Dynamic metastable long-living droplets formed by sticker-spacer proteins
- Department of Chemistry and Chemical Biology, Harvard University, United States
Figures
Figure 1
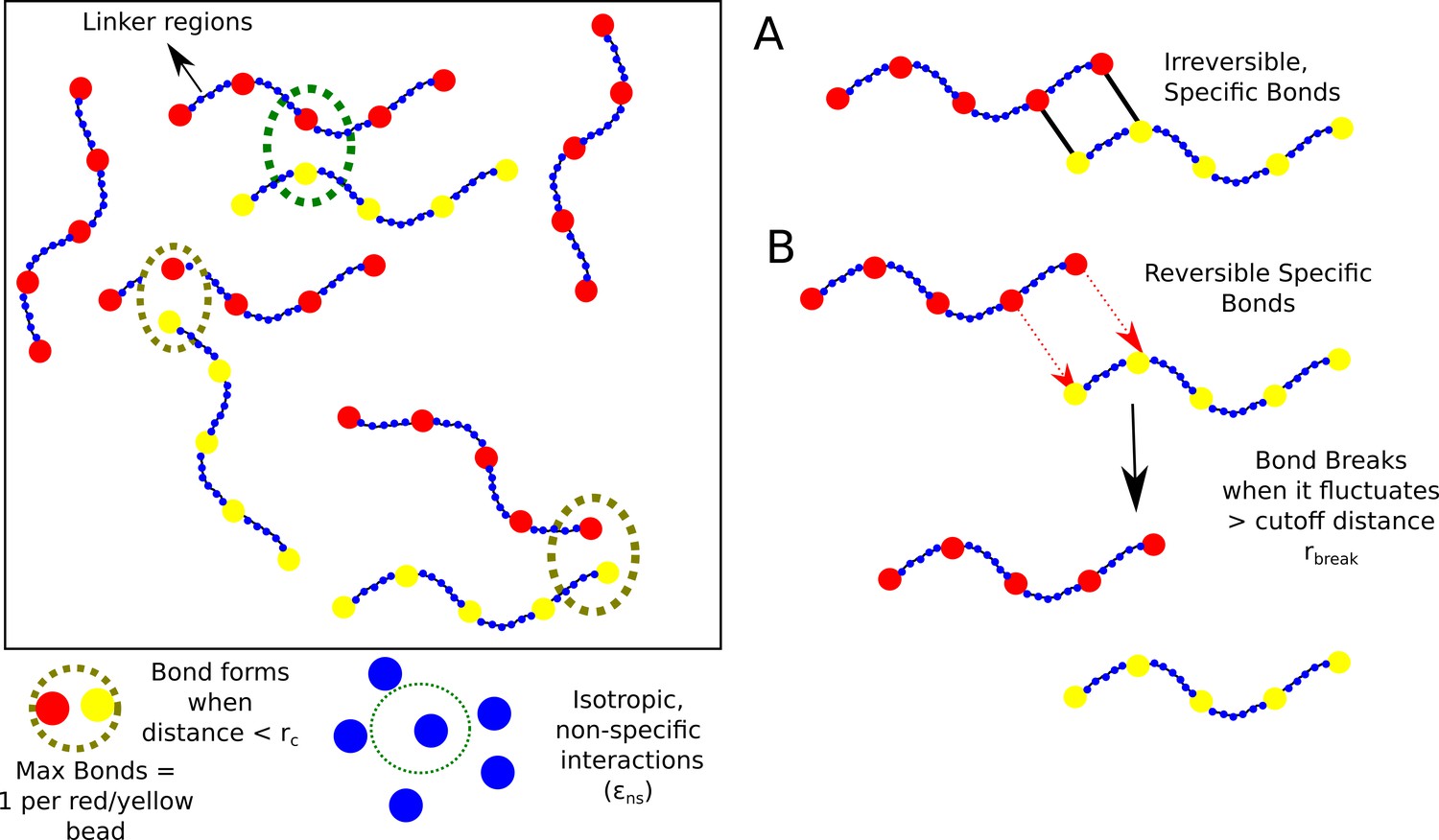
Model.
Schematic of the polymer model for studying phase-separation by multivalent biopolymers.
Figure 2 with 1 supplement
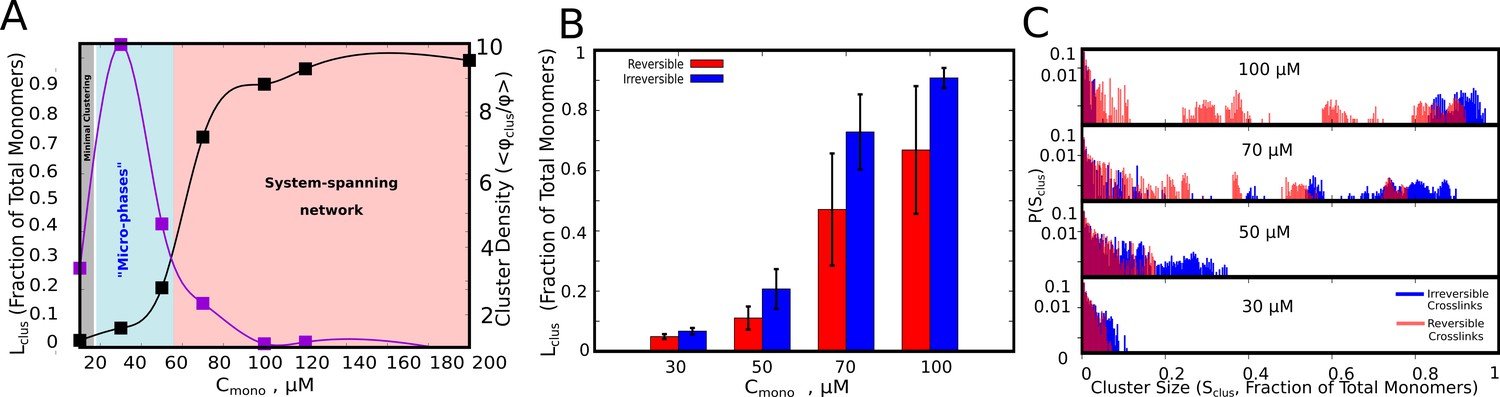
Cluster Sizes in Langevin Dynamics simulations.
(A) Black Curve – The single largest cluster as a function of free monomer concentration (in μM) for irreversible functional interactions. The largest cluster size is shown as a fraction of the total number of monomers in the simulation box. Purple Curve – The mean density of the cluster, = , normalized by the bulk density of monomers in the simulation, = . Sclus and Ntot refer to the size of the cluster and the total number of monomers in the simulation box, respectively. and refer to the radius of gyration of the cluster, and the radius of gyration of proteins in their randomly located initial configuration at the start of the simulation, respectively. The quantity / shows the degree of enrichment of polymer chains within the cluster upon self-assembly. The smooth curves are plotted as a guide to the eye, using the cspline curve fitting. (B) Comparison of the mean sizes of the single largest cluster for reversible and irreversible specific interactions, for varying free monomer concentrations. (C) Cluster size distributions for varying free monomer concentrations for reversible and irreversible specific interactions. Darker shades of red in the distributions indicate verlapping regions of the distribution while the blue and light red shades indicate regions with no overlap in the presence and absence of breakable interactions. The linker stiffness for the self-assembling polymer chains in this plot is 2 kcal/mol while the strength of inter-linker interaction is 0.1 kcal/mol (per pair of interacting beads). The mean and distributions of the largest cluster sizes were computed using 500 different configurations from five independent simulation runs of 16 μs.
-
Figure 2—source data 1
Compressed zip file containing the source data for cluster sizes and cluster size distributions (along with raw unprocessed data) plotted in Figure 2.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig2-data1-v2.zip
Figure 2—figure supplement 1
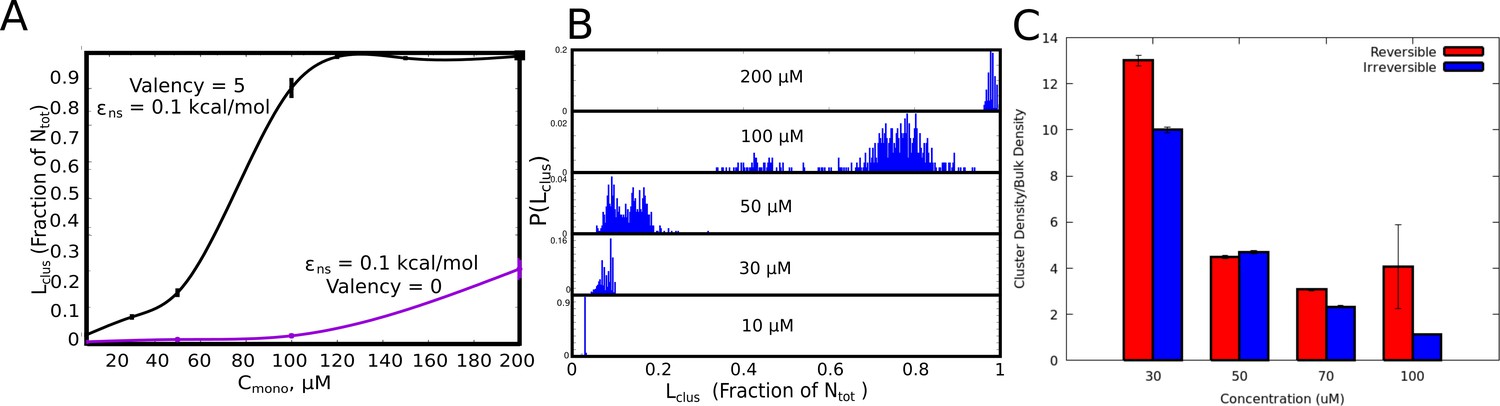
Cluster Sizes in Langevin Dynamics simulations.
