The naive T-cell receptor repertoire has an extremely broad distribution of clone sizes
- Theoretical Biology and Bioinformatics, Utrecht University, Netherlands
- Division of Infection and Immunity, University College London, United Kingdom
- Department of Pathology, Yale School of Medicine, United States
Figures
Figure 1 with 2 supplements
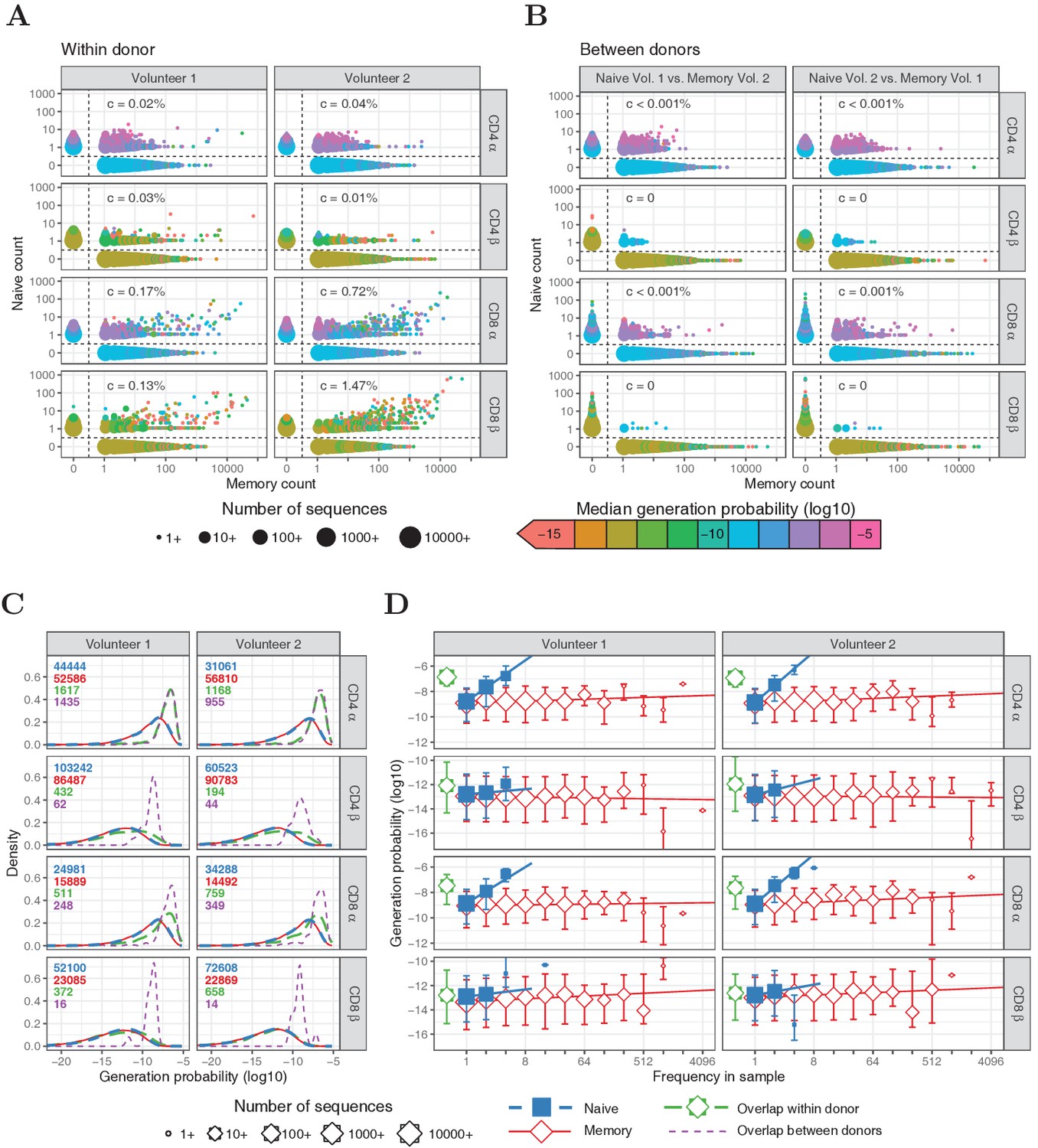
Frequencies and generation probabilities of TCRα and TCRβ sequences from memory and naive T cells.
(A) Frequency of TCRα and TCRβ sequences in naive versus total frequency in memory repertoires sampled from the same volunteer. Symbol sizes represent number of sequences with these frequencies and colour represents their median generation probability , as determined using IGoR (Marcou et al., 2018). The value is the slope of linear regression on sequences with a memory count > 100 and indicates the estimated probability that a given TCR sequence from a memory cell appears in the naive sample. (B) As A., but comparing frequency in naive sample from one volunteer with frequency in memory from the other volunteer. (C) Distributions of generation probabilities (log10) for TCR α and β sequences from CD4+ and CD8+ from two volunteers. Blue dashed: naive, red solid: memory, green long-dashed: overlap (i.e., sequences observed in both naive and memory within a volunteer), purple dashed: overlap between volunteers (i.e., sequences observed in the naive subset of Volunteer 1 and a memory subset of Volunteer 2, or vice versa). The total number of sequences for each group are indicated in corresponding colors. (D) The median is shown for each observed frequency class (log2 bins) of sequences exclusively observed in naive (blue squares) or memory T-cell (red diamonds) samples. of the overlapping chains is shown in green for reference (irrespective of frequency). Symbol sizes indicate numbers of sequences for each frequency class. Error bars represent the 25% and 75% quartiles, solid lines indicate linear regression between observed frequency and , weighted by the number of sequences with that frequency.
-
Figure 1—source data 1
Memory and naive counts in Experiment 1.
Number of TCRα and TCRβ sequences exclusively occurring in naive or memory samples within log2-frequency bins, as in Figure 1D. Bin intervals are excluding the left, but including the right number.
