlocks with the double mutations as templates (Integrated DNA Technologies). FRET biosensor ICUE3, CAAX-ICUE3 and LYN-ICUE3 were described before (DiPilato and Zhang, 2009). To make the β2AR-ICUE3 fusion biosensor, ICUE3 was fused to the C-terminal of FLAG-β2AR with Gly-Ser linker. HA-Gsα was made by replacing CFP with HA tag, using Gsα-CFP as template (a gift from Dr. Catherine Berlot, Addgene plasmid # 55793).
Antibodies and chemicals
Request a detailed protocolMouse monoclonal antibodies against β2AR at serine 261/262 (clone 2G3) and at serine 355/356 (clone 10A5) were kindly provided by Dr. Richard Clark (UT Huston). Polyclonal antibodies against β2AR (sc-570) and phosphorylated β2AR at serine 355/356 (sc-16719R) were purchased from Santa Cruz Biotechnology. Polyclonal antibodies against α11.2 residues 754–901 for total α11.2 (FP1), residues 1923–1935 for phosphorylated serine 1928 site (LGRRApSFHLECLK, pS1928) and residues 1694–1709 for phosphorylated serine 1700 site (EIRRAIpSGDLTAEEEL, pS1700) were described before (Patriarchi et al., 2016). Polyclonal antibodies against GluA1 residues 894–907 for total GluA1, residues 826–837 for phosphorylated serine 831 site (LIPQQpSINEAIK, pS831) and residues 840–851 for phosphorylated serine 845 site (TLPRNpSGAGASK, pS845) were described before (Patriarchi et al., 2016). Other antibodies used in the experiments include: anti-FLAG (F3040, Sigma), anti-HA (MMS-101R, Covance), Alexa fluor 488 conjugated goat anti-rabbit IgG and Alexa fluor 594 conjugated goat anti-mouse IgG (A-11034 and A-11032, Thermo Fisher), DyLight 680 conjugated goat anti-mouse IgG and anti-rabbit IgG (35518 and 35568, Thermo Fisher), IRDye 800CW conjugated goat anti-mouse IgG and anti-rabbit IgG (926–32210 and 926–32211, Li-cor).
Isoproterenol (I2760), timolol (T6394), alprenolol (A8676), propranolol (P0884), metoprolol (M5391), CGP12177A (C125), CGP20712A (C231), ICI118551 (I127), 3-isobutyl-1-methylxanthine (I5879) and 2',5'-dideoxyadenosine (D7408) were purchased from Sigma. Carvedilol (15418) was from Cayman Chemical, H89 (H-5239) was from LC Labs, pertussis toxin (179B) was from List Labs.
Cell culture and transfection
Request a detailed protocolHuman embryonic kidney HEK293 cells were from American Type Culture Collection (ATCC) and were maintained in Dulbecco’s modified Eagle medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS, Sigma). HEK293 cells stably expressing FLAG-β2AR was from previous study (De Arcangelis et al., 2009). HEK293 cells stably expressing FLAG-mutant β2AR was generated in this study. Briefly, cells transfected with β2AR-mutant were selected by G418 resistance (Corning) and cell clones were obtained by limiting serial dilution, monoclonal cells were analyzed by western blots and the one with comparable β2AR expression to FLAG-β2AR stable cells was chosen.
Mouse embryonic fibroblasts (MEFs) from β1AR/β2AR double knockout (DKO) mouse were described in previous study (Cervantes et al., 2010) and were maintained in DMEM supplemented with 10% FBS. Primary mouse hippocampal neurons were isolated and cultured from P0-P1 early postnatal DKO mouse pups, and primary rat hippocampal neurons were prepared from E17-E19 embryonic rats using methods described previously (Chen et al., 2008; Beaudoin et al., 2012). Briefly, dissected hippocampi were dissociated by 0.25% trypsin (Corning) and trituration. Neurons were plated on poly-D-lysine-coated (Sigma) glass coverslips in 24-well plate for imaging and in 6-well plate for biochemistry at a cell density of 50,000/well and 1 million/well, respectively. Neurons were cultured in Neurobasal medium supplemented with GlutaMax and B-27 (Thermo Fisher).