(A) The single largest cluster as a function of free monomer concentration (in μM). The largest cluster size is shown as a fraction of the total number of monomers in the simulation box. The smooth curves are plotted as a guide to the eye, using the cspline curve fitting. The vertical bars represent the standard error. (B) Cluster size distributions for increasing free monomer concentrations. The linker stiffness for the self-assembling polymer chains in this plot is two kcal/mol while the strength of inter-linker interaction is 0.1 kcal/mol (per pair of interacting beads). The simulations were performed for an instantaneous bond formation assumption (=1). (C) Comparison of mean intra-cluster density (/) as a function of concentration for reversible and irreversible crosslinks.
Figure 3 with 1 supplement
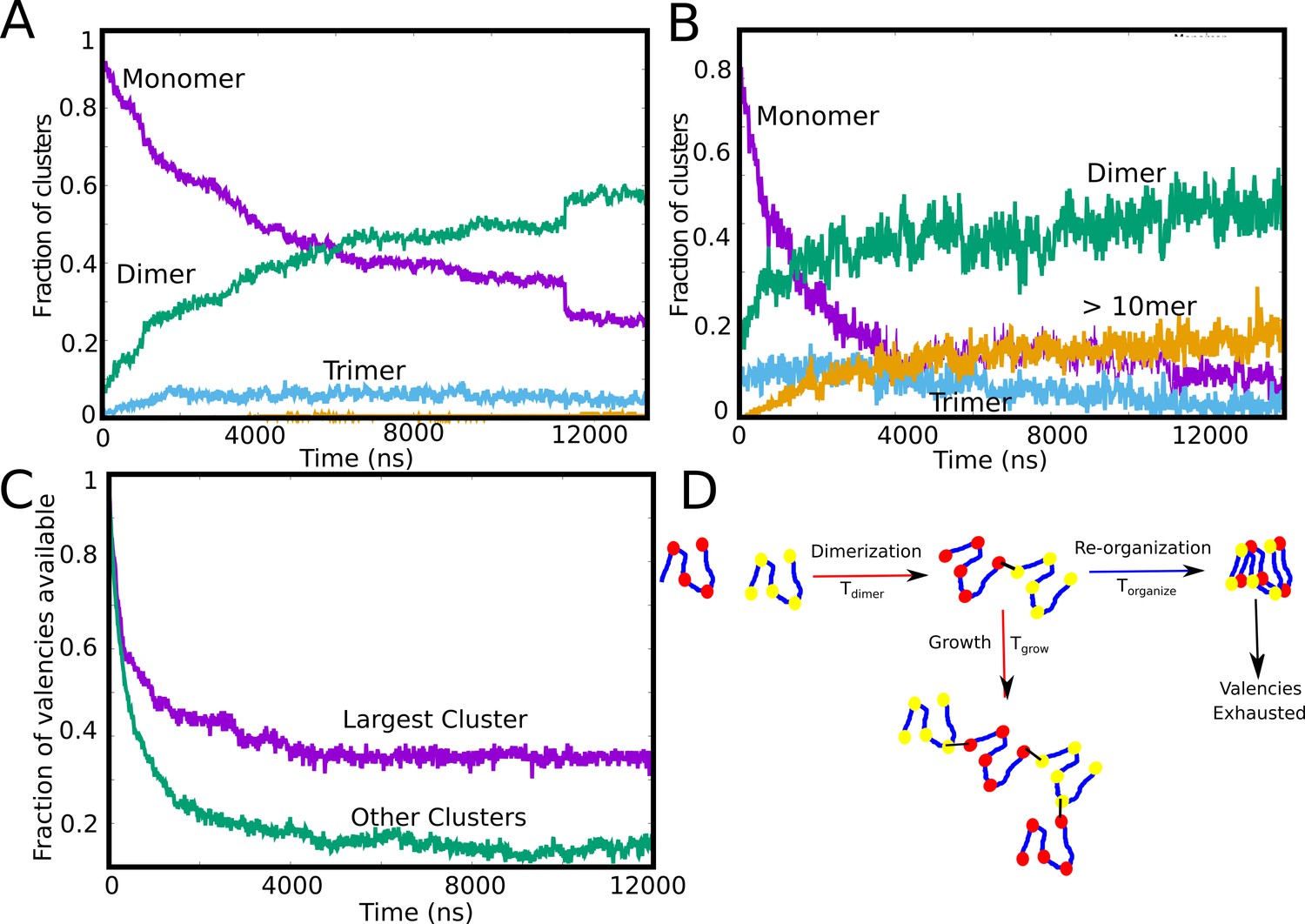
Tracking cluster formation at early timescales.
A and B show the temporal evolution of specific contacts for a free monomer concentration of 10, and 50 μM, respectively. For a low concentration of 10 μM, there is an initial decrease in the monomer population (purple curve) which is concomitant with an increase in the dimer population. A negligible fraction of the clusters is in the form of large-mers (size > = 10, orange curve) at these low concentration since the available valencies for growth are consumed by the smaller aggregated species. An increase in concentration from 10μM to 50μM results in an increase in the large-mer (orange curve, 50 μM) population as the monomer fraction decreases during the simulation. A higher free monomer concentration allows the larger clusters to grow due to consumption of free monomers (with unsatisfied valencies) before they get converted into smaller clusters (dimers, trimers) with satisfied valencies. (C) Time evolution of available valencies within the single largest cluster and outside the single largest cluster, for a of 50 μM, of 0.1 kcal/mol and linker bending rigidity of 2 kcal/mol. (D) A schematic figure showing the possible mechanisms of cluster growth and arrest and the competing timescales that could punctuate the process.
-
Figure 3—source data 1
Compressed zip file containing the source data for temporal evolution of different species (dimer,trimer,monomer etc) for different concentrations plotted in Figure 3.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig3-data1-v2.zip
Figure 3—figure supplement 1
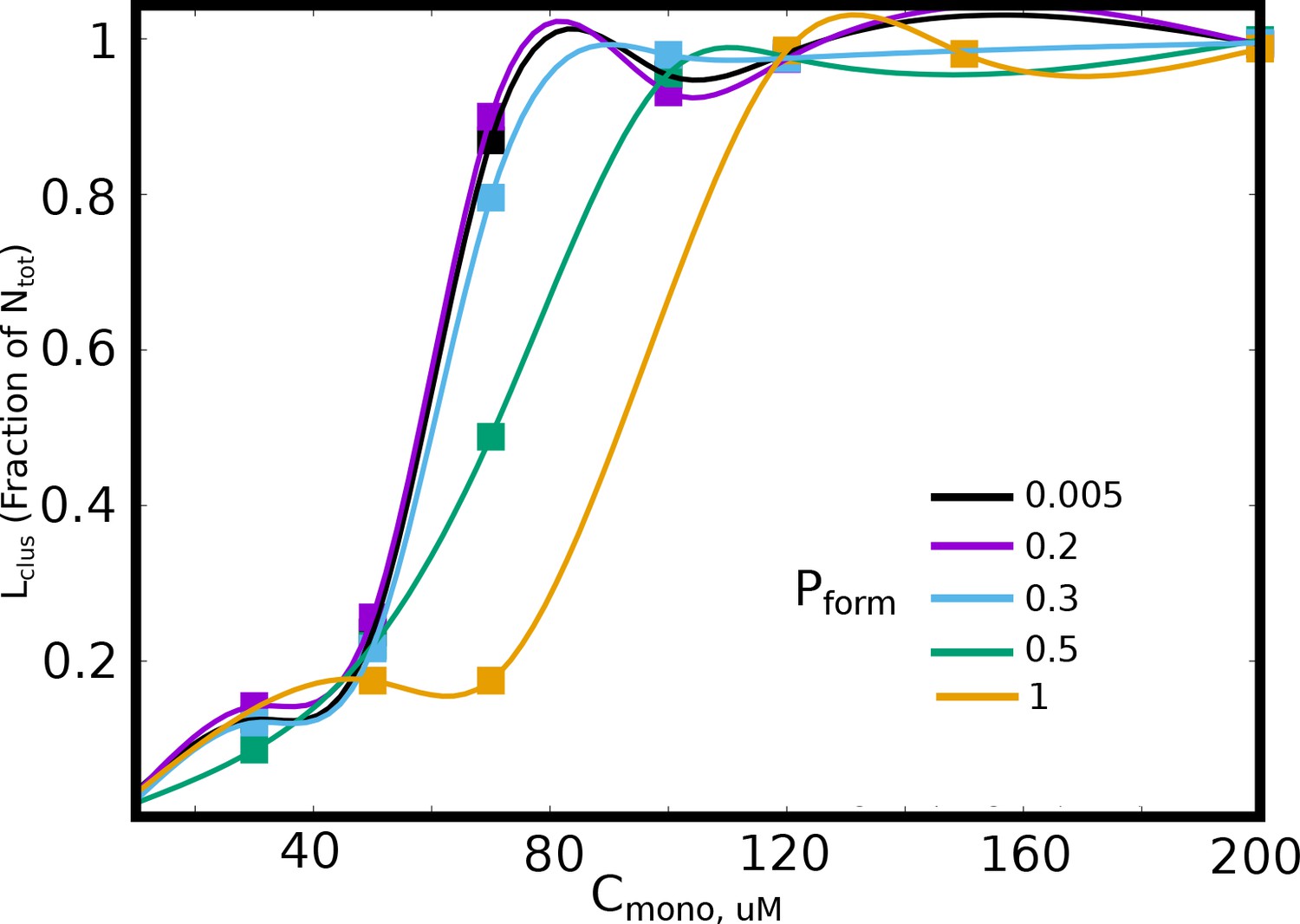
The size of the largest cluster, for different values of bond formation probability, .
