Discovery of several thousand highly diverse circular DNA viruses
- National Cancer Institute, National Institutes of Health, United States
- National Institute of Allergy and Infectious Diseases, National Institutes of Health, United States
- University of California, Davis, United States
- Harvard Medical School, The Harvard Stem Cell Institute, Brigham and Women's Hospital, United States
- Broad Institute of MIT and Harvard, United States
- Harvard University, United States
- National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, United States
- University of Cambridge, United Kingdom
- Mililani Mauka Elementary, United States
- Johns Hopkins University School of Medicine, United States
- National Institute on Aging, National Institutes of Health, United States
- Ohio State University, United States
- Arizona State University, United States
- University of Cape Town, South Africa
- San Diego State University, United States
Figures
Figure 1 with 3 supplements
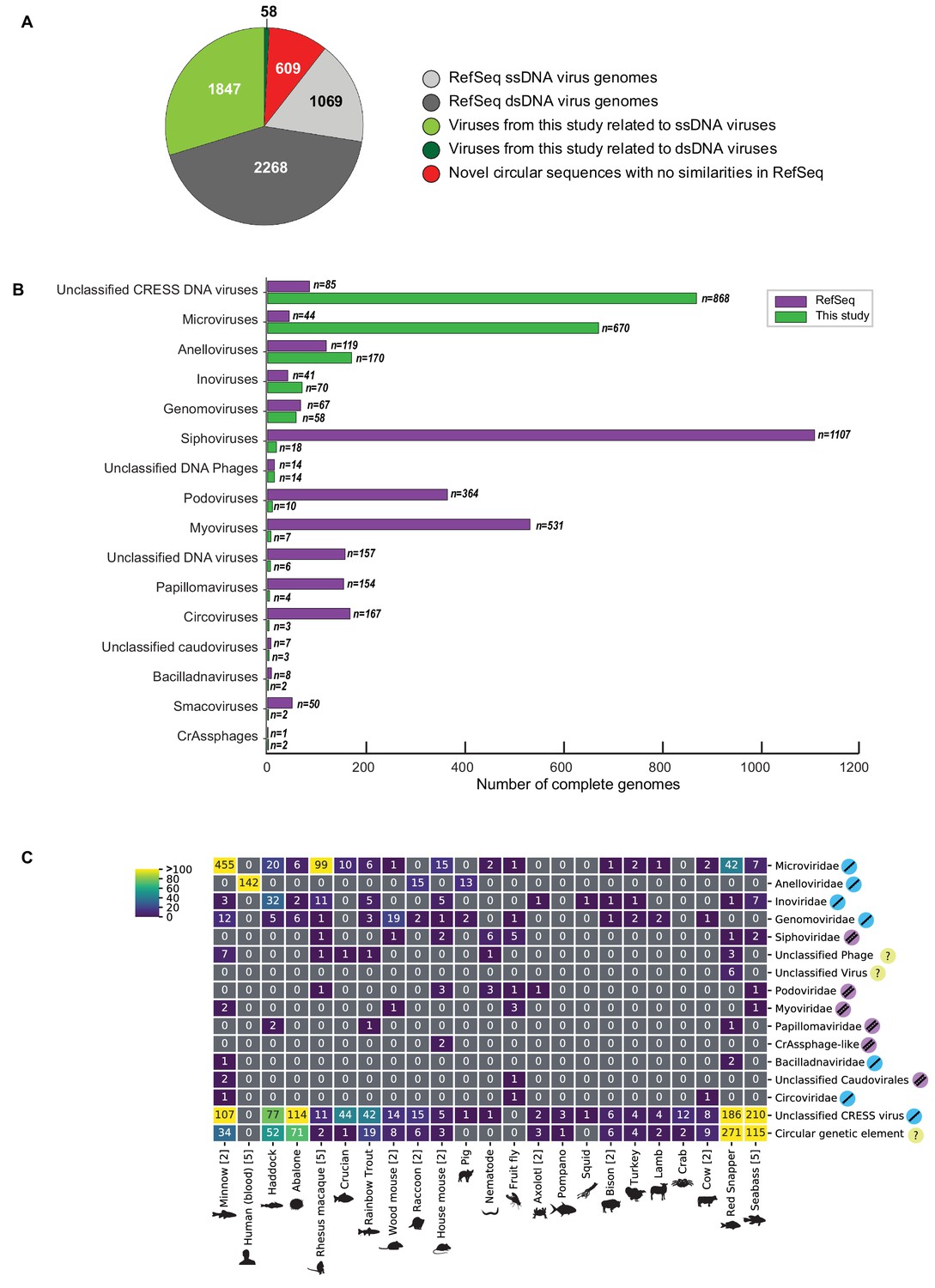
Novel viruses associated with animal samples.
Gross characterization of viruses discovered in this project compared to NCBI RefSeq virus database entries. (A) Pie chart representing the number of viral genomes in broad categories. (B) Bar graph showing the number of new representatives of known viral families or unclassified groups. (C) Heatmap reporting number of genomes found associated with each animal species. Number of samples per species in brackets. Note that genomes in this study were assigned taxonomy based on at least one region with a BLASTX hit with an E value <1 × 10−5, suggesting commonality with a known viral family. Some genomes may ultimately be characterized as being basal to the assigned family.
Figure 1—figure supplement 1
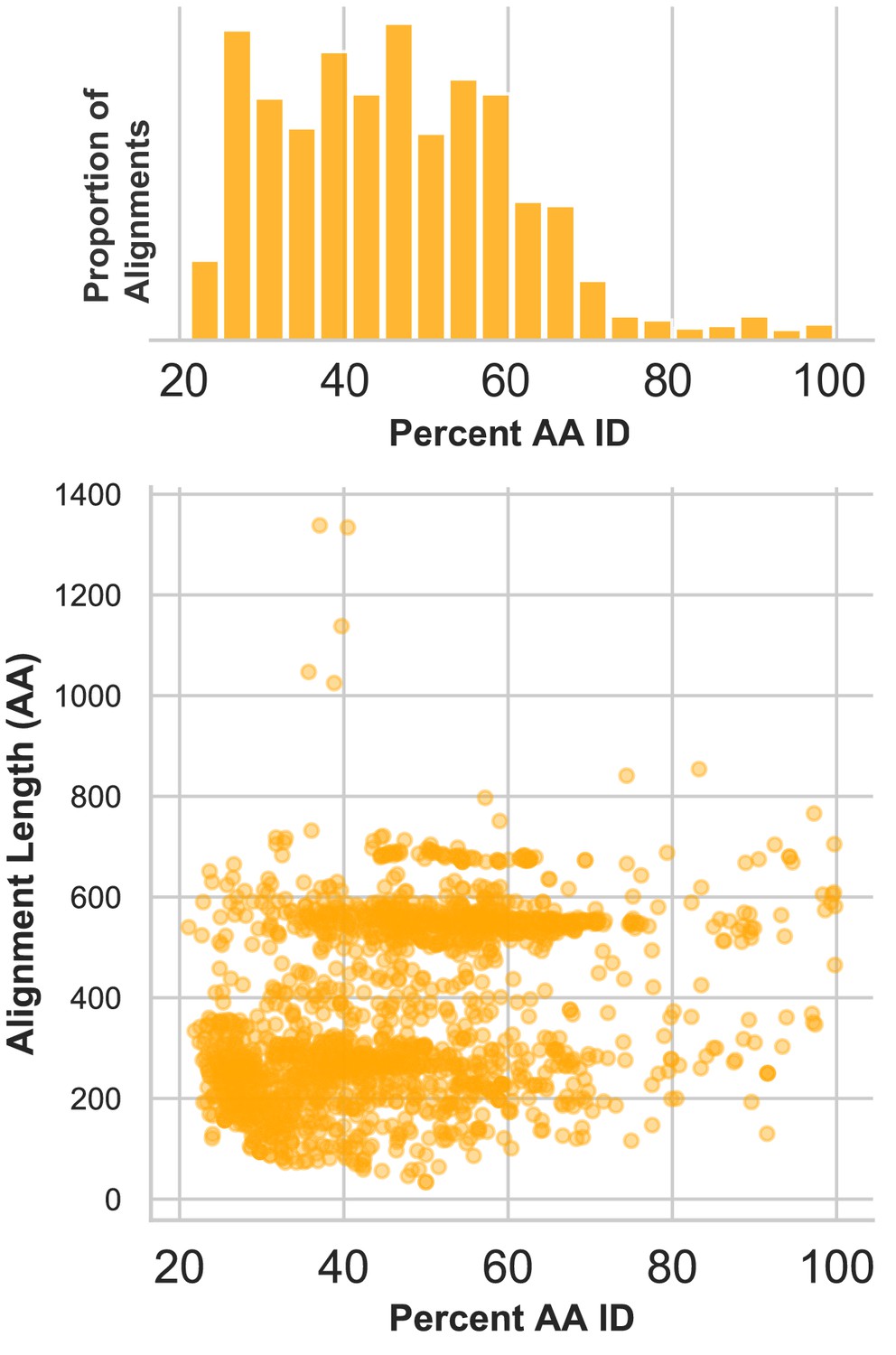
Divergence of proteins encoded by circular contigs.