- https://cdn.elifesciences.org/articles/49900/elife-49900-fig1-data1-v1.txt
Figure 1—figure supplement 1
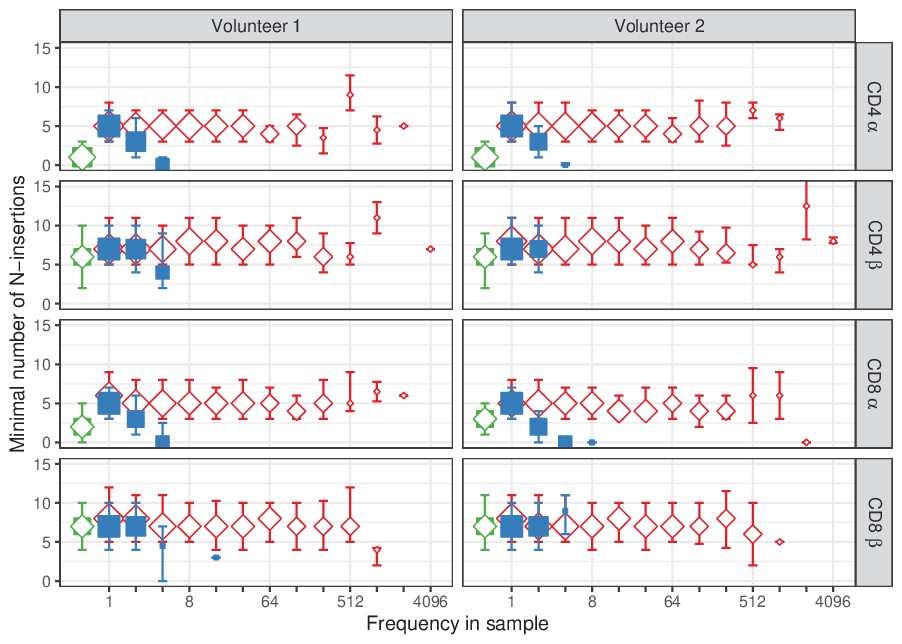
TCRα and TCRβ sequences abundant in naive tend to have less N-insertions.
For each sequence in our dataset of single samples, the minimal number of N-insertions was determined by the length of the CDR3 nucleotide sequence not matching germline TRAV and TRAJ (for TCRα), or TRBV, TRBD and TRBJ (for TCRβ). The median insertion length is shown for each observed frequency class (log2 bins) in naive (blue squares) and memory T-cell (red diamonds) samples. Insertion length of the overlapping TCR sequences is shown in green for reference (irrespective of frequency). Symbol sizes indicate numbers of sequences for each frequency class. Error bars represent the 25% and 75% quartiles.
Figure 1—figure supplement 2

Similar to Figure 1, but for HTS data processed with RTCR.
(A) Frequency in of TCRα and TCRβ sequences in naive versus total frequency in memory samples of the same volunteer. Symbol sizes represent number of sequences with these frequencies and their median generation probability , as determined using IGoR (Marcou et al., 2018). The value is the slope of linear regression on sequences with a memory count > 100 and indicates the estimated probability that a given TCR sequence from a memory cell is FACS-sorted as naive. (B) As A., but comparing frequency in naive sample from one volunteer with frequency in memory from the other volunteer. (C) Distributions of generation probabilities (log10) for TCR α and β sequences from CD4+ and CD8+ from two volunteers. Blue dashed: naive, red solid: memory, green long-dashed: overlap (i.e., sequences observed in both naive and memory within a volunteer), purple dashed: overlap between volunteers (i.e., sequences observed in the naive subset of Volunteer one and a memory subset of Volunteer 2, or vice versa). The total number of sequences for each group are indicated in corresponding colors. (D) The median is shown for each observed frequency class (log2 bins) of sequences exclusively observed in naive (blue squares) or memory T-cell (red diamonds) samples. of the overlapping chains is shown in green for reference (irrespective of frequency). Symbol sizes indicate numbers of sequences for each frequency class. Error bars represent the 25% and 75% quartiles, solid lines indicate linear regression between observed frequency and , weighted by the number of sequences with that frequency.
Figure 2 with 3 supplements

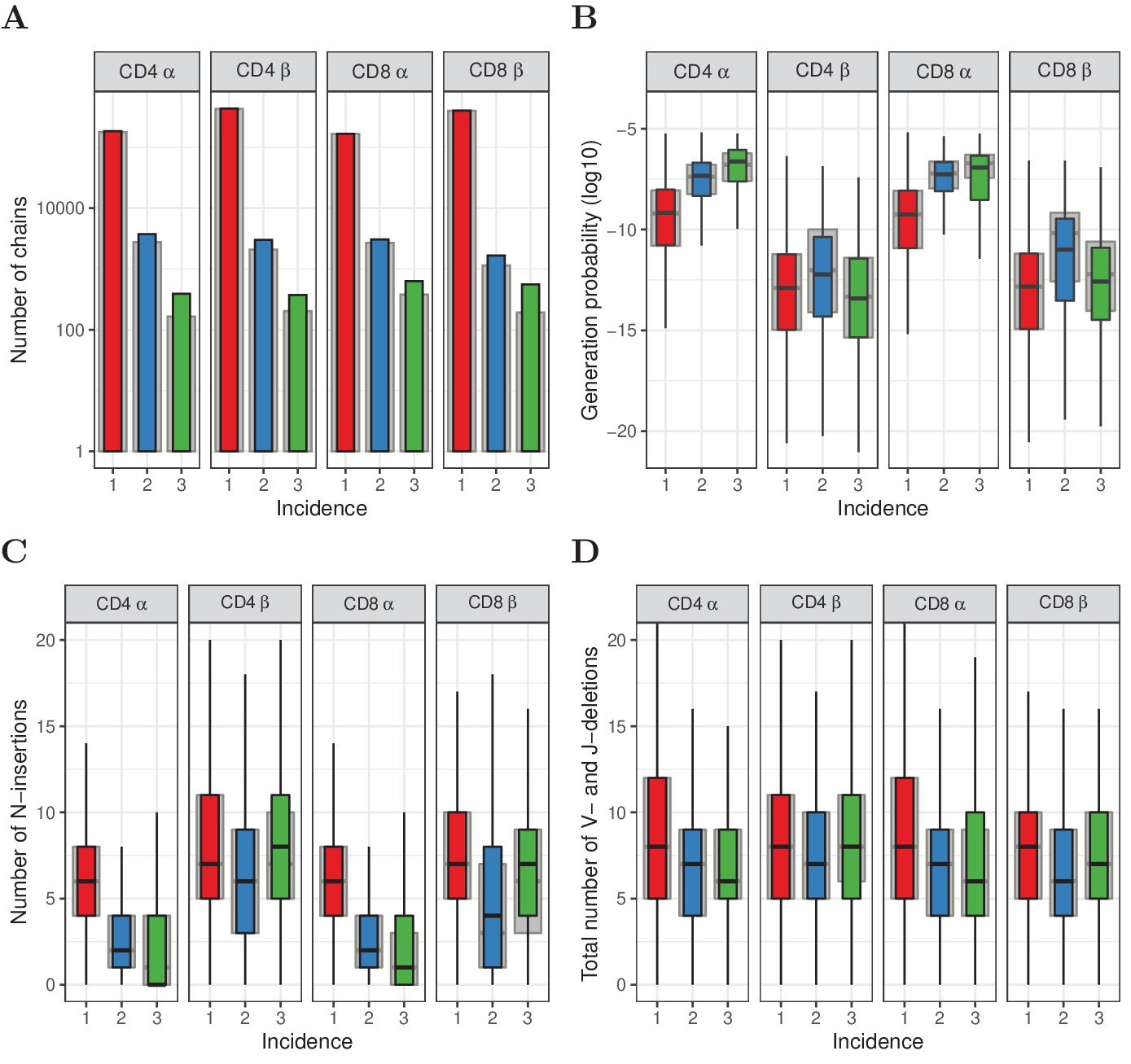
Subsampling naive T cells confirms that frequently observed TCRα but not TCRβ sequences have high generation probabilities.