A lower results in a slower arrest of the clusters, and thereby results in increased cluster sizes for smaller free monomer concentration, .
Figure 4

Factors influencing the key timescales.
(A) The mean first passage time for the first specific interaction between a pair of polymer chains as a function of the bulk density, , (see Table 1 for definition). (B) The mean first passage time for a pair of polymer chains to exhaust all the available valencies within the dimer.
-
Figure 4—source data 1
Compressed zip file containing the source data for Figure 4.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig4-data1-v2.zip
Figure 5 with 3 supplements

Linkers as modulators of self-assembly propensity.
(A) The size of the largest cluster for flexible linker regions(=2 kcal/mol) with varying inter-linker interaction strength (black curve, 0.1 kcal/mol and purple curve, 0.5 kcal/mol). Sticky inter-linker interactions result in smaller cluster sizes. (B). Purple Curve – Mean Radius of gyration for the individual-polymer chains (<>) within a self-assembled cluster as a function of increased inter-linker interactions. Black Curve— Mean density of the clusters as a function of inter-linker interaction strength. (C) The mean re-organization times, as a function of linker-stiffness, for different values of inter-linker interaction strengths.
-
Figure 5—source data 1
Compressed zip file containing the source data for largest cluster size, density of the cluster and radius of gyration of constituent chains, for different values of plotted in Figure 5.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig5-data1-v2.zip
Figure 5—figure supplement 1
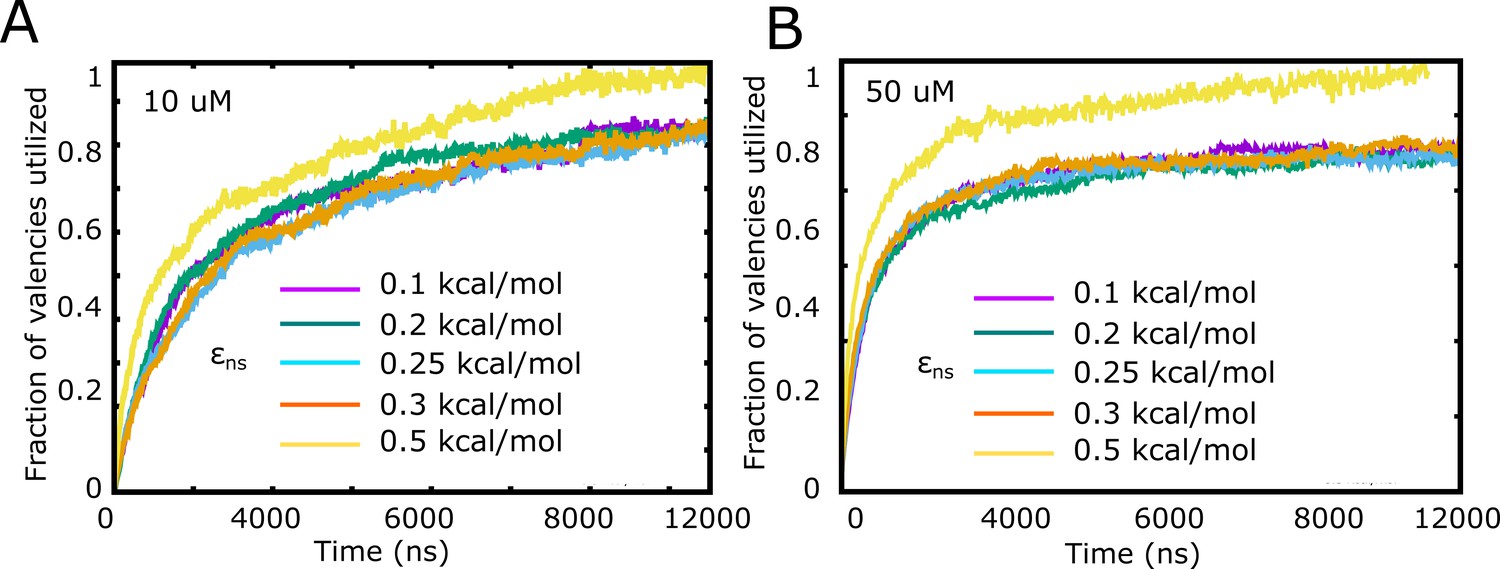
Fraction of valencies utilized as a function of increasing inter-linker interaction strength, for (A) 10 μM and (B) 50 μM free monomer concentration.
Figure 5—figure supplement 2
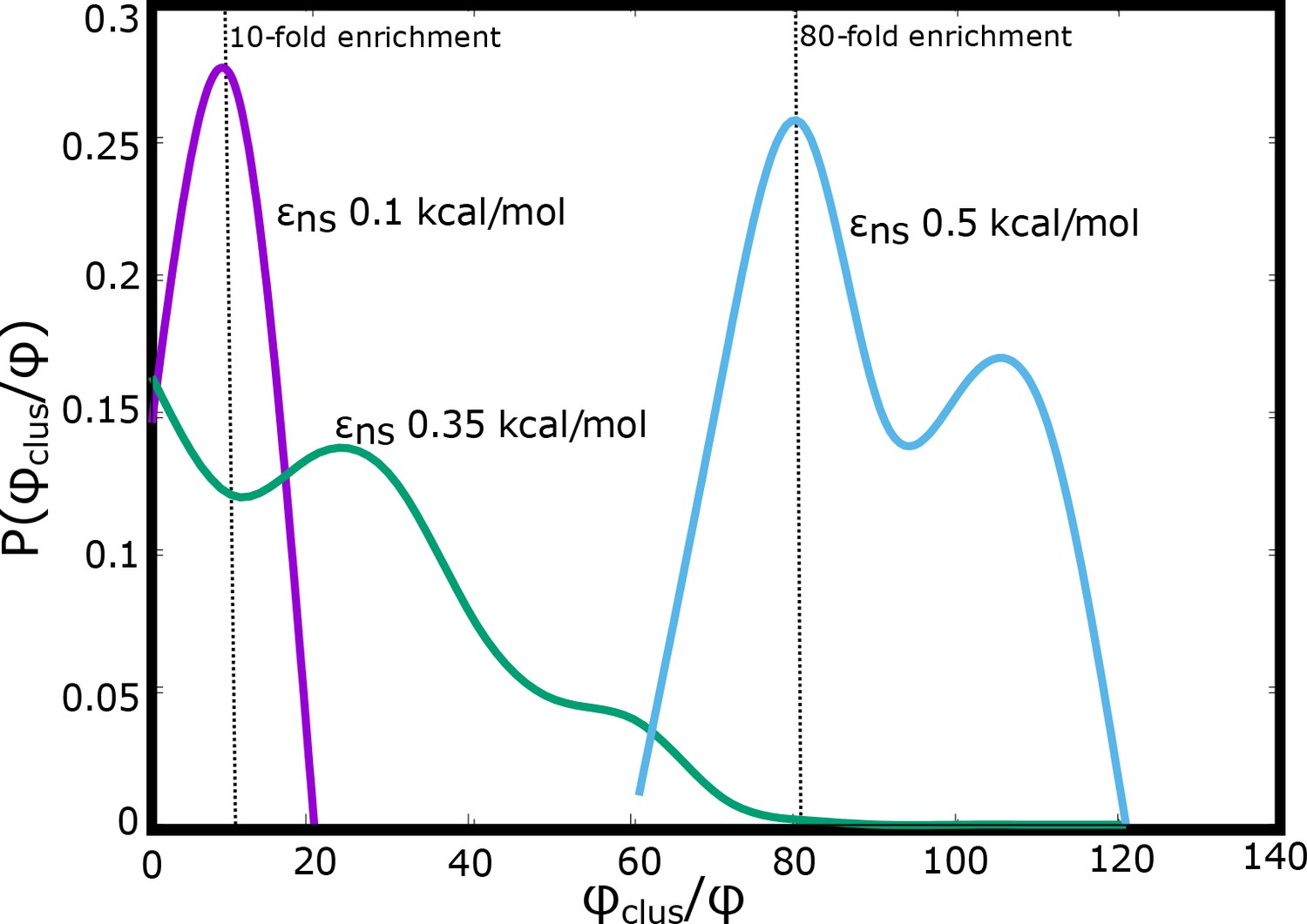
The probability of finding clusters with varying densities (normalized by the bulk densities) for different values of inter-linker interactions.
As the inter-linker interactions increase, the degree of enrichment can go from 10-fold to 100-fold.