BLASTX summary of each circular DNA molecule recovered from virus-enriched samples. Sequences were queried against a database of viral and plasmid sequences. Only hits with E values < 10−5 were plotted. Here, BLASTX only reports the most significant stretch of amino acid sequence from each circular contig, and, therefore, other regions of each contig can be assumed to be equally or less conserved.
Figure 1—figure supplement 2
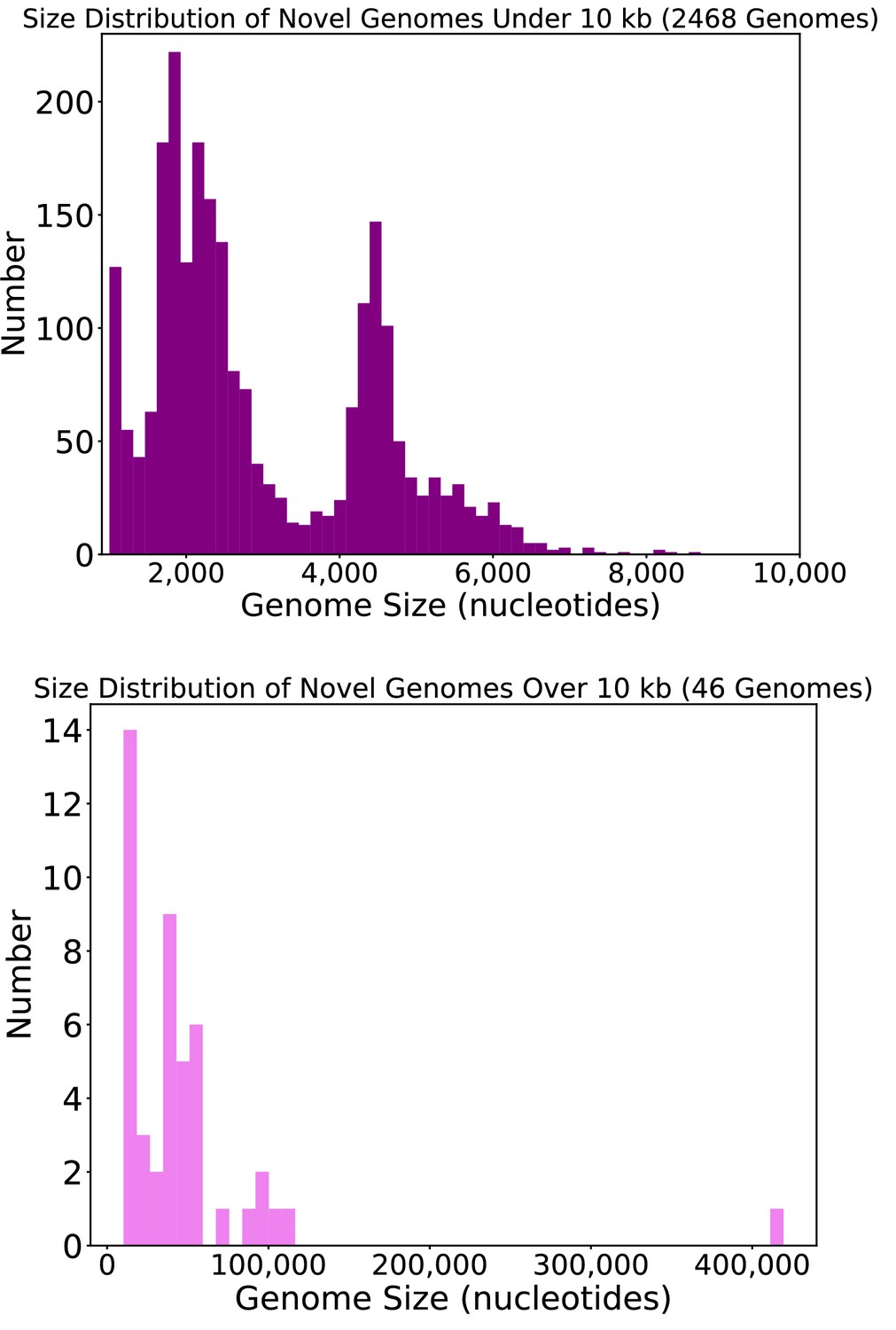
Size distribution of circular DNA sequences from this study.
Length, in nucleotides, of circular DNA sequences representing putative viral genomes from this study.
Figure 1—figure supplement 3
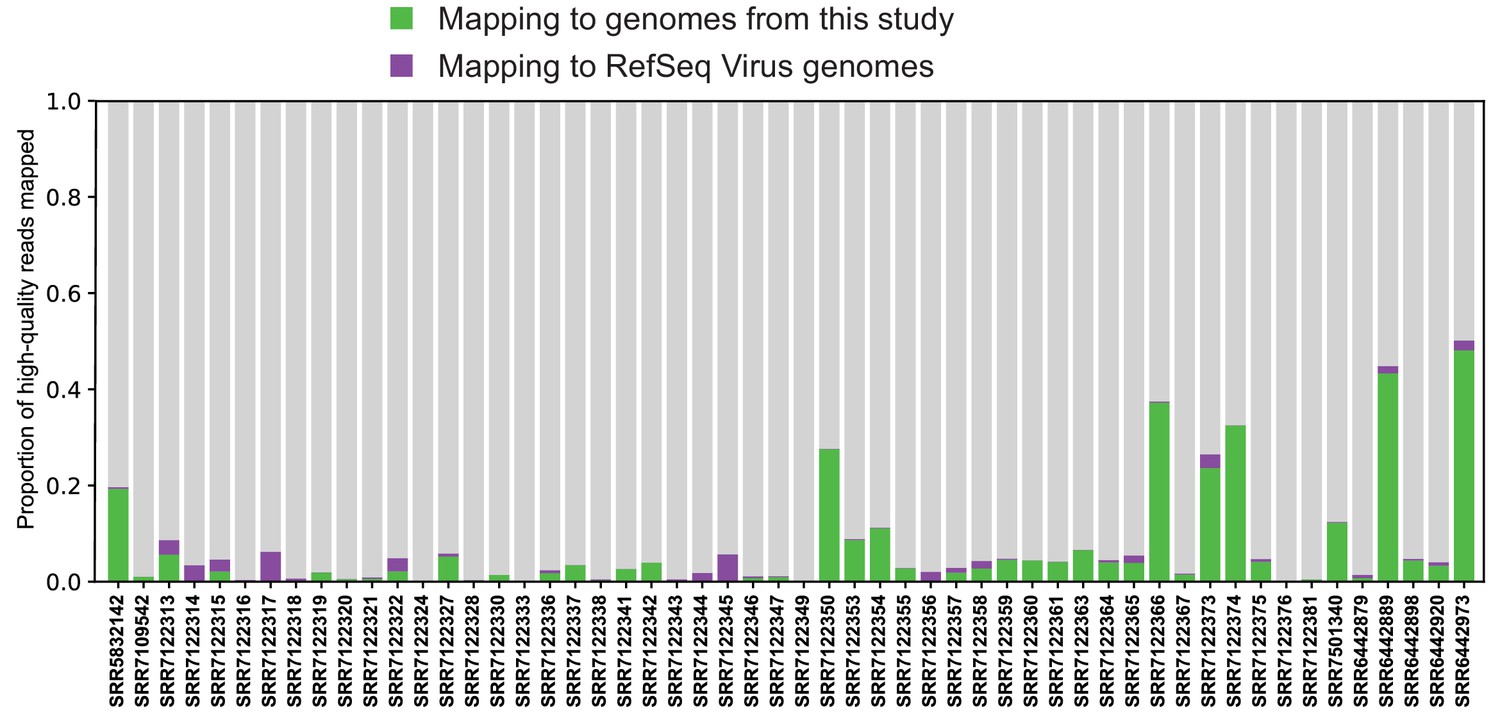
Mapping reads to complete viral genome references.