(A) The number of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples (experiment 2). The grey background bars show the results after removing all sequences that were also observed in the corresponding memory samples. (B) Generation probabilities (log10) of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. (C) Minimal number of N-additions of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. (D) Number of V- and J-deletions of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. The plot shows median (black horizontal line), interquartile range (filled bar) and the range from the bar up to 1.5 times the interquartile range (black vertical range, outliers not shown).
-
Figure 2—source data 1
Naive TCRα and TCRβ abundance in the subsamples of Experiment 2.
Per dataset, the number of TCR sequences is shown per incidence and abundance across subsamples. Abundance is log2 binned according to the total UMI count across the three naive subsamples. Pearson correlation coefficient between incidence and total UMI count is shown for each dataset.
- https://cdn.elifesciences.org/articles/49900/elife-49900-fig2-data1-v1.txt
Figure 2—figure supplement 1
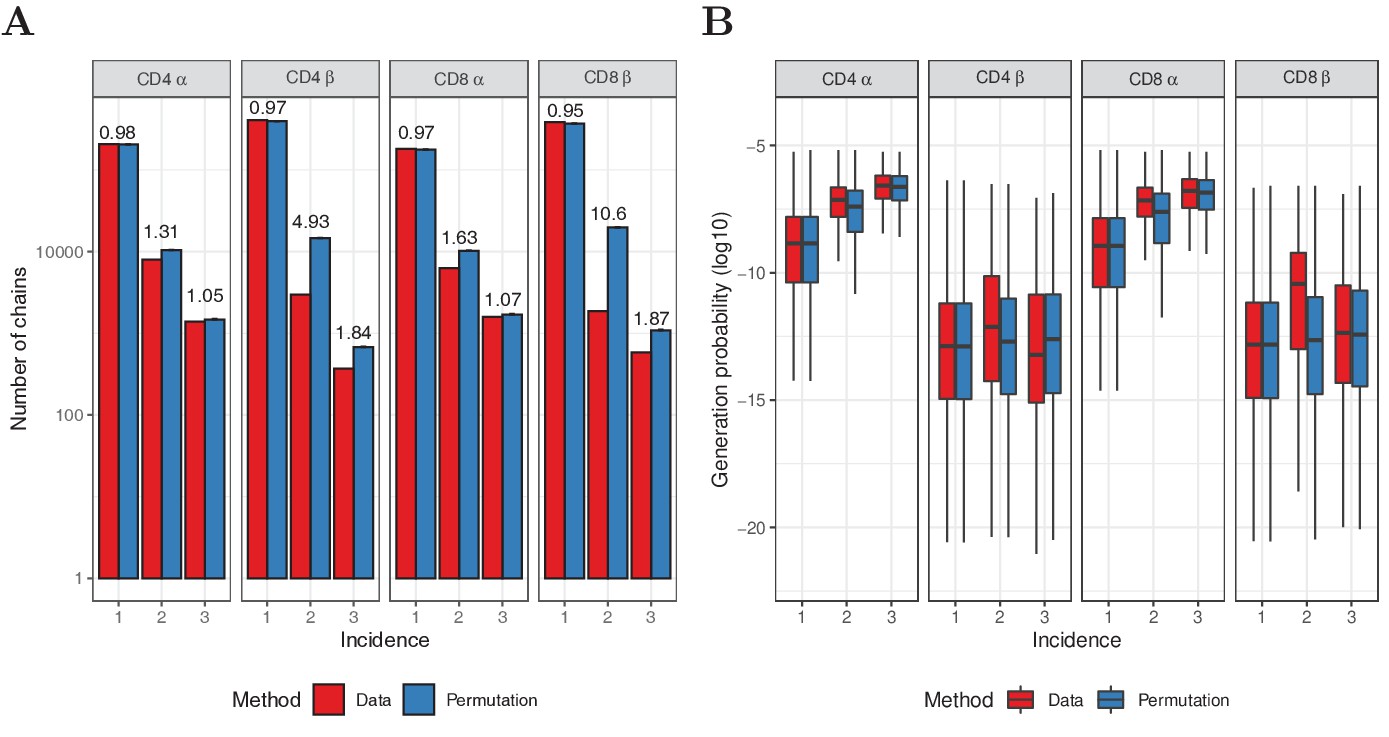
Permutation of subsampling experiment.
(A) Number of chains observed in 1, 2 or 3 subsamples (red) and after redistributing the sequences over the samples (blue). For the permutation test, mean values of 10 iterations are shown, with error bars indicating one standard deviation. The fold-change between data and permutation is indicated on top of the bars. (B) Generation probabilities of the sequences in A, as determined with IGoR (Marcou et al., 2018). The plot shows median (black horizontal line), interquartile range (filled bar) and the range from the bar up to 1.5 times the interquartile range (black vertical range, outliers not shown).
Figure 2—figure supplement 2
Similar to Figure 2, but for HTS data processed with RTCR.
(A) The number of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. The grey background bars show the results after removing all sequences that were also observed in the corresponding memory samples. (B) Generation probabilities (log10) of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. (C) Minimal number of N-additions of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. (D) Number of V- and J-deletions of TCRα and TCRβ sequences observed in 1, 2 or 3 subsamples. The plot shows median (black horizontal line), interquartile range (filled bar) and the range from the bar up to 1.5 times the interquartile range (black vertical range, outliers not shown).