Figure 5—figure supplement 3
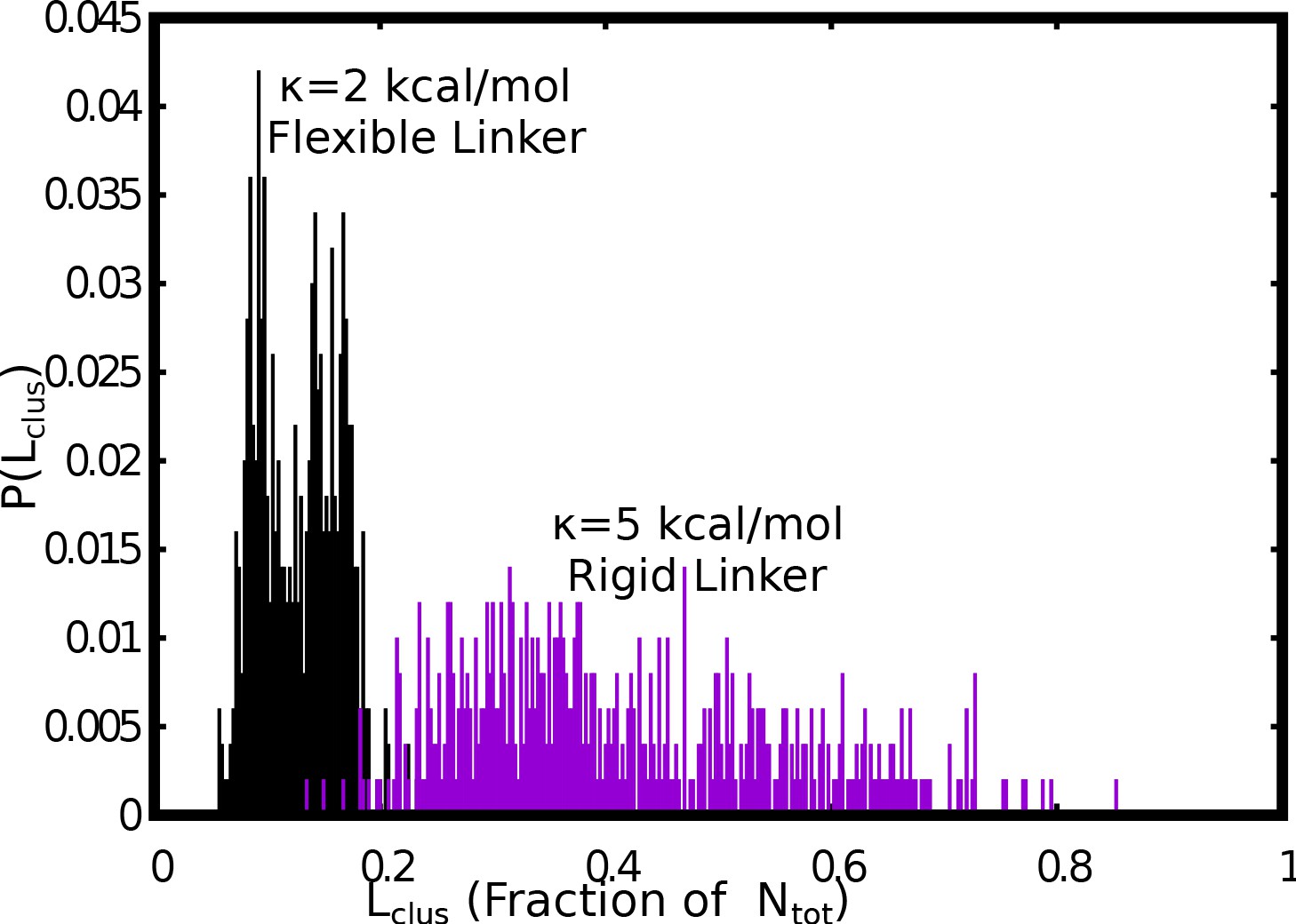
Comparision between the size distributions of the largest cluster, for flexible (=2 kcal/mol) versus stiff (=5 kcal/mol) linker regions.
The free monomer concentration used for this plot was 50 μM and a weak interlinker interaction strength of = 0.1 kcal/mol was used.
Figure 6
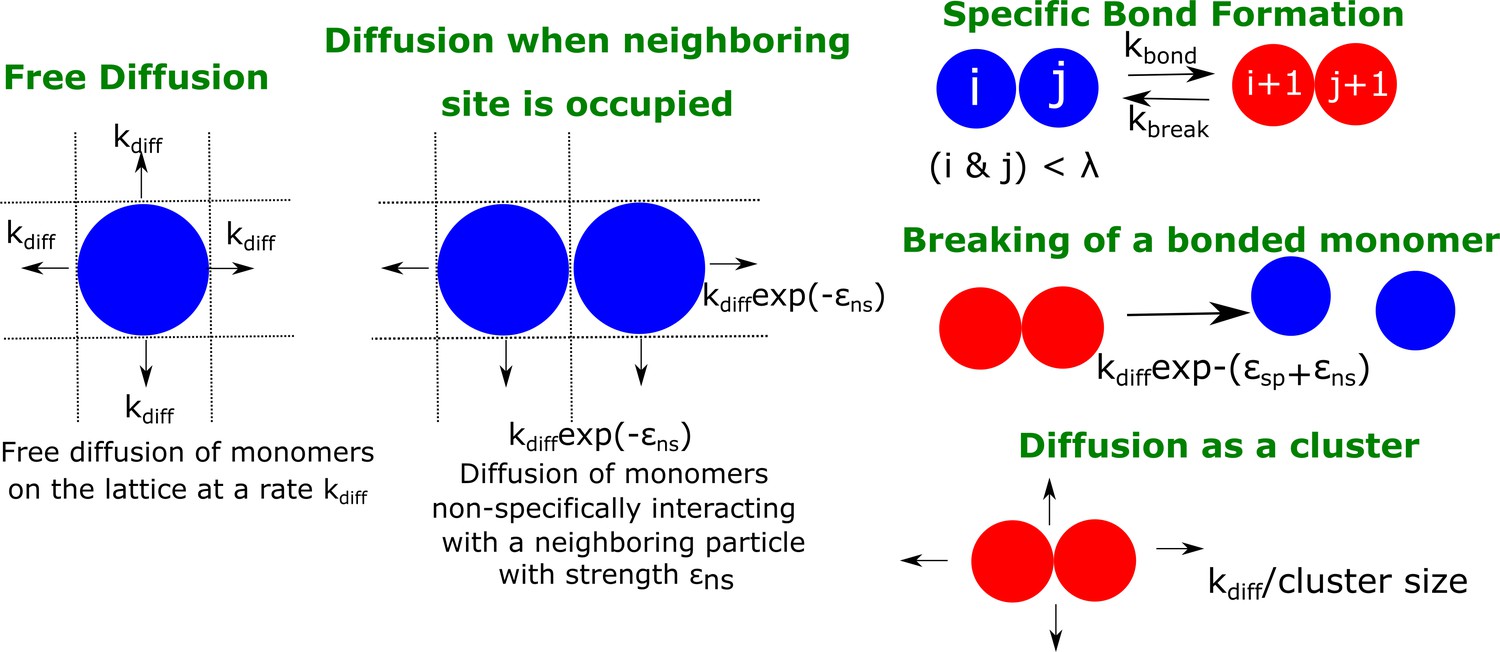
A schematic figure detailing the different rates in our phenomenological kinetic model simulated using the Gillespie algorithm.
The particles on the lattice can diffuse freely (when there are no neighboring particles) with a rate . In the presence of a neighboring particle, a non-specifically interacting monomer can diffuse away with a rate .exp(- ). Neighboring particles can also form specific interactions (with fixed valency ) at a rate or break an existing interaction with a rate . Clusters could diffuse at a rate that is scaled by their sizes Sclus. and refer to the strength of non-specific and specific interactions, respectively.
Figure 7 with 6 supplements
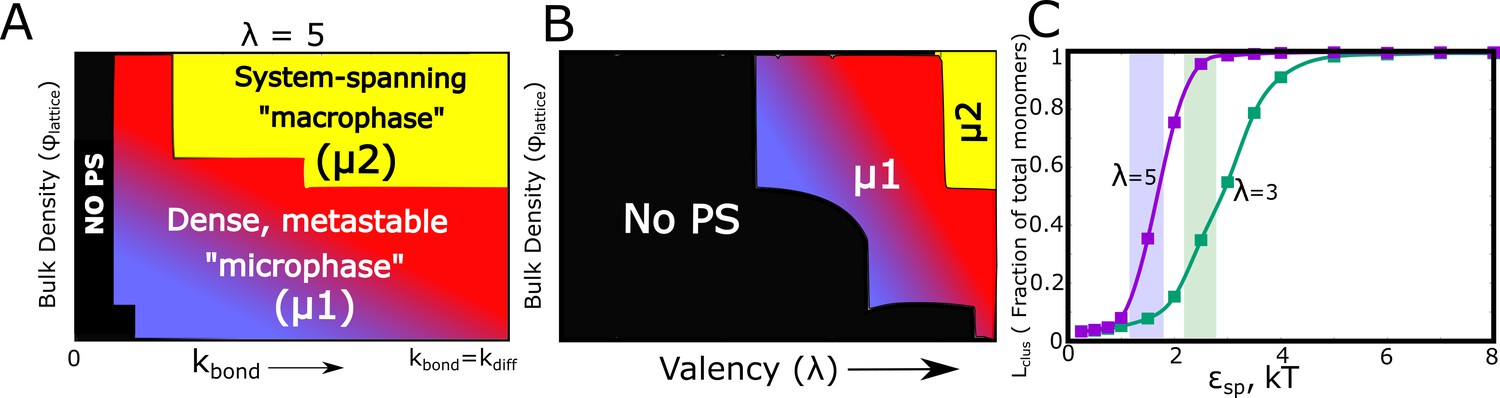
Kinetic Monte Carlo Simulations.