Quality-trimmed reads were aligned with Bowtie2 to reference genomes from RefSeq and this study. Genomes were masked for low-complexity regions.
Figure 2 with 2 supplements
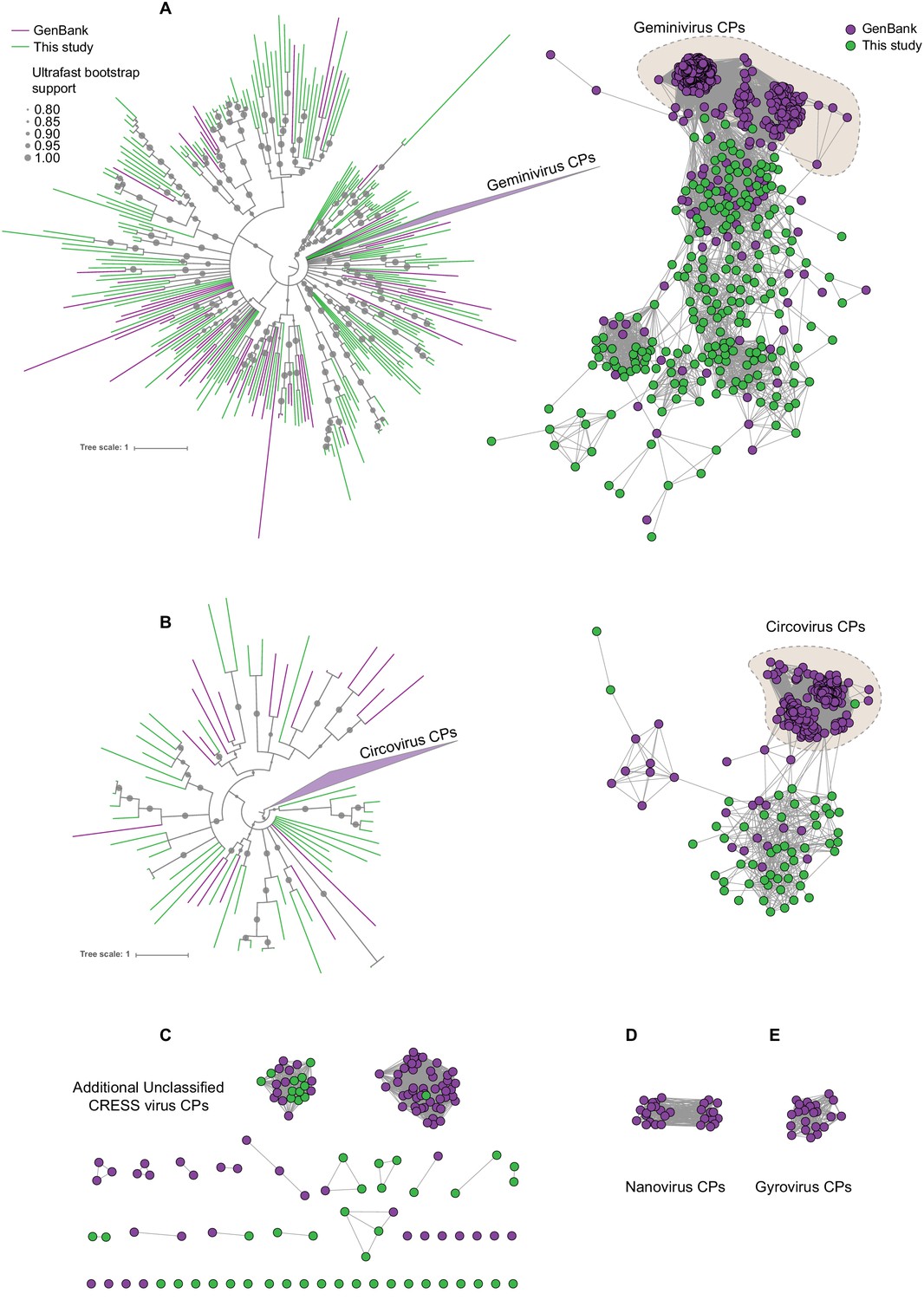
Sequence similarity network analysis of CRESS virus capsid proteins.
EFI-EST was used to conduct pairwise alignments of amino acid sequences from this study and GenBank with predicted structural similarity to CRESS virus capsid proteins. The E value cutoff for the analysis was 10−5. (A) Cluster consisting of proteins with predicted structural similarity to geminivirus-like capsids and/or STNV-like capsids. The phylogenetic tree was made from all sequences in this cluster. (B) A cluster consisting of sequences with predicted structural similarity to Circovirus capsid proteins. The phylogenetic tree was made from all sequences in this cluster. (C) Assorted clusters and singletons from unclassified CRESS virus proteins that were modeled to be capsids. (D) Nanovirus capsids. (E) Gyrovirus capsids.