Figure 2—figure supplement 3
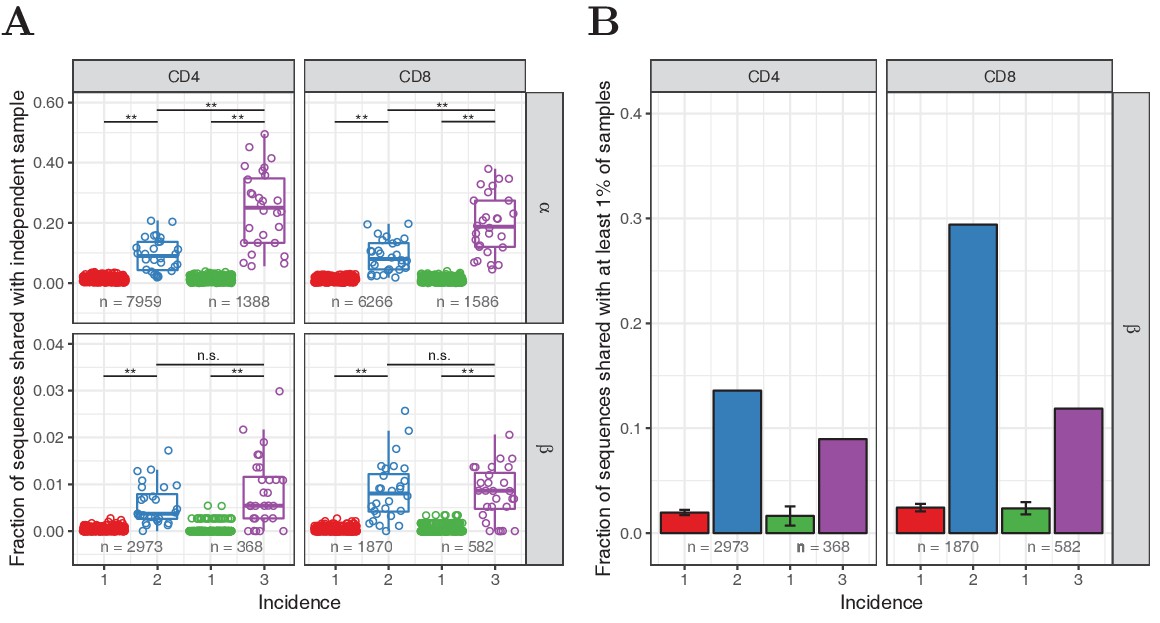
Observed frequency predicts sharing for TCRα but not TCRβ sequences.
(A) We compared the occurrence of TCRα and TCRβ sequences observed in two or three subsamples (incidence 2 or 3, respectively), and equalsize samples of sequences observed in one subsample (incidence 1), in unfractionated blood samples collected from 28 healthy donors. Symbols depict the number of shared TCRα or TCRβ sequences for each whole blood repertoire, as a proportion of the total number in the samples being tested (the latter is indicated at the bottom). The boxplot depicts the median value and 25th and 75th percentiles. Shared fractions were compared by Wilcoxon-Mann-Whitney test, **: , n.s.: not significant (). (B) Fraction of each set of sequences from A that was observed in at least 1% of the samples from a large cohort of 786 individuals (Emerson et al., 2017). Error bars show the standard deviation for the multiple sets of sequences with incidence 1. A smaller fraction of the most frequently observed β chains (incidence 3) are shared than those with incidence 2, which is in line with the observations using IGoR.
Figure 3

Schematic representation of the neutral model and various clone-size distributions.
(A) Schematic representation of the dynamics of the neutral model for the naive T-cell pool. Each event starts with removal of one randomly selected cell from the pool, followed by peripheral division of another cell (with probability 1- θ), or a chance for thymic production (probability θ). After of these thymus events, a clone of cells is generated and added to the peripheral pool, reflecting the divisions of T cells before entering the periphery. (B) Schematic representations of the various clone-size distributions that were used to predict the naive repertoire. The green, orange and blue colored lines depict three parameter choices for each distribution, resulting in a low, medium and high mean clone size, respectively.
Figure 4 with 3 supplements
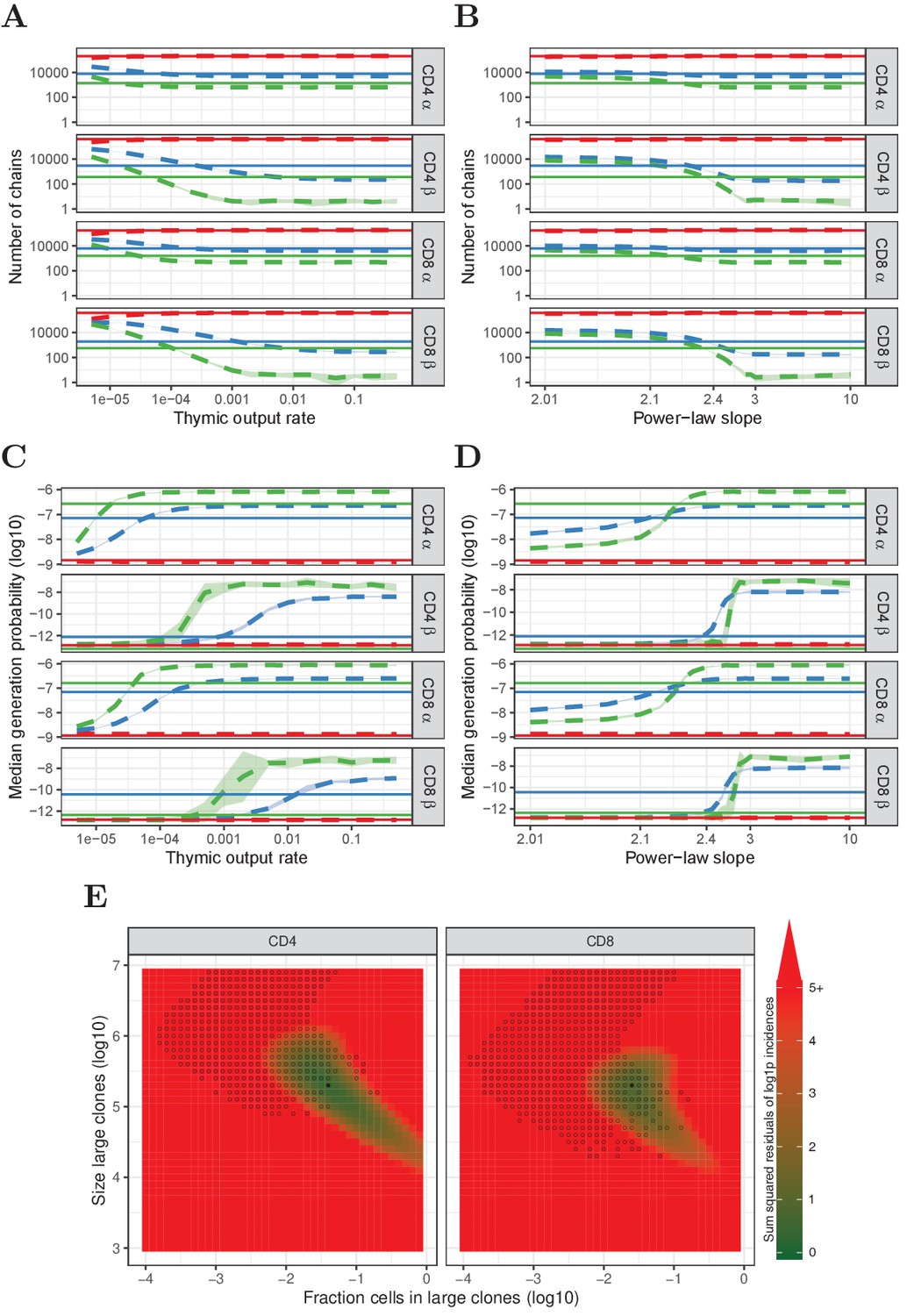
Predictions of the neutral, power-law and two-population model compared with HTS data.