(A) Phase diagram highlighting the different phases (metastable microphase (μ1) or system-spanning macrophase (μ2), and the non-phase separated state (No PS) ) encountered upon increasing (between 0 and ), and the bulk density of monomers ( ) within the box. The assembling particles have a valency () of 5 in these simulations. The region shaded yellow represents part of the phase-space where we observe a system-spanning network whereas the bluish-red region represents the metastable micro-phase with a distribution of cluster sizes (see Figure 7—figure supplement 3 for distributions). As we move from the blue to the red regions in this dynamic phase diagram, the size of the single largest cluster increases (also see Figure 7—figure supplement 1). (B) Phase diagram highlighting the different phases encountered upon varying valency and bulk density as the phase parameters. The system-spanning macrophase is only encountered for larger valency particles at higher densities. This phase diagram was computed for = and of 0.35 kT. (C) The fraction of monomers in the largest cluster as a function of epsilon, for = and = 0.04. The single largest cluster sizes in all sub-panels of this Figure were computed for a simulation time-scale of 2 hr (with the fundamental timsecale of diffusion being set to = 1 ).
-
Figure 7—source data 1
Compressed zip file containing the source data for kinetic phase diagrams in Figure 7.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig7-data1-v2.zip
Figure 7—figure supplement 1
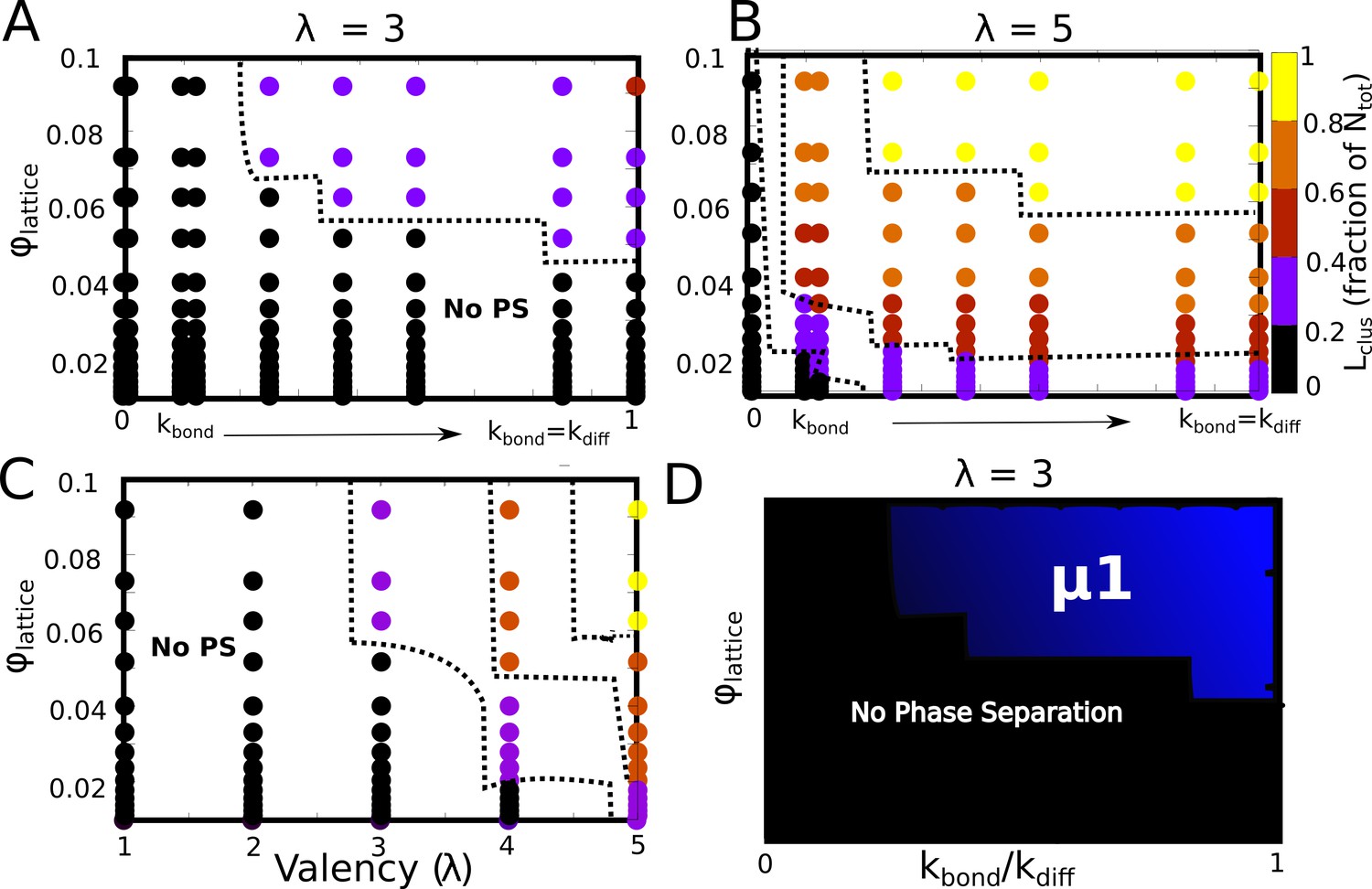
Detailed phase diagrams for (A) and (B) -, (C) - as the phase parameters.
The cluster sizes were computed at the end of a simulation run of 2 hr (actual time), setting the rate of diffusion to 1 . (D) The bonding rate was varied to identify the relationship between and .
Figure 7—figure supplement 2
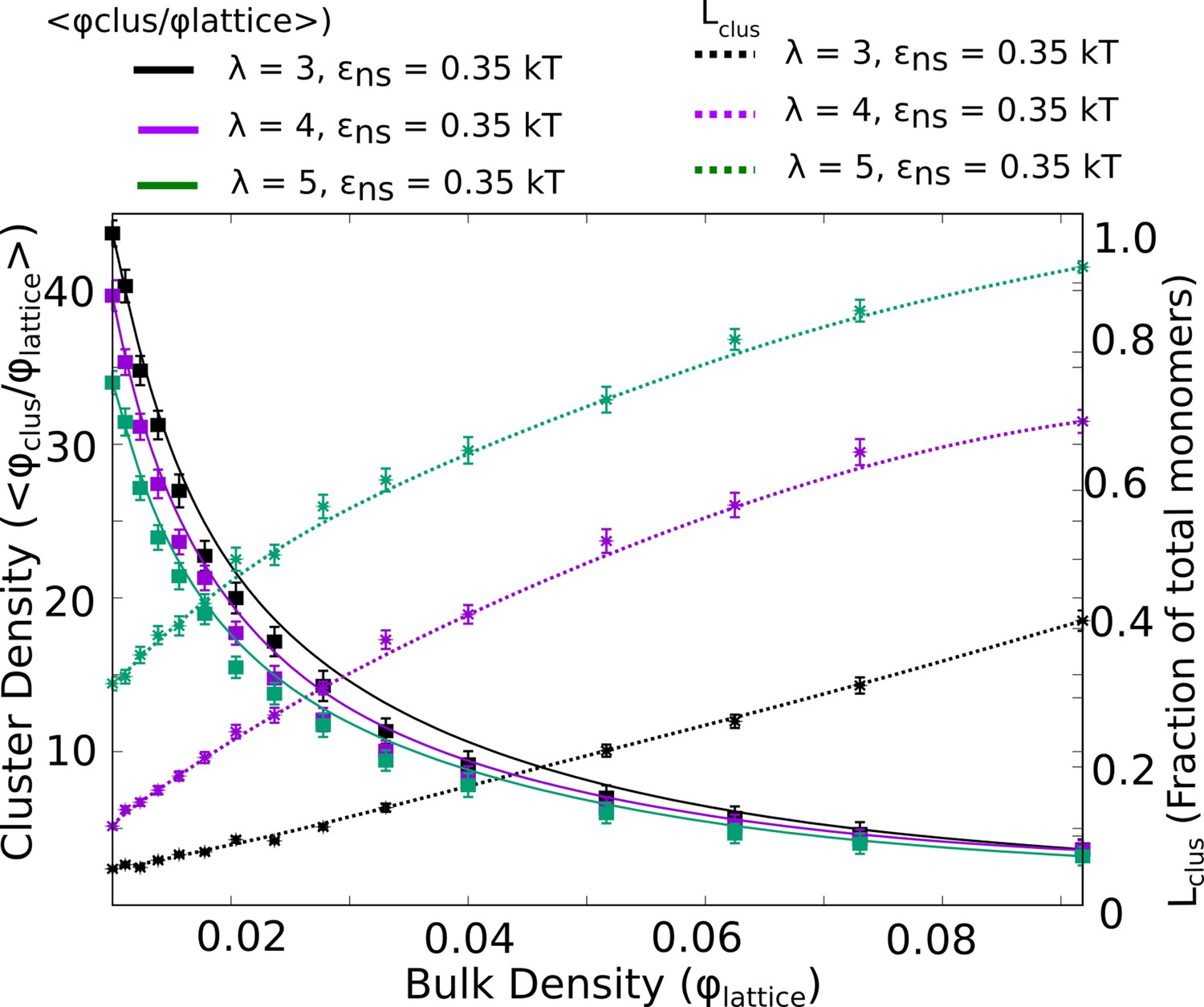
Intra-cluster densities of largest cluster (solid curves) and the corresponding sizes of the single largest cluster (dashed curves) as a function of bulk density, for three different values of .