-
Figure 2—source data 1
Phylogenetic tree file of circovirus-like capsid protein sequences, corresponding to Figure 2, Panel B.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-data1-v1.tre
-
Figure 2—source data 2
Sequence similarity network of CRESS virus capsid protein sequences, corresponding to Figure 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-data2-v1.cys
-
Figure 2—source data 3
Phylogenetic tree file of gemini- and STNV-like capsid protein sequences, corresponding to Figure 2, Panel A.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-data3-v1.tre
Figure 2—figure supplement 1
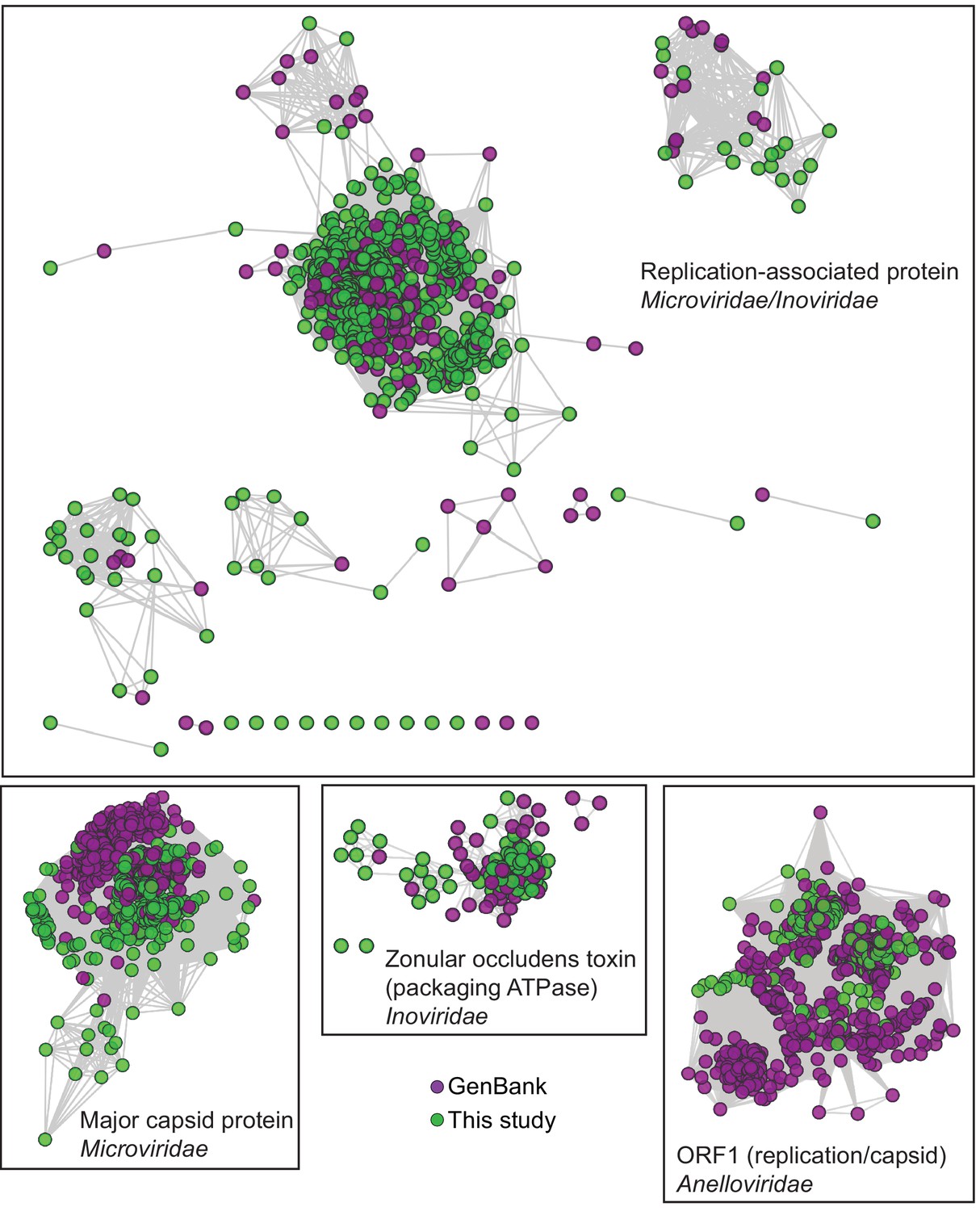
Network Analysis of additional viral hallmark genes.
Depiction of additional viral hallmark genes from this study and GenBank as sequence similarity networks. E value cutoff = 10−5. See Figure 2 and Materials and methods.
-
Figure 2—figure supplement 1—source data 1
Sequence similarity network of anellovirus ORF1 protein sequences, corresponding to Figure 2—figure supplement 1.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp1-data1-v1.cys
-
Figure 2—figure supplement 1—source data 2
Sequence similarity network of inovirus ZOT protein sequences, corresponding to Figure 2—figure supplement 1.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp1-data2-v1.cys
-
Figure 2—figure supplement 1—source data 3
Sequence similarity network of microvirus Major Capsid protein sequences, corresponding to Figure 2—figure supplement 1.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp1-data3-v1.cys
-
Figure 2—figure supplement 1—source data 4
Sequence similarity network of inovirus and microvirus Replication-associated protein sequences, corresponding to Figure 2—figure supplement 1.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp1-data4-v1.cys
Figure 2—figure supplement 2

Phylogenetic trees of viral hallmark genes.
Sequences were aligned with PROMALS3D using structure guidance when possible. Trees were drawn using IQ-Tree with automatic determination of substitution model. See Materials and methods. Branches are labeled with bootstrap percent support after 1000 ultrafast bootstrapping events. (A) Microviridae major capsid protein. (B) Inoviridae zonular occludens toxin. (C) CRESS virus Rep. (D) Anelloviridae ORF1 (E) Microviridae/Inoviridae Replication-associated protein I. (F) Microviridae/Inoviridae Replication-associated protein II. (G) Microviridae/Inoviridae Replication-associated protein III.
-
Figure 2—figure supplement 2—source data 1
Phylogenetic tree file of anellovirus ORF1 protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data1-v1.tre
-
Figure 2—figure supplement 2—source data 2
Phylogenetic tree file of CRESS virus Rep protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data2-v1.tre
-
Figure 2—figure supplement 2—source data 3
Phylogenetic tree file of inovirus ZOT protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data3-v1.tre
-
Figure 2—figure supplement 2—source data 4
Phylogenetic tree file of microvirus Major Capsid protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data4-v1.tre
-
Figure 2—figure supplement 2—source data 5
Phylogenetic tree file (1 of 3) of inovirus and microvirus Replication-associated protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data5-v1.tre
-
Figure 2—figure supplement 2—source data 6
Phylogenetic tree file (2 of 3) of inovirus and microvirus Replication-associated protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data6-v1.tre
-
Figure 2—figure supplement 2—source data 7
Phylogenetic tree file (3 of 3) of inovirus and microvirus Replication-associated protein sequences, corresponding to Figure 2—figure supplement 2.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig2-figsupp2-data7-v1.tre
Figure 3
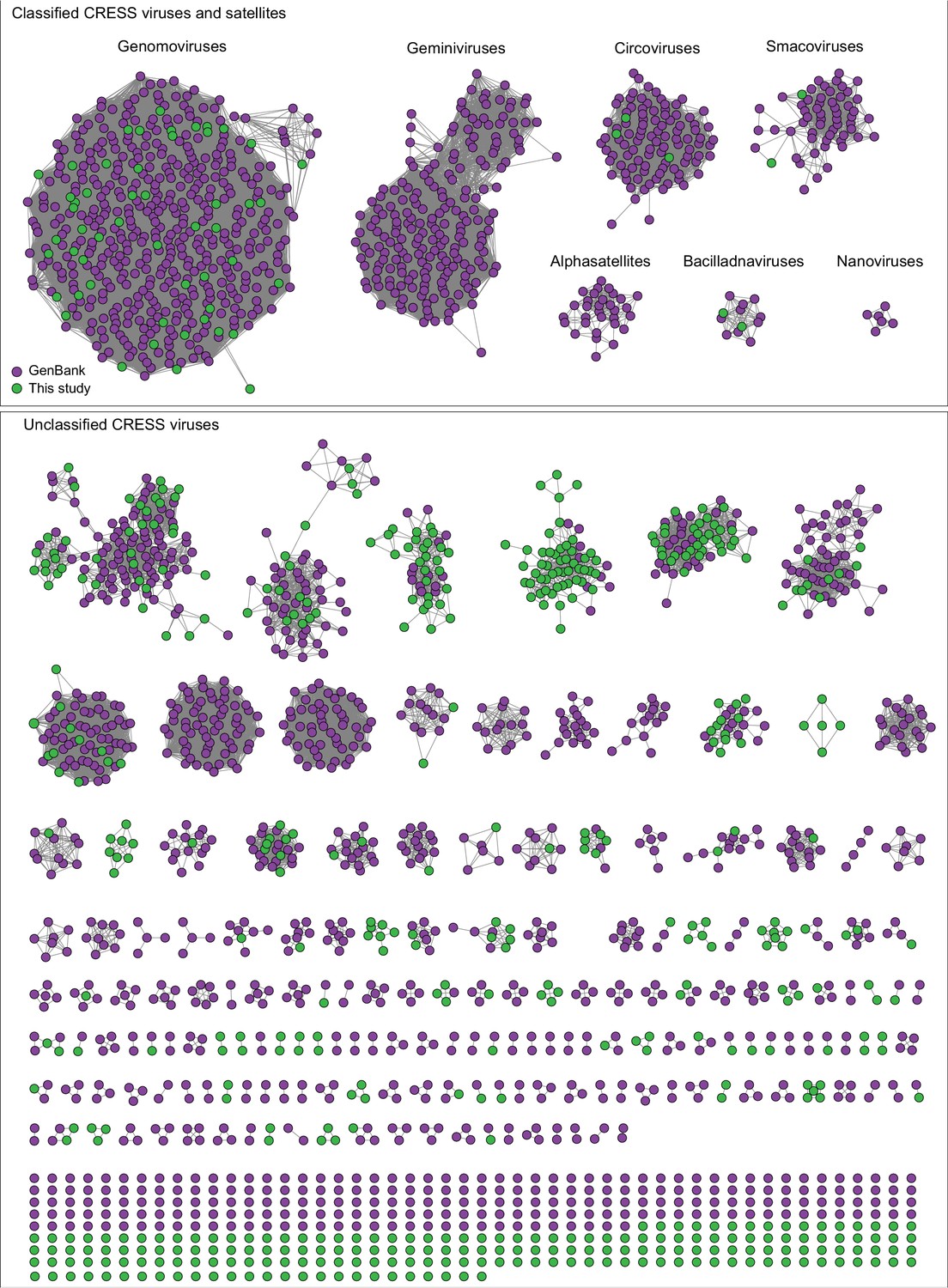
Network analysis of CRESS virus Rep proteins.