(A) Number of TCRα and TCRβ sequences which are predicted to be shared between 1 (red), 2 (blue) and 3 (green) subsamples as a function of the thymic output rate θ for the neutral model. (B) As A., but as a function of the slope of the power-law distribution. (C) The median generation probability of TCRα and TCRβ sequences predicted by the neutral model. Dashed lines depict the mean of 10 model prediction repeats, shaded area indicates the standard deviation, solid lines show observed results in HTS data. (D) As C., but as a function of the slope of the power-law distribution. (E) Graphical representation of parameter sweep results for prediction of CD4+ and CD8+ repertoires from αβ clone-size distributions following a mixture model consisting of singleton clones and a small fraction of large clones. The color represents goodness of fit, with dark green being better predictions for number of sequences per incidence in samples. Empty circles indicate parameter combinations resulting in qualitatively correctly predicted , that is 3 > 2 > 1 for TCRα and 2 > 1 for TCRβ and 2 > 3 for TCRβ. Filled circles indicate parameter combinations with the smallest distance to the incidence data and a correct prediction.
Figure 4—figure supplement 1
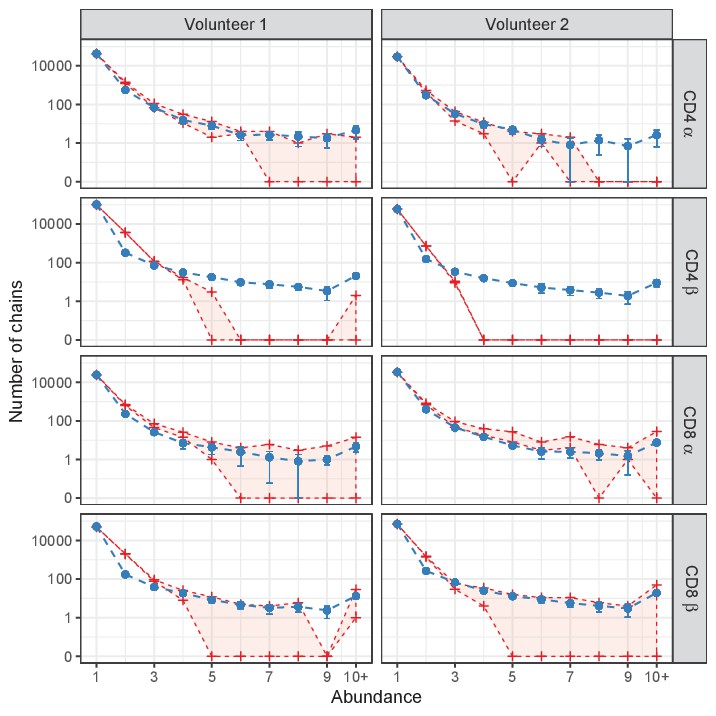
Prediction of power-law model (exponent 2.3) for single sample data.
Red lines indicate abundance of TCRα and TCRβ sequences, both without (top) and with cleaning of overlap with memory (bottom). Blue lines represent model prediction for the abundance of α and β chains (blue dashed line, error bars indicate standard deviation over 10 simulations, note the different predictions between the volunteers due to different sample sizes). Sequences represented with 10 or more UMIs are grouped ('10+'). Due to the impact of sampling multiple RNAs from the same cell (Section 'Subsampling to exclude inflated abundance through multiple RNA contributions by single cells'), the predictions under-predict the number of doublets, especially for β chains. Apart from this, the model fits well to the ‘uncleaned’ data in most cases, but the cleaned data under-represents abundant clones, suggesting that several clones shared between memory and naive represent genuine abundant naive clones. The only exception is for CD4+ TCRβ, where we observe much fewer large clones than the model predicts.
Figure 4—figure supplement 2

Similar to Figure 4, but for HTS data from which TCRα and TCRβ sequences were removed that also occurred in the corresponding memory samples.
(A) Number of α and β chains predicted to occur in 1 (red), 2 (blue) and 3 (green) subsamples as a function of the thymic output rate θ for the neutral model. (B) As A., but as a function of the slope of the power-law distribution. (C) The median generation probability of TCRα and TCRβ sequences predicted by the neutral model. Dashed lines depict the mean of 10 model prediction repeats, shaded area indicates the standard deviation, solid lines show observed results in HTS data. (D) As C., but as a function of the slope of the power-law distribution. (E) Graphical representation of parameter sweep results for prediction of CD4+ and CD8+ repertoires from αβ clone-size distributions consisting of singleton clones and a small fraction of large clones. The color represents goodness of fit, with dark green being better predictions for number of sequences per incidence in samples. Symbols indicate parameter combinations resulting in qualitatively correctly predicted , that is 3 > 2 > 1 for TCRα and 2 > 1 for TCRβ and 2 > 3 for TCRβ. Filled circles indicate parameter combinations with the smallest distance to the incidence data and a correct prediction.