In the regime where cluster sizes approach the total number of monomers, the density of the cluster is at its lowest.
Figure 7—figure supplement 3
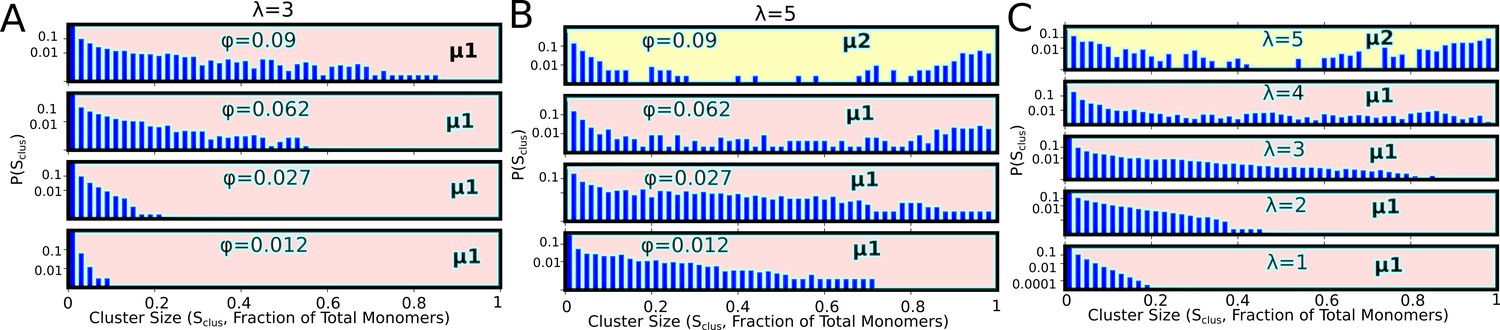
Cluster size distributions for varying densities for λ=3 (A), and λ=5 (B).
(C) Effect of valency on size distributions at a fixed of 0.09. These distributions were computed at the end of 100-independent simulation runs of 2 hr (actual time) each. μ1 and μ2, in these figures refer to the macro-phase separated state (with large network-spanning clusters), and the micro-phase separated state (with coexisting clusters of various sizes), respectively. These distributions are representative of the μ1 and μ2 phases shown in the Figure 7 of the main text.
Figure 7—figure supplement 4
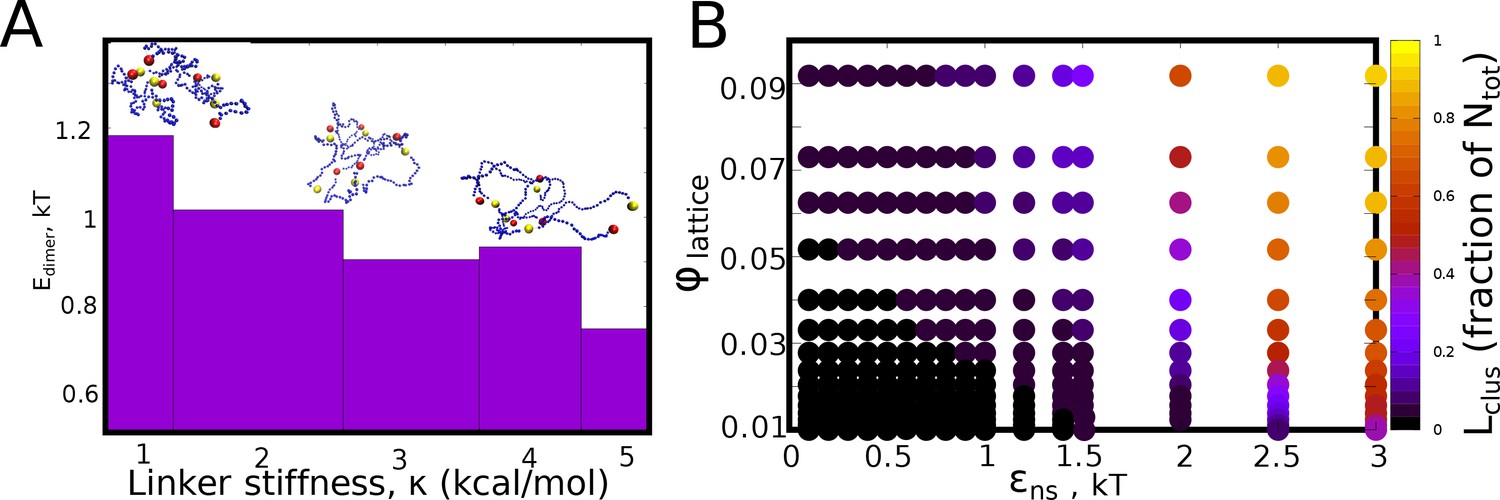
Inter-protein interaction strengths.
(A) The mean pair-wise interaction energy for 100 different dimeric structures (from the LD simulations), for an inter-linker interaction strength of 0.1 kcal/mol, for different values of linker bending rigidity. (B) The - phase diagram (for =0) showing no phase separation for low values of isotropic interaction strength. However, for values of 1kT, phase separation is observed at the end of the simulation timescale of 2 hr (actual time).
Figure 7—figure supplement 5
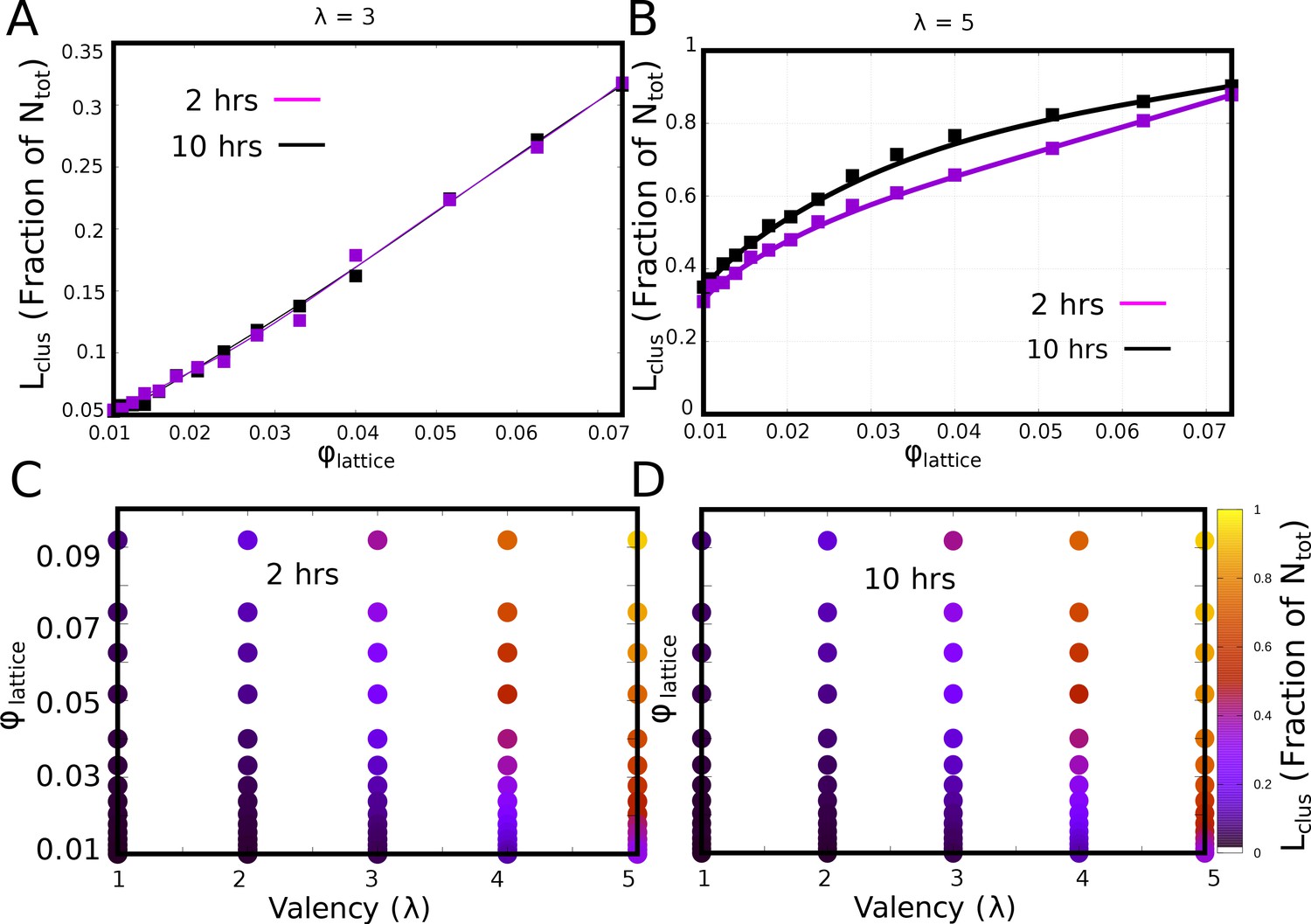
Convergence of phase diagrams.
(A and B) The fraction of monomers in the largest cluster for 2 and 10 hr of actual time, for of 3 and 5, respecively. (C and D). The - phase diagram at the end of 2 and 10 hr of simulation time, respectively, showing very little difference.