EFI-EST was used to conduct pairwise alignments of amino acid sequences from this study and. GenBank that were structurally modeled to be a rolling-circle replicase (Rep). The analysis used an E value cutoff of 10−60 to divide the data into family-level clusters.
-
Figure 3—source data 1
Sequence similarity network of CRESS virus Rep proteins, corresponding to Figure 3.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig3-data1-v1.cys
Figure 4 with 2 supplements
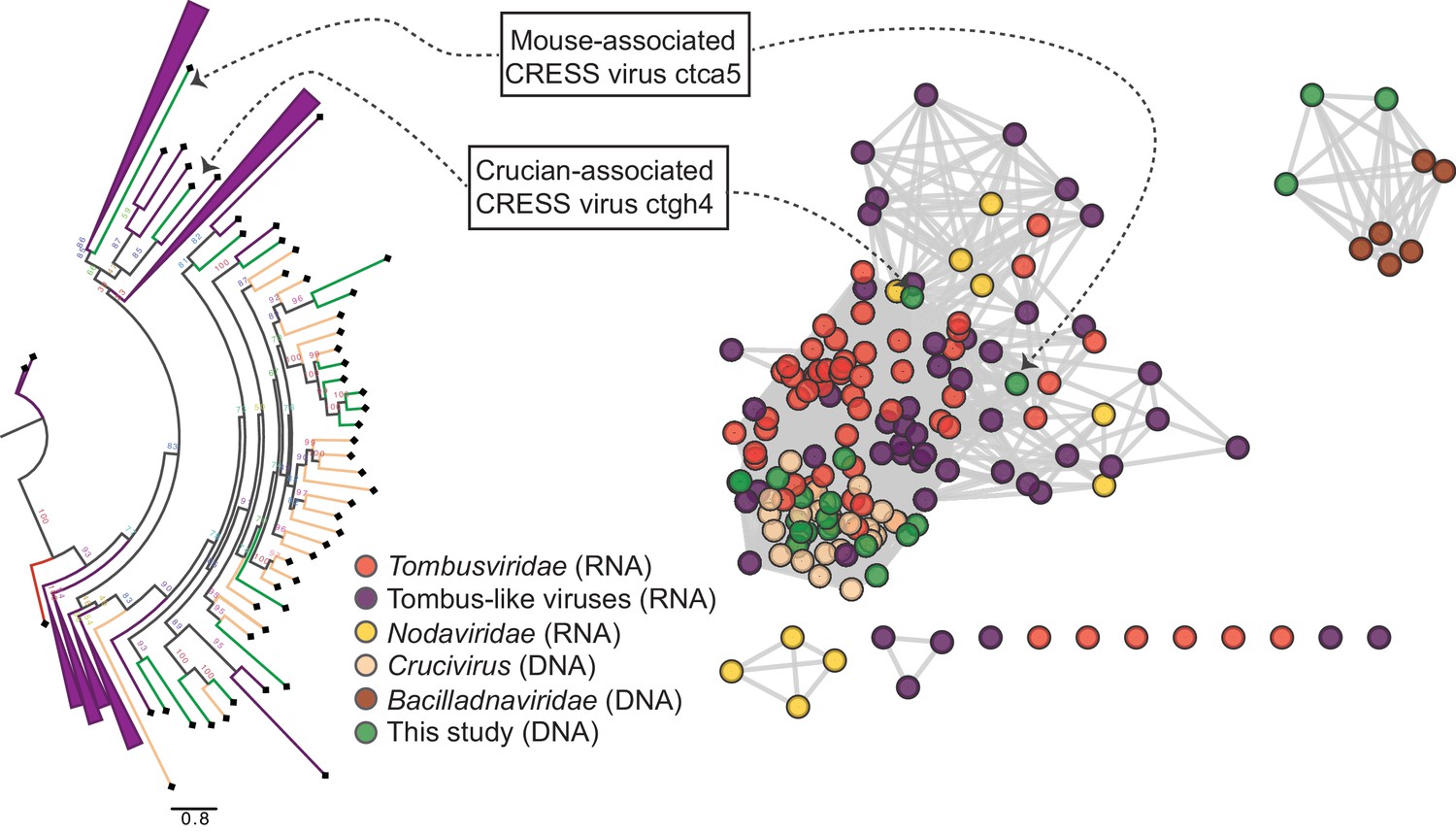
RNA virus capsid-like proteins.
Sequence similarity network generated with EFI-EST (E value cutoff of 10−5) showing capsid protein sequences of select ssRNA viruses (Nodaviridae, Tombusviridae, tombus-like viruses) and ssDNA viruses (Bacilladnaviridae and crucivirus) together with protein sequences from DNA virus genomes observed in the present study with predicted structural similarity to an RNA virus capsid protein domain (PDB: 2IZW). Predicted capsid proteins for CRESS virus ctca5 and CRESS virus ctgh4 have no detectable similarity to any known DNA virus sequences. On the left, a phylogenetic tree representing the large cluster is displayed. Collapsed branches consist of Tombusviridae, tombus-like viruses, and Nodaviridae capsid genes.
-
Figure 4—source data 1
Sequence similarity network of RNA virus-like S-domain-containing capsid protein sequences, corresponding to Figure 4.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig4-data1-v1.cys
-
Figure 4—source data 2
Phylogenetic tree file of RNA virus-like S-domain-containing capsid protein sequences, corresponding to Figure 4.