Figure 4—figure supplement 3
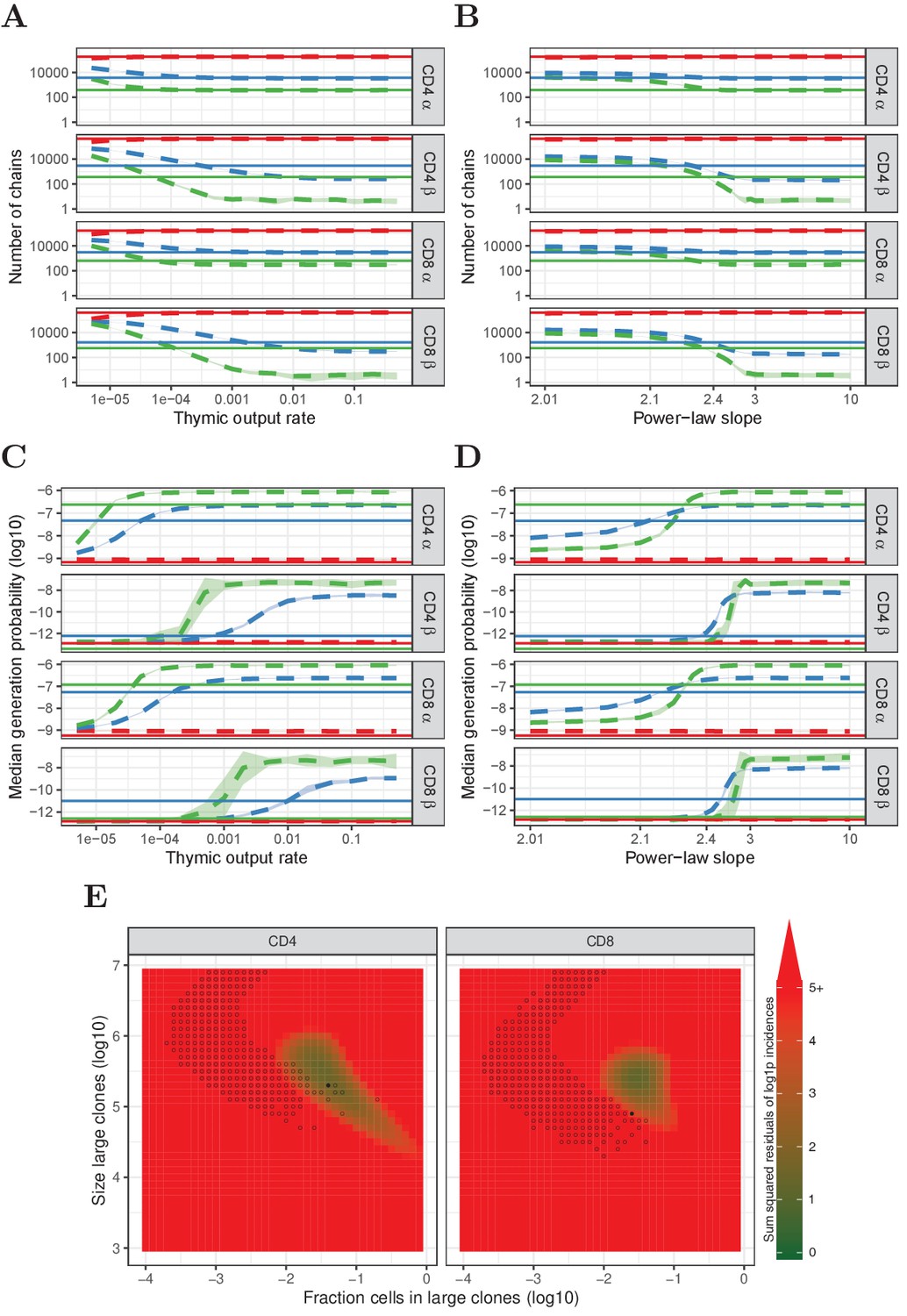
Similar to Figure 4, but for HTS data processed with RTCR.
(A) Number of TCRα and TCRβ sequences which are predicted to be shared between 1 (red), 2 (blue) and 3 (green) subsamples as a function of the thymic output rate θ for the neutral model. (B) As A., but as a function of the slope of the power-law distribution. (C) The median generation probability of TCRα and TCRβ sequences predicted by the neutral model. Dashed lines depict the mean of 10 model prediction repeats, shaded area indicates the standard deviation, solid lines show observed results in HTS data. (D) As C., but as a function of the slope of the power-law distribution. (E) Graphical representation of parameter sweep results for prediction of CD4+ and CD8+ repertoires from αβ clone-size distributions following a mixture model consisting of singleton clones and a small fraction of large clones. The color represents goodness of fit, with dark green being better predictions for number of sequences per incidence in samples. Empty circles indicate parameter combinations resulting in qualitatively correctly predicted , i.e., 3 > 2 > 1 for TCRα and 2 > 1 for TCRβ and 2 > 3 for TCRβ. Filled circles indicate parameter combinations with the smallest distance to the incidence data and a correct prediction.