Figure 7—figure supplement 6
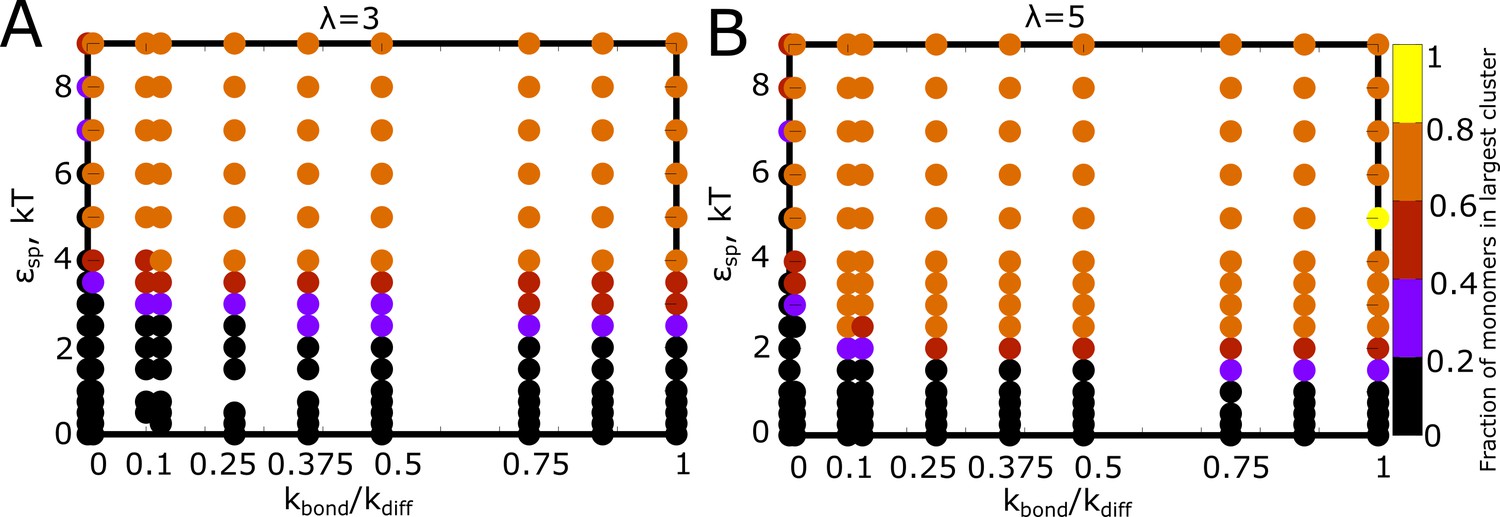
Detailed phase diagrams for (A) and (B) - as the phase parameters.
The was set to 0.04, an intermediate density identified from the previous phase diagrams with density as a phase parameter. The cluster sizes were computed at the end of a simulation run of 2 hr (actual time), setting the rate of diffusion to 1 .
Figure 8 with 1 supplement
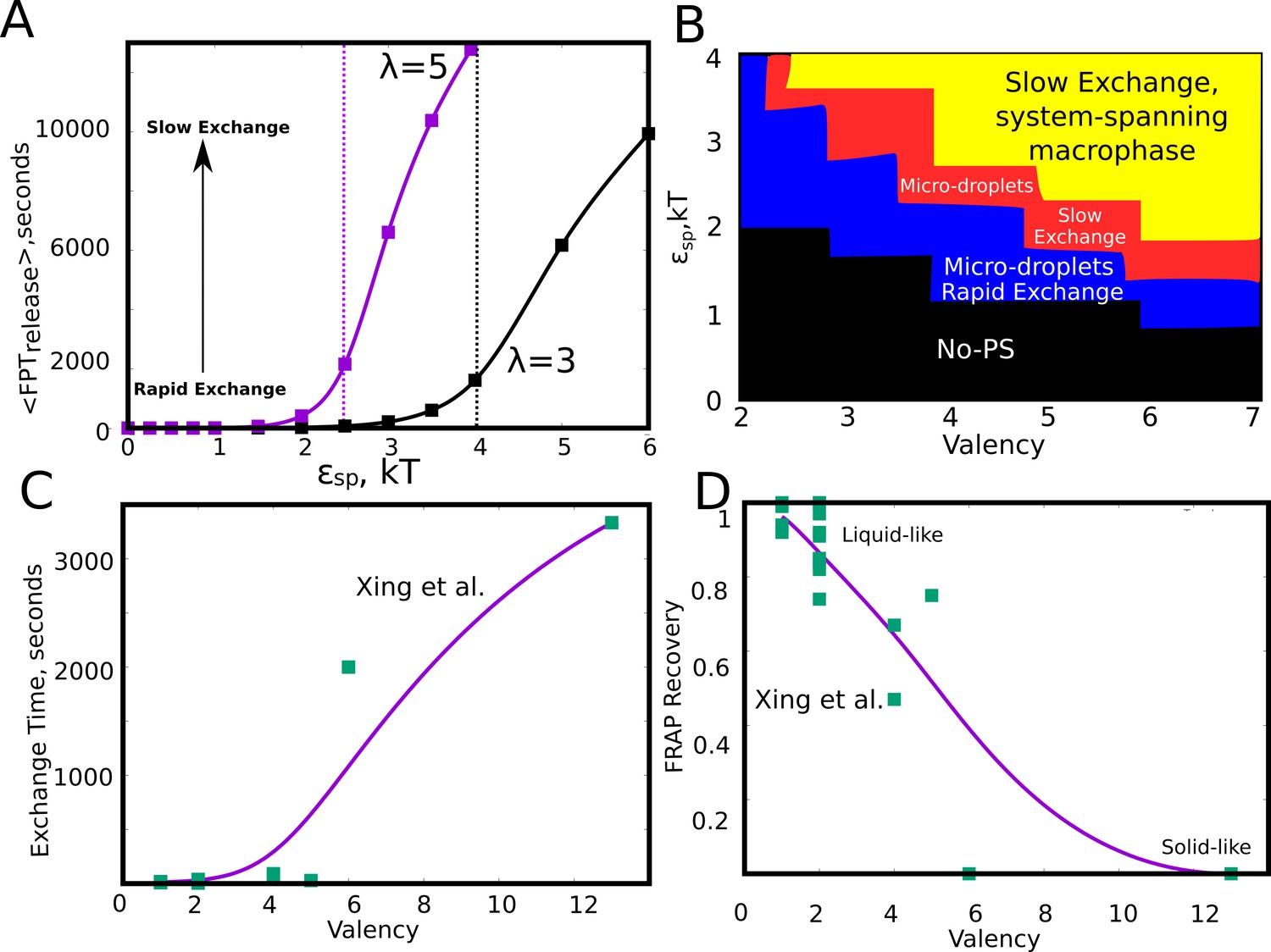
Effect of model parameters on the exchange times between monomers and the aggregates.
The parameter values used in panels A and B are =0.04, / = 1. (A) Mean first passage time for the monomers to go from the buried state (with four neighbors) to the free state (with no neighbors) in response to varying values of specific interaction strength. The two curves show the trends for species with different interaction valencies. The dashed solid lines refer to value of beyond which the largest cluster consumes 50% or more of available monomers. (B) The state of the system, for variation in and suggests that the system displays remarkable malleability in dynamicity and size distributions. Weak and a low results in no phase separation (shaded black). For higher values of both these parameters, the system can access the single large macro-phasic state (shaded yellow), however with a dramatic slowdown in exchange times. For an intermediate range, the system adopts a metastable microphase separated state, with either slow (shaded red) or fast-exchange (shaded blue) dynamics. (C) The experimentally determined molecular exchange times for molecules of varying interaction valencies. (Xing et al., 2018). (D) The extent of recovery after a photobeaching experiment, for interacting species with varying valencies. The data in panel C) and D) has been obtained from a study by Xing et al., 2018. The solid curves in C and D are added to guide the eye.
-
Figure 8—source data 1
Compressed zip file containing the source data for kinetic phase diagrams in Figure 8.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig8-data1-v2.zip
Figure 8—figure supplement 1
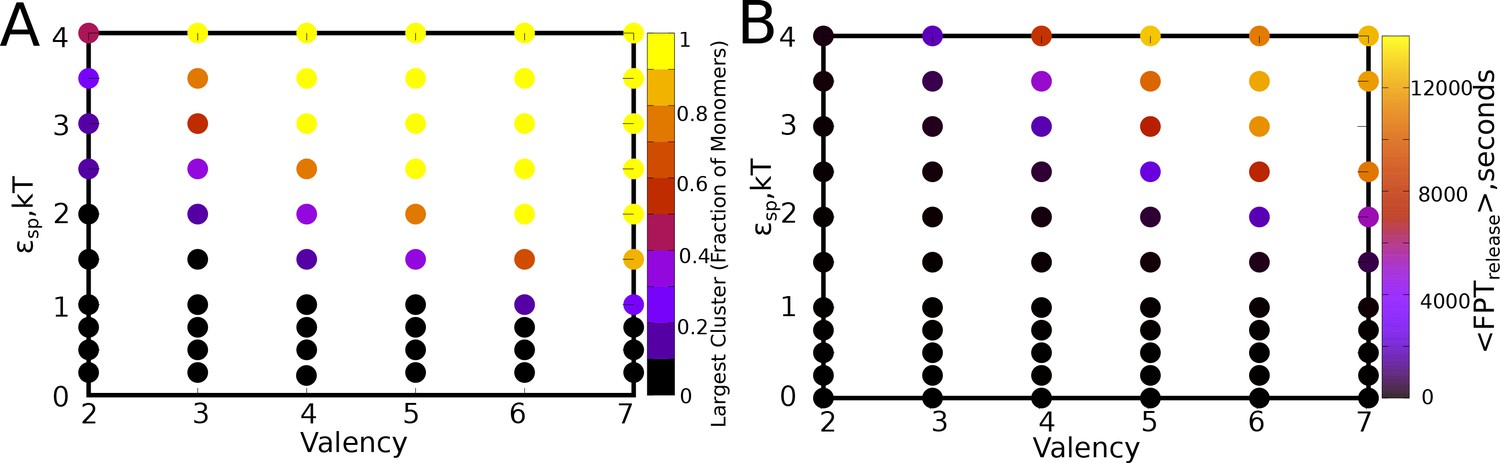
<> for varying values of and , for a of 0.04, and a ratio of 1.