- https://cdn.elifesciences.org/articles/51971/elife-51971-fig4-data2-v1.tre
Figure 4—figure supplement 1
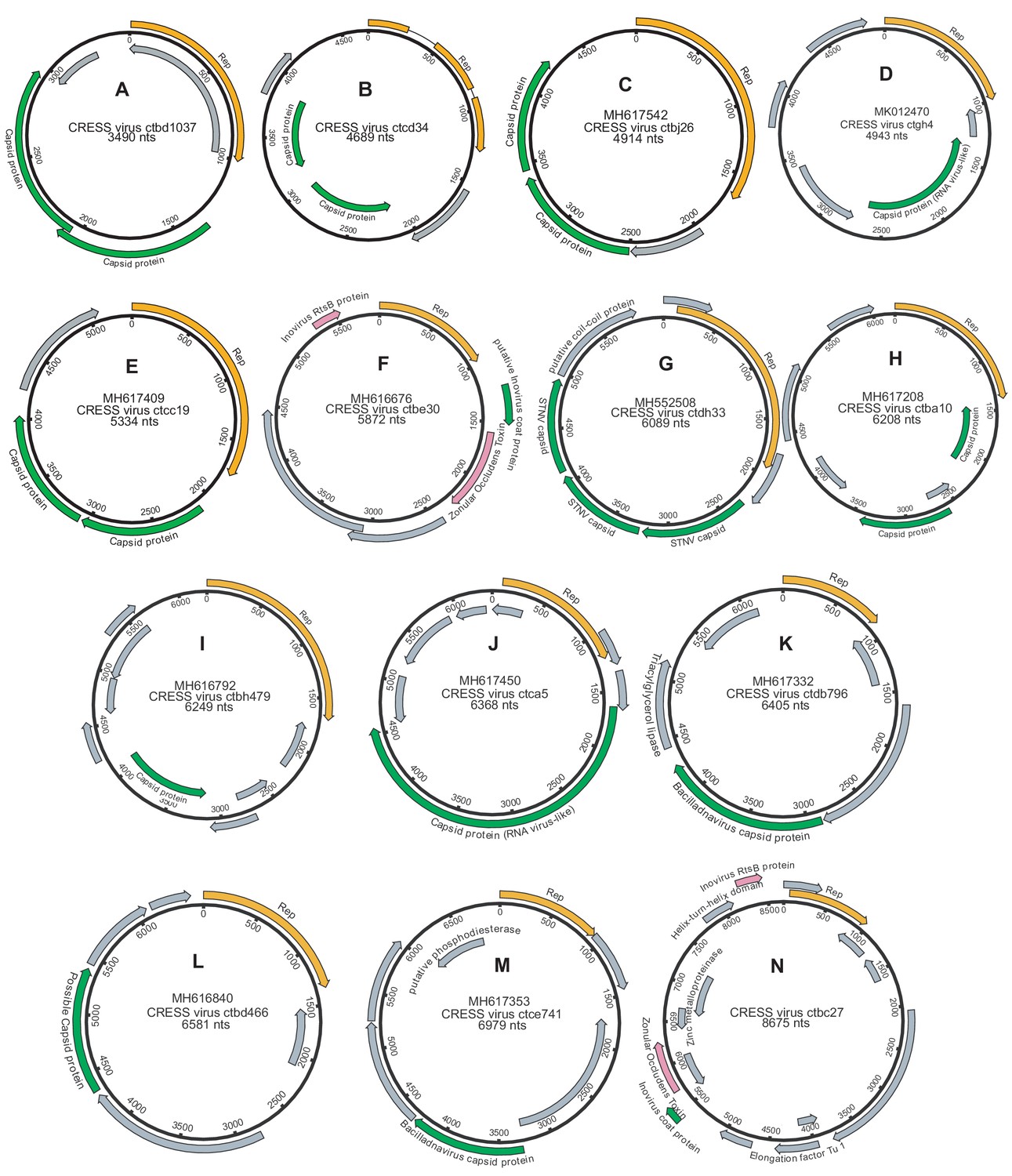
Genome maps of large CRESS virus genomes.
Predicted CRESS Rep-like genes are displayed in orange, virion structural genes shown in green, other identifiable viral genes shown in pink, other genes in gray. GenBank accession numbers are displayed above the virus name.
Figure 4—figure supplement 2

Validation of proteins with predicted similarity to RNA virus capsid proteins.
(A) First order neighbors for Crucian-associated CRESS virus ctgh4 capsid protein were extracted from the network shown in Figure 5 and aligned using Muscle. (B) The same approach was applied to CRESS virus ctbd466 capsid protein. (C) A visualization (Integrative Genomics Viewer) of a read alignment to CRESS virus isolate ctca5. The visualization shows no evidence of artifactual chimerization in the contig assembly process.
Figure 5 with 2 supplements
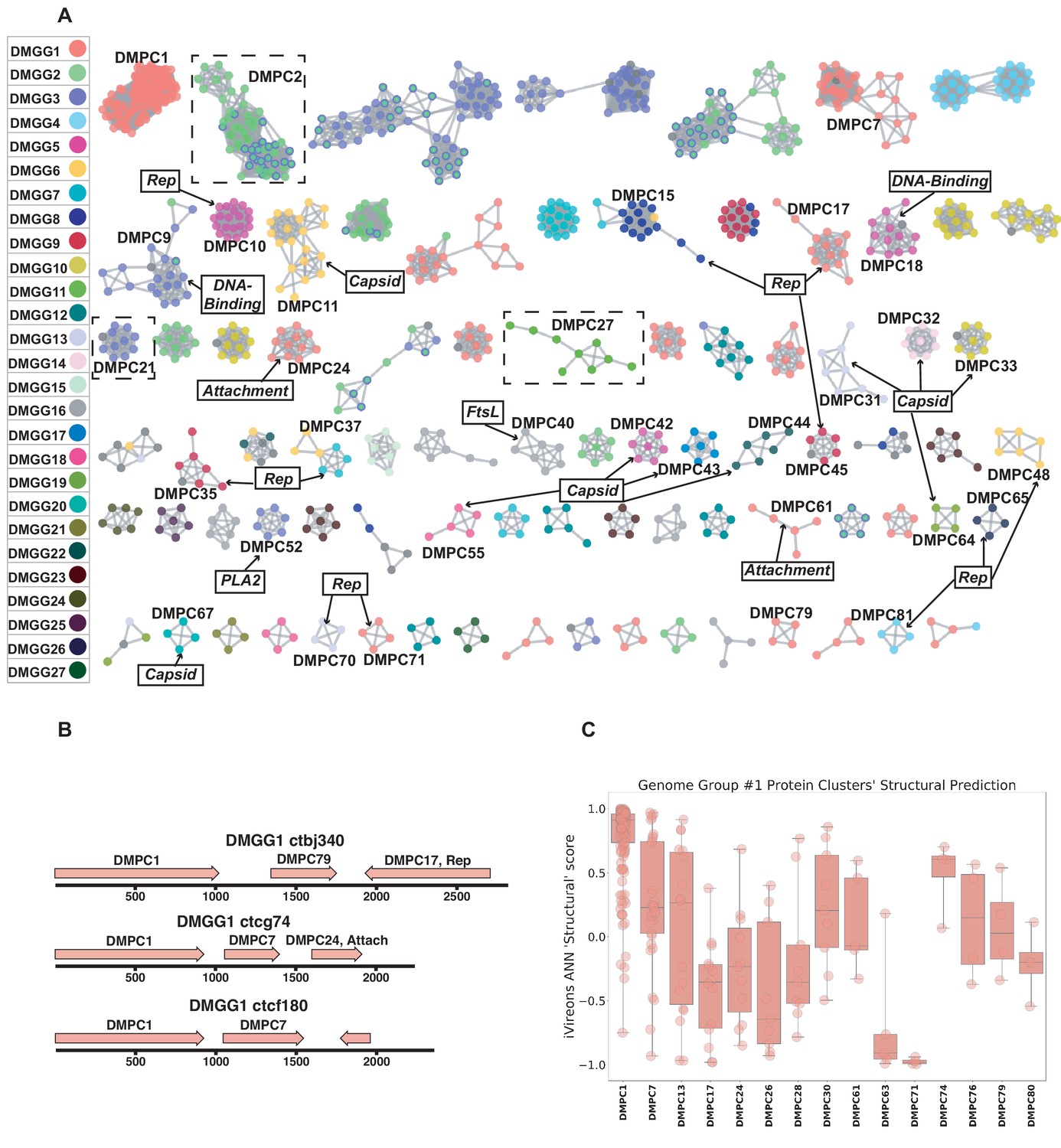
Dark matter analysis.
(A) Sequence similarity network analysis for genes from dark matter circular sequences (minimum cluster size = 4). Clusters are colored based on assigned dark matter genome group (DMGG). Structural predictions from HHpred are indicated (>85% probability). Rep = rolling circle replicases typical of CRESS viruses or ssDNA plasmids. Capsid = single jellyroll capsid protein. Attachment = cell attachment proteins typical of inoviruses. DNA-Binding = DNA binding domain. PLA2 = phospholipase A2. FtsL = FtsL like cell division protein. Clusters that contain a representative protein that was successfully expressed as a virus-like particle are outlined by a dashed rectangle (See Figure 6). (B) Maps of three examples of DMGG1 with DMPCs labeled (linearized for display). (C) DMGG1 iVireons 'structure' score summary by protein cluster. Scores range from −1 (unlikely to be a virion structural protein) to 1 (likely to be a virion structural protein). Additional iVireons score summaries can be found in Figure 5—figure supplement 2.