HEK293 cells were transfected with plasmids using polyethylenimine according to manufacturer’s instructions (Sigma). Neurons were transfected by the Ca2+-phosphate method (Jiang and Chen, 2006). Briefly, cultured neurons on 6–10 DIV were switched to pre-warmed Eagle’s minimum essential medium (EMEM, Thermo Fisher) supplemented with GlutaMax 1 hr before transfection, conditioned media were saved. DNA precipitates were prepared by 2x HBS (pH 6.96) and 2 M CaCl2. After incubation with DNA precipitates for 1 hr, neurons were incubated in 10% CO2 pre-equilibrium EMEM for 20 min, then replaced with conditioned medium and cultured in 5% CO2 incubator until use.
Confocal microscopy imaging
Request a detailed protocolRat hippocampal neurons were transfected with FLAG-β2AR on 10 DIV, treated for 5 min with 10 nM or 1 μM indicated drugs on 12 DIV. Mouse DKO hippocampal neurons were transfected with FLAG-β2AR or FLAG-mutant and HA-α11.2 at 1:1 ratio on 6–8 DIV, and stimulated with indicated drugs and times 24 hr after transfection. Treated cells were fixed, permeabilized, and co-stained with indicated antibodies with a final concentration of 1 μg/ml for each antibody, which were revealed by a 1:1000 dilution of Alexa fluor 488 conjugated goat anti-rabbit IgG or Alexa fluor 594 conjugated goat anti-mouse IgG, respectively. Fluorescence images were taken by Zeiss LSM 700 confocal microscope with a 63×/1.4 numerical aperture oil-immersion lens.
Proximity ligation assay
Request a detailed protocolHEK293 cells growing on poly-D-lysine coated coverslips were transfected with FLAG-β2AR or FLAG-mutant, HA-Gsα and pEYFP-N1 at 8:1:1 ratio. 24 hr after transfection, cells were serum-starved 2 hr, treated 100 nM indicated drugs for 5 min. Following stimulation, cells were fixed, permeabilized, and co-stained with anti-β2AR antibody (1:100 dilution) from rabbit in conjunction with anti-HA antibody (1:1000 dilution) from mouse. The proximity ligation reaction was performed according to the manufacturer’s protocol using the Duolink in situ detection orange reagents (Sigma). Images were recorded with Zeiss LSM 700 confocal microscope with a 63×/1.4 numerical aperture oil-immersion lens. To quantify the PLA signals, the number of red fluorescent objects in each image was quantified using the Squassh plug-in for ImageJ software (Rizk et al., 2014), and divided by the number of transfected cells.
Fluorescence resonance energy transfer (FRET) measurement
Request a detailed protocolFRET measurement was performed as previously described (De Arcangelis et al., 2009). Briefly, HEK 293 cells were transfected with ICUE3 or β2AR-ICUE3, DKO MEFs were co-transfected with ICUE3 and FLAG-β2AR or FLAG-mutant. Cells were imaged on a Zeiss Axiovert 200M microscope with a 40×/1.3 numerical aperture oil-immersion lens and a cooled CCD camera. Dual emission ratio imaging was acquired with a 420DF20 excitation filter, a 450DRLP diachronic mirror, and two emission filters (475DF40 for cyan and 535DF25 for yellow). The acquisition was set with 0.2 s exposure in both channels and 20 s elapses. Images in both channels were subjected to background subtraction, and ratios of yellow-to-cyan were calculated at different time points.
Western blot
Request a detailed protocolHEK293 cells stably expressing FLAG-β2AR or FLAG-mutant were serum-starved for 2 hr and treated with indicated drugs and times, then harvested by lysis buffer (10 mM Tris pH 7.4, 1% NP40, 150 mM NaCl, 2 mM EDTA) with protease and phosphatase inhibitor cocktail. Rat hippocampal neurons on 10–14 DIV were treated with indicated drugs and times, then harvested by lysis buffer (10 mM Tris pH 7.4, 1% TX-100, 150 mM NaCl, 5 mM EGTA, 10 mM EDTA, 10% glycerol) with protease and phosphatase inhibitor cocktail. Protein samples were analyzed by western blot using antibodies as indicated at a 1:1000 dilution and signals were detected by Odyssey scanner (Li-cor).