(B) Mean first passage times for a particle to exchange between a cluster and the bulk. The parameter values are same as in panel A.
Figure 9 with 1 supplement

Effect of solvent viscosity.
Langevin Dynamics Simulations. (A and B) Cluster size distributions from Langevin dynamics simulations for different values of solvent viscosity and different free monomer concentrations. The shaded region in the two subpanels (A) and (B) is to highlight that a more viscous solvent results in larger clusters disappearing from the distribution. (C) The size of the single largest cluster as a function of solvent viscosity for a free monomer concentration of 70 μM. (D and E) Monte Carlo Simulations. The size of the single largest cluster as a function of density for varying rates of diffusion, for specific interaction valencies =3 (D) and = 5 (E).
-
Figure 9—source data 1
Compressed zip file containing the source data for mean largest cluster sizes (and size distributions) from LD and MC simulations studying the effect of solvent viscosity.
- https://cdn.elifesciences.org/articles/56159/elife-56159-fig9-data1-v2.zip
Figure 9—figure supplement 1
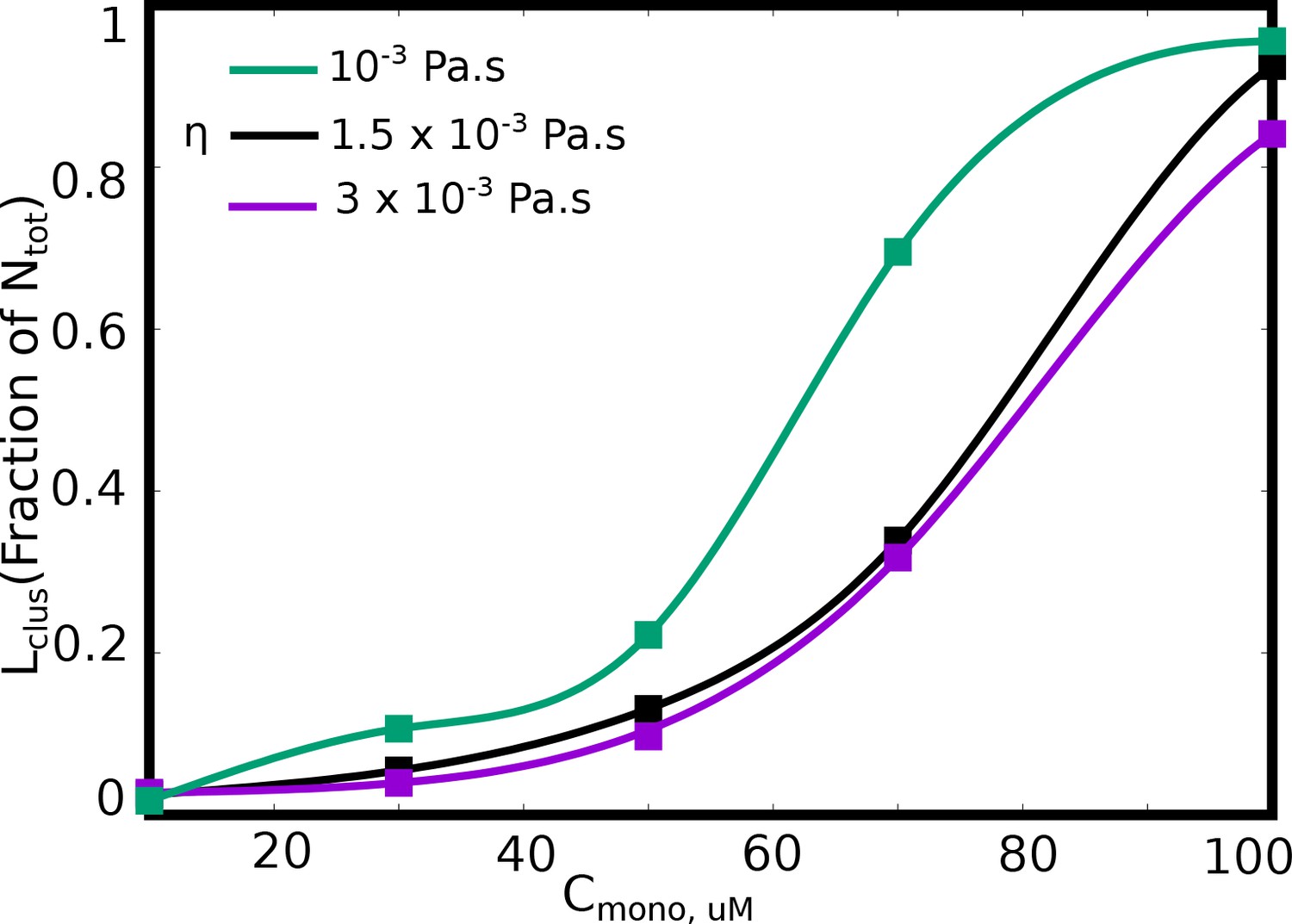
as a function of free monomer concentrations in LD simulations performed with various solvent viscosities.
Figure 10
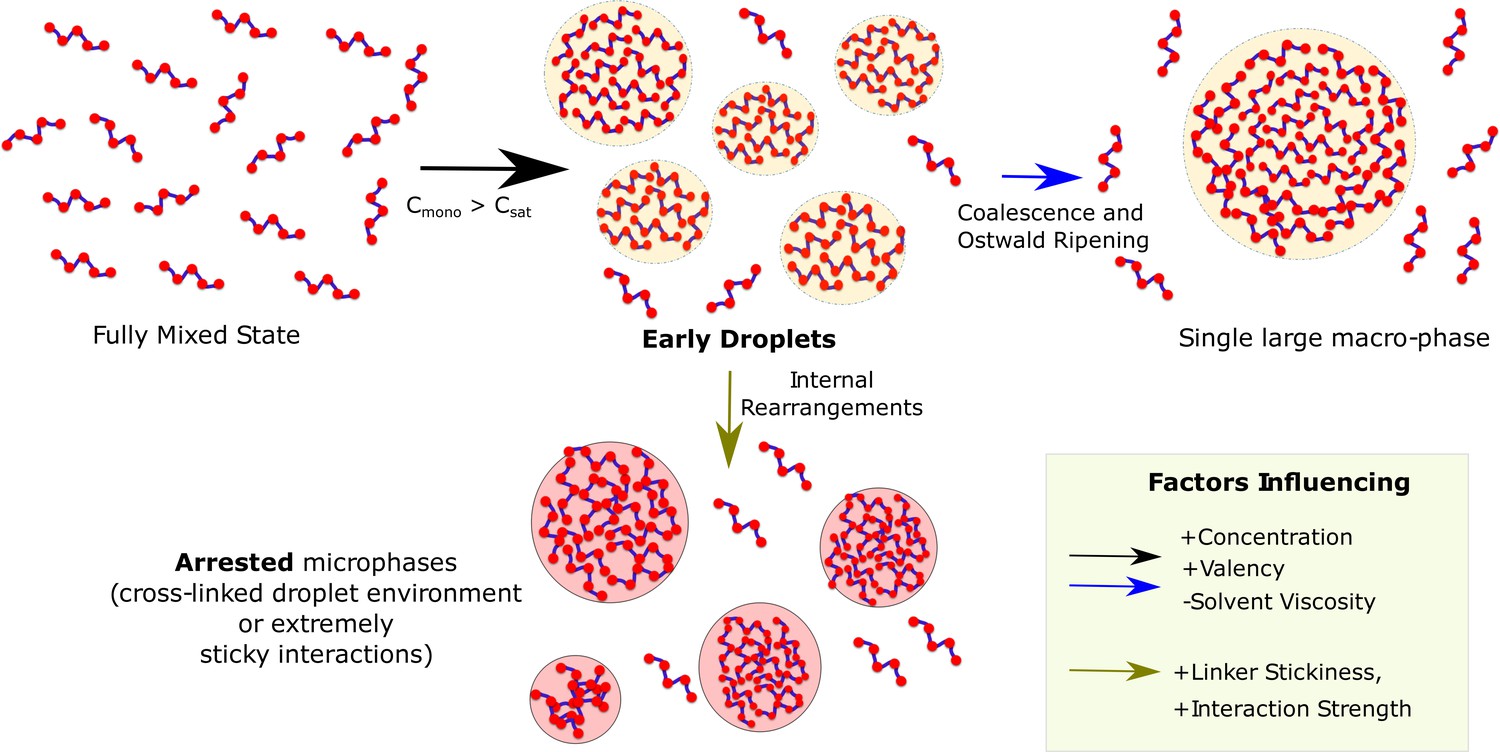
Graphical Summary.
Muli-valent proteins can exist in two Flory-like equilibrium states, the fully mixed solvated state and a single large macro-phase separated state (when free monomer concentration () exceeds the critical concentration ()