Figure 5—figure supplement 1
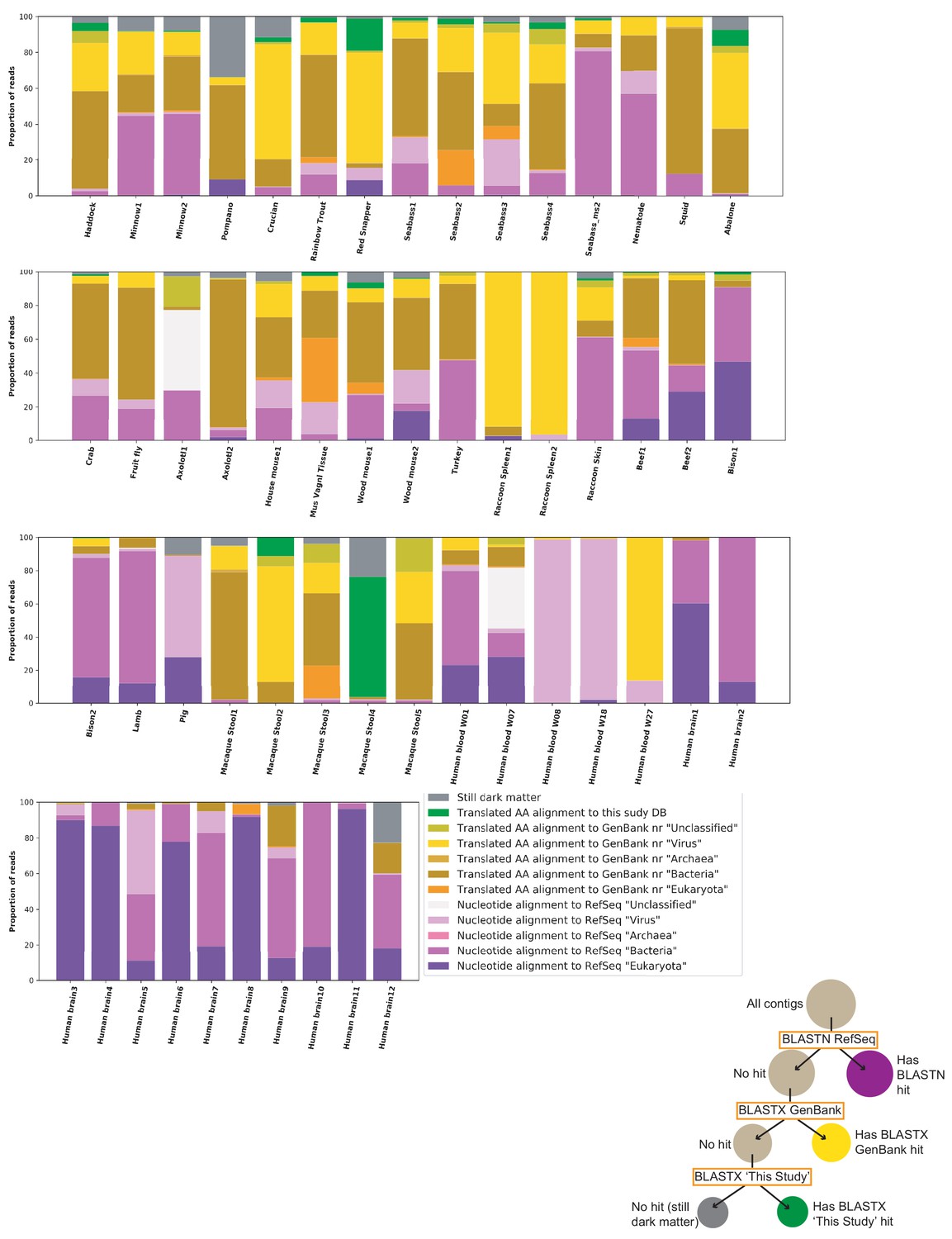
Sample characterization by iterative BLAST Searches.
Contigs of over 1000 nts from each sample were subject to iterative BLAST searches. First, BLASTN was performed against the RefSeq database. Contigs without hits were then queried by BLASTX against all of GenBank ‘nr’ database. Contigs without hits were then queried by BLASTX against a database of proteins from genomes reported in this study. The proportion of total reads mapping to each contig was calculated and used for this plot. Individual inspection of contigs shows that most hits in the ‘Translated AA alignment to GenBank’ nr’ ‘Bacteria’’ were likely plasmid or prophage proteins. The proportions of hits in each category are sensitive to stringency settings and to which databases are chosen for the analysis. The key aims of the figure are to display the proportion of reads the current survey rendered classifiable and the fraction of remaining dark matter reads in various samples.
Figure 5—figure supplement 2
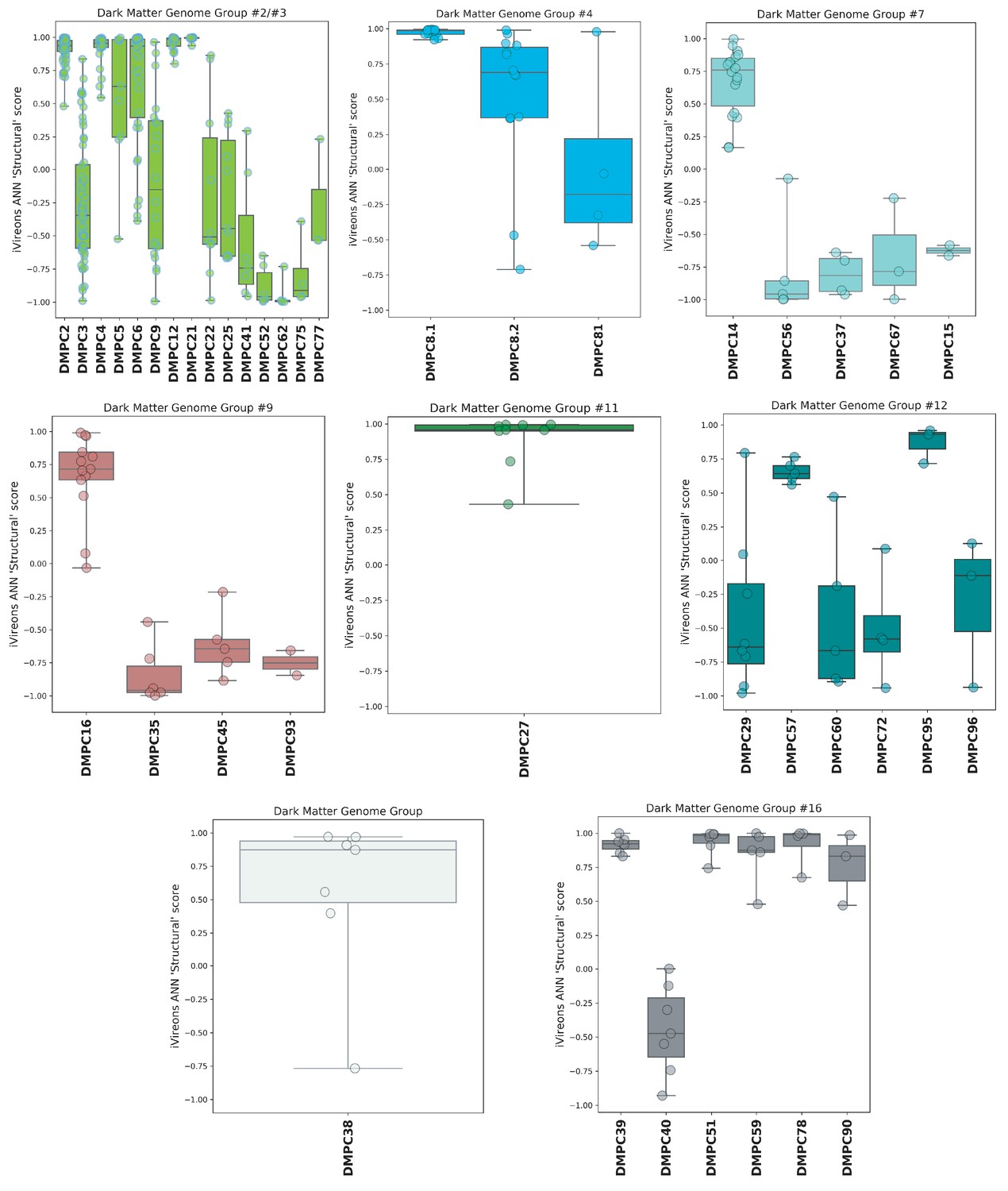
iVireons scores of DMGGs with candidate viral structural gene(s).