Cell-attached patch clamp electrophysiology
Request a detailed protocolPrimary rat and mouse hippocampal neurons were used on 7–10 DIV. Cell-attached patch clamp recordings were performed on an Olympus IX70 inverted microscope in a 15 mm culture coverslip at room temperature (22–25°C). Signals were recorded at 10 kHz and low-pass filtered at 2 kHz with an Axopatch 200B amplifier and digitized with a Digidata 1440 (Molecular Devices). Recording pipettes were pulled from borosilicate capillary glass (0.86 OD) with a Flaming micropipette puller (Model P-97, Sutter Instruments) and polished (polisher from World Precision Instruments). Pipette resistances were strictly maintained between 6–7 MΩ to ameliorate variations in number of channels in the patch pipette. The patch transmembrane potential was zeroed by perfusing cells with a high K+ extracellular solution containing (in mM) 145 KCl, 10 NaCl, and 10 HEPES, pH 7.4 (NaOH). The pipette solution contained (in mM) 20 tetraethylammonium chloride (TEA-Cl), 110 BaCl2 (as charge carrier), and 10 HEPES, pH 7.3 (TEA-OH). This pipette solution was supplemented with 1 µM ω-conotoxin GVIA and 1 µM ω-conotoxin MCVIIC to block N and P/Q-type Ca2+ channels, respectively, and (S)-(-)-BayK-8644 (500 nM) was included in the pipette solution to promote longer open times and resolve channel openings as previously performed by our group and others (Shen et al., 2018; Davare et al., 2001; Wang et al., 2001; Qian et al., 2017; Hess et al., 1986; Schuhmann et al., 1997; Costantin et al., 1998; Yue and Marban, 1990; Dzhura and Neely, 2003; Navedo et al., 2005). In a subset of experiments, BayK was left out of the pipette solution. Note that ISO and CAR had similar effects on channel activity whether BayK was included or not in the pipette solution. To examine the effects of β-adrenergic stimulation on the L-type CaV1.2 single-channel activity, 1 μM isoproterenol was added to the pipette solution in independent experiments. Note that we have previously used the L-type CaV1.2 channel blocker nifedipine (1 μM) to confirm the recording of L-type CaV1.2 currents under control conditions and in the presence of isoproterenol (Patriarchi et al., 2016). Single-channel activity was recorded during a single pulse protocol (2 s) from a holding potential of −80 mV to 0 mV every 5 s. An average of >50 sweeps were collected with each recording file under all experimental conditions. The half-amplitude event-detection algorithm of pClamp10 was used to measure overall single-channel L-type CaV1.2 activity as nPo, where n is the number of channels in the patch and Po is the open probability. Because the variability of nPo can be a critical element to interpret single channel data due to overstating open probability based on a high n number, we corrected this parameter by the number of channels (n) describing channel open probability and availability as well as calculating the mean ensemble average current. Data were pooled for each condition and analyzed with GraphPad Prism software.
Statistical analysis
Request a detailed protocolData were analyzed using GraphPad Prism software and expressed as mean ± s.e.m. Differences between two groups were assessed by appropriate two-tailed unpaired Student’s t-test or nonparametric Mann-Whitney test. Differences among three or more groups were assessed by One-way ANOVA with Tukey’s post hoc test or the Kruskal-Wallis test with Dunn’s post hoc test. p<0.05 was considered statistically significant (denoted by * or # in figures).
Data availability
Request a detailed protocolAll data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for all main figures.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for all main figures.