Figure 5 with 1 supplement
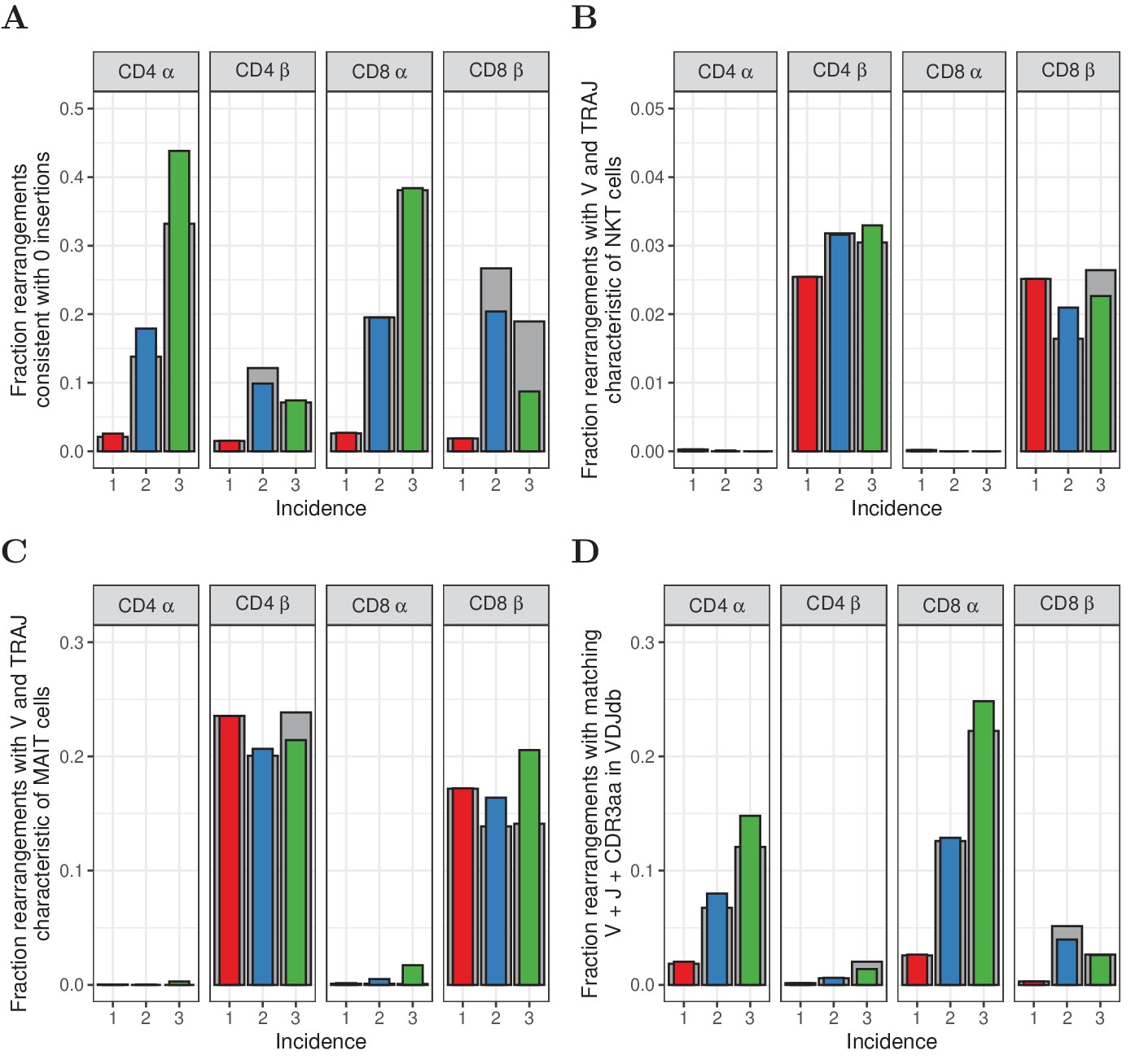
Characterization of abundant TCRα and TCRβ sequences.
(A) The fraction of rearrangements with zero minimal N-additions for sequences observed in 1, 2 or 3 naive subsamples. Data are shown without (colored bars) and with cleaning of overlap with memory (grey bars). (B) Fraction of TCRα and TCRβ sequences with V(J) usage characteristic of NKT cells (TRAV24-TRAJ18 for TCRα; TRBV11 for TCRβ). (C) Fraction of TCRα and TCRβ sequences with V(J) usage characteristic of MAIT cells (TRAV1-2 with TRAJ33, TRAJ12 or TRAJ20 for TCRα; TRBV20 or TRBV6 for TCRβ). (D) Fraction of sequences having at least one match (CDR3 amino acid sequence as well as V and J annotation) with the VDJdb (Shugay et al., 2018).
Figure 5—figure supplement 1
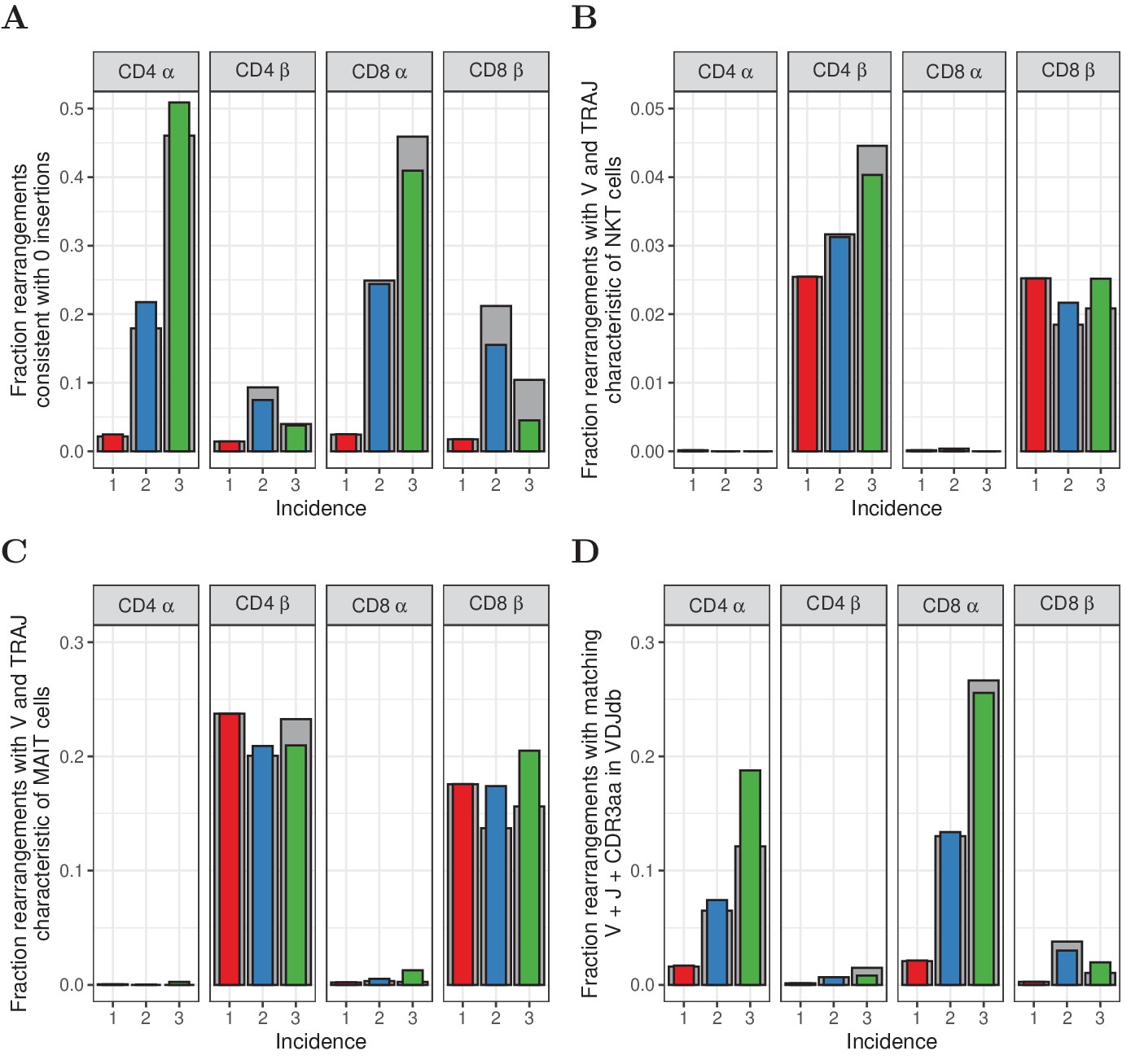
Similar to Figure 5, but for HTS data processed with RTCR.