Box-and-whisker plots of iVireons ‘Structural' scores for individual DMPCs (numbers on x-axes) grouped by DMGG. Scores (y-axes) range from −1 (unlikely to be a virion structural protein) to 1 (likely to be a virion structural protein). DMGG2 and DMGG3 have been combined due to inferred chimerism.
Figure 6
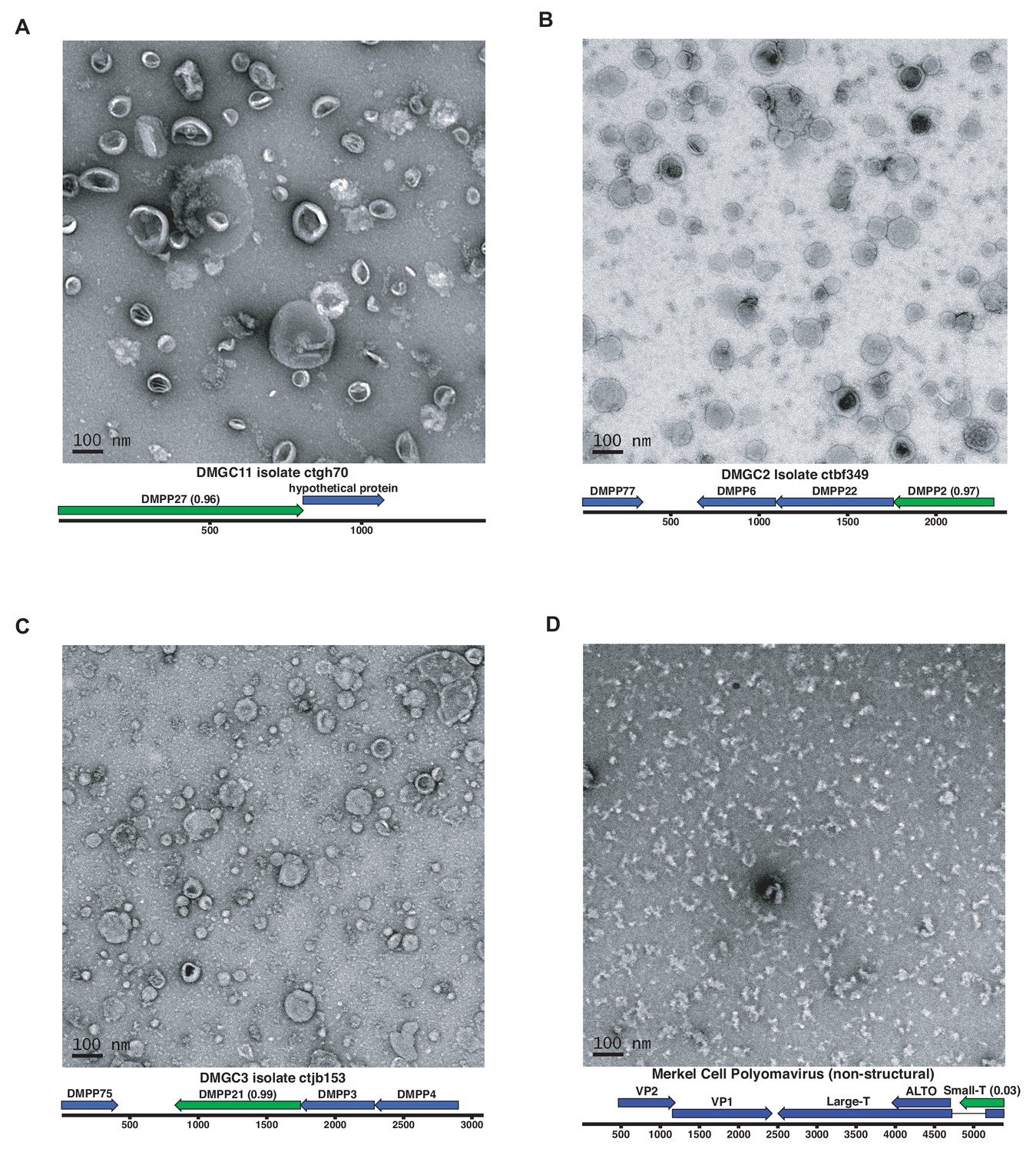
Expression of putative capsid proteins Images taken by negative stain electron microscopy.
Genome maps are linearized for display purposes. Expressed genes are colored green. iVireons scores are listed in parentheses. (A-C) Images represent virus-like particles from iVireons-predicted viral structural genes. (D) Merkel cell polyomavirus small T antigen (a viral non-structural protein) is shown as a negative control.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Escherichia coli) | T7 Express lysY/Iq E. coli | NEB | Cat#: C3013I | |
| Cell line (Homo-sapiens) | 293TT cells | https://dtp.cancer.gov/repositories/ | NCI-293TT | Deposition to ATCC in progress |
| Recombinant DNA reagent | Dark matter capsid expression plasmids | Generated here | Lead contact | |
| Commercial assay or kit | TempliPhi 100 Amplification Kit | Sigma | Cat#: GE25-6400-10 | |
| Chemical compound, drug | Optiprep Density Medium | Sigma | Cat#: D1556-250ML | |
| Chemical compound, drug | Sepharose 4B beads | Sigma | Cat#: 4B200-100ML | |
| Software, algorithm | Cenote-Taker | http://www.cyverse.org/discovery-environment | Cenote-Taker 1.0.0 | github: https://github.com/mtisza1/Cenote-Taker |
| Software, algorithm | EFI-EST | https://efi.igb.illinois.edu/efi-est/ | EFI-EST | |
| Software, algorithm | NCBI BLAST | NCBI | RRID:SCR_004870 | |
| Software, algorithm | SPAdes assembler | http://cab.spbu.ru/software/spades/ | RRID:SCR_000131 | |
| Software, algorithm | A Perfect Circle (APC) | https://github.com/mtisza1/Cenote-Taker/blob/master/apc_ct1.pl | APC | |
| Software, algorithm | EMBOSS suite (getorf) | http://emboss.sourceforge.net/ | RRID:SCR_008493 | |
| Software, algorithm | Circlator | http://sanger-pathogens.github.io/circlator/ | RRID:SCR_016058 | |
| Software, algorithm | HHSuite | https://directory.fsf.org/wiki/Hhsuite | RRID:SCR_016133 | |
| Software, algorithm | tbl2asn | https://www.ncbi.nlm.nih.gov/genbank/tbl2asn2/ | RRID:SCR_016636 | |
| Software, algorithm | MacVector | http://macvector.com | RRID:SCR_015700 | |
| Software, algorithm | Bandage | https://rrwick.github.io/Bandage/ | Bandage |
Additional files
-
Supplementary file 1
ViromeQC enrichment scores.
- https://cdn.elifesciences.org/articles/51971/elife-51971-supp1-v1.xls
-
Supplementary file 2
Sample metadata and virus summary.
- https://cdn.elifesciences.org/articles/51971/elife-51971-supp2-v1.xlsx
-
Supplementary file 3
Sequences associated with brain samples.
- https://cdn.elifesciences.org/articles/51971/elife-51971-supp3-v1.fasta
-
Supplementary file 4
Viruses observed in multiple samples.
- https://cdn.elifesciences.org/articles/51971/elife-51971-supp4-v1.xlsx
-
Supplementary file 5
Read counts for abundant viruses.
- https://cdn.elifesciences.org/articles/51971/elife-51971-supp5-v1.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/51971/elife-51971-transrepform-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Discovery of several thousand highly diverse circular DNA viruses
eLife 9:e51971.
https://doi.org/10.7554/eLife.51971




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}