References
-
An in-depth analysis of vasodilation in the management of hypertension: focus on adrenergic blockadeJournal of Cardiovascular Pharmacology 53:379–387.https://doi.org/10.1097/FJC.0b013e31819fd501
-
Nebivolol, bucindolol, metoprolol and carvedilol are devoid of intrinsic sympathomimetic activity in human myocardiumBritish Journal of Pharmacology 133:1330–1338.https://doi.org/10.1038/sj.bjp.0704188
-
NS21: re-defined and modified supplement B27 for neuronal culturesJournal of Neuroscience Methods 171:239–247.https://doi.org/10.1016/j.jneumeth.2008.03.013
-
Facilitation by the beta2a subunit of pore openings in cardiac Ca2+ channelsThe Journal of Physiology 507 ( Pt 1:93–103.https://doi.org/10.1111/j.1469-7793.1998.093bu.x
-
Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytesThe Journal of Biological Chemistry 284:33824–33832.https://doi.org/10.1074/jbc.M109.020388
-
Differential modulation of cardiac Ca2+channel gating by beta-subunitsBiophysical Journal 85:274–289.https://doi.org/10.1016/S0006-3495(03)74473-7
-
Cross-Talk between insulin signaling and G Protein-Coupled receptorsJournal of Cardiovascular Pharmacology 70:74–86.https://doi.org/10.1097/FJC.0000000000000481
-
β-Blockers in hypertension: studies and meta-analyses over the yearsCanadian Journal of Cardiology 30:S16–S22.https://doi.org/10.1016/j.cjca.2014.02.012
-
DREADDs: novel tools for drug discovery and developmentDrug Discovery Today 19:469–473.https://doi.org/10.1016/j.drudis.2013.10.018
-
The superfamily of heptahelical receptorsNature Cell Biology 2:E133–E136.https://doi.org/10.1038/35017152
-
The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptorThe Journal of Biological Chemistry 275:37779–37788.https://doi.org/10.1074/jbc.M002092200
-
Different intrinsic activities of bucindolol, carvedilol and metoprolol in human failing myocardiumBritish Journal of Pharmacology 130:1131–1139.https://doi.org/10.1038/sj.bjp.0703400
-
Noradrenergic modulation of emotional memory in agingAgeing Research Reviews 27:61–66.https://doi.org/10.1016/j.arr.2016.03.004
-
β-adrenergic receptor signalling and its functional consequences in the diseased heartEuropean Journal of Clinical Investigation 46:362–374.https://doi.org/10.1111/eci.12598
-
β2-Adrenergic receptor supports prolonged theta tetanus-induced LTPJournal of Neurophysiology 107:2703–2712.https://doi.org/10.1152/jn.00374.2011
-
GRKs and beta-arrestins: roles in receptor silencing, trafficking and signalingTrends in Endocrinology & Metabolism 17:159–165.https://doi.org/10.1016/j.tem.2006.03.008
-
beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptorJournal of Biological Chemistry 281:1261–1273.https://doi.org/10.1074/jbc.M506576200
-
Identification of two serine residues involved in agonist activation of the beta-adrenergic receptorThe Journal of Biological Chemistry 264:13572–13578.
-
Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivityFASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 24:3511–3521.https://doi.org/10.1096/fj.10-156661
-
Recent developments in biased agonismCurrent Opinion in Cell Biology 27:18–24.https://doi.org/10.1016/j.ceb.2013.10.008
-
Myocyte adrenoceptor signaling pathwaysScience 300:1530–1532.https://doi.org/10.1126/science.1079206
-
Permeation in the dihydropyridine-sensitive calcium channel. Multi-ion occupancy but no anomalous mole-fraction effect between Ba2+ and Ca2+The Journal of General Physiology 95:911–939.https://doi.org/10.1085/jgp.95.5.911
-
Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to gs and gi. Demonstration in a reconstituted systemThe Journal of Biological Chemistry 277:31249–31256.https://doi.org/10.1074/jbc.M202753200
-
Bias in chemokine receptor signallingTrends in Immunology 35:243–252.https://doi.org/10.1016/j.it.2014.02.004
Article and author information
Author details
Funding
National Institutes of Health (GM129376)
- Yang K Xiang
U.S. Department of Veterans Affairs (BX002900)
- Yang K Xiang
National Institutes of Health (HL098200)
- Manuel F Navedo
National Institutes of Health (HL121059)
- Manuel F Navedo
National Institutes of Health (HL149127)
- Manuel F Navedo
American Heart Association (Postdoctoral fellowship)
- Ao Shen
- Qian Shi
American Heart Association
- Yang K Xiang
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by NIH grant GM129376 and VA Merit grant BX002900 to YKX, and NIH grants HL098200, HL121059 and HL149127 to MFN. AS and QS were recipients of AHA postdoctoral fellowship. YKX is an established AHA investigator.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#20234) of the University of California at Davis. Every effort was made to minimize suffering.
Copyright
© 2019, Shen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,008
- views
-
- 296
- downloads
-
- 16
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 16
- citations for umbrella DOI https://doi.org/10.7554/eLife.49464
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
β-blockers augment L-type Ca2+ channel activity by targeting spatially restricted β2AR signaling in neurons
eLife 8:e49464.
https://doi.org/10.7554/eLife.49464




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}