(A) The fraction of rearrangements with zero minimal N-additions for sequences observed in 1, 2 or 3 naive subsamples. Data are shown without (colored bars) and with cleaning of overlap with memory (grey bars). (B) Fraction of TCRα and TCRβ sequences with V(J) usage characteristic of NKT cells (TRAV24-TRAJ18 for TCRα; TRBV11 for TCRβ). (C) Fraction of TCRα and TCRβ sequences with V(J) usage characteristic of MAIT cells (TRAV1-2 with TRAJ33, TRAJ12 or TRAJ20 for TCRα; TRBV20 or TRBV6 for TCRβ). (D) Fraction of sequences having at least one match (CDR3 amino acid sequence as well as V and J annotation) with the VDJdb (Shugay et al., 2018).
Figure 6
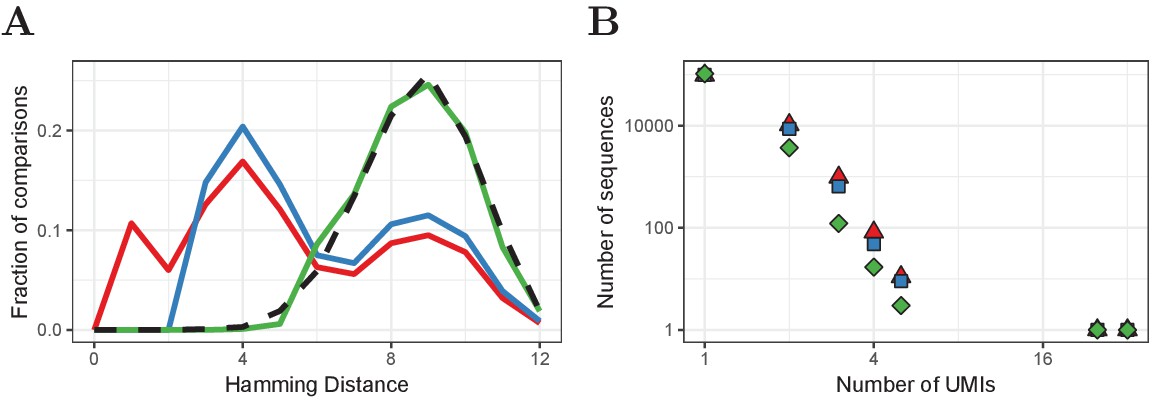
Improved UMI correction leads to more reliable estimation of sequence frequencies.
(A) Distribution of Hamming Distances of UMIs within TCRβ sequences (naive CD4+ sample of volunteer 1) before correction (red), after default correction (blue) and after improved correction (green), in comparison with the distribution of UMIs between sequences (black dashed). (B) Distributions of the same TCRβ sequences after the different correction strategies. Frequently observed TCRβ sequences remain at the same frequency after correction, whereas the frequency of other sequences tends to be overestimated due to mutated UMIs, which is compensated for by improved UMI correction.
Figure 7
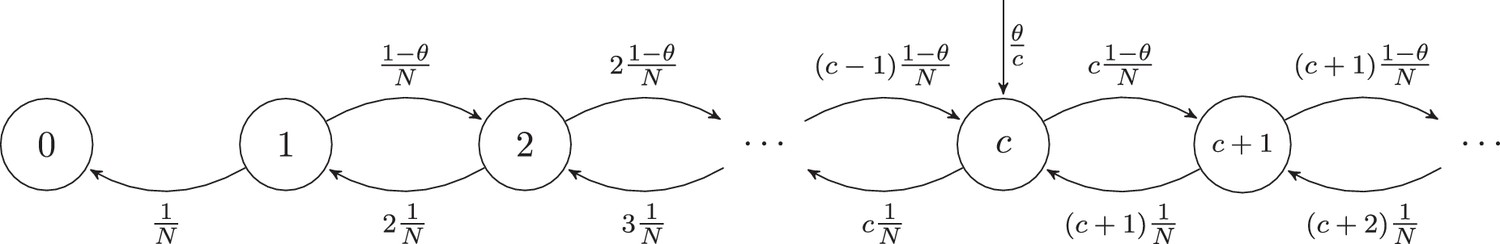
Markov chain representation of the neutral model with thymic introduction size .
Figure 8

Pre- and post-selection densities and -dependent selection factors for α and β chains.
(A) Relative frequency of generation probabilities of TCRα (red) and TCRβ (blue) sequences in the combined HTS data (solid) and IGoR output (dashed). (B) The bin-specific selection factors are determined by division of the density of a given bin in the HTS data by the density in the pre-selection IGoR output. A value of 1 means that a sequence with this has an average probability to be selected in the thymus, whereas lower values indicate stronger selection and higher values weaker selection (i.e., a higher probability to pass selection).
Tables
Author response table 1
| Fraction of TCR sequences with incidence > | ||||
|---|---|---|---|---|
| Data | Permutation | Fold difference | % due to RNA | |
| CD4α | 4.3% | 5.5% | 1.3x | 21.6% |
| CD4β | 0.8% | 3.7% | 4.6x | 78.2% |
| CD8α | 4.1% | 6.3% | 1.5x | 34.0% |
| CD8β | 0.6% | 5.4% | 8.5x | 88.3% |
Additional files
-
Supplementary file 1
Quantitative details about each dataset.
Number of FACS-sorted cells in each sample, number of reads obtained for both α and β and the number of UMIs that were accepted after Decombinator processing and UMI clustering. The naive subsamples were FACS-sorted and then divided into four equal aliquots. For the CD8+ EMRA samples, TCRα and TCRβ were sequenced together for both volunteers in Experiment 1.
- https://cdn.elifesciences.org/articles/49900/elife-49900-supp1-v1.txt
-
Supplementary file 2
Fraction of TCR sequences with incidence > 1.
For each dataset, the fraction of sequences observed in multiple subsamples is given, both in the data and after permuting the samples. From the fold-differences, the effect of single cells contributing multiple RNA molecules is estimated.
- https://cdn.elifesciences.org/articles/49900/elife-49900-supp2-v1.txt
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/49900/elife-49900-transrepform-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The naive T-cell receptor repertoire has an extremely broad distribution of clone sizes
eLife 9:e49900.
https://doi.org/10.7554/eLife.49900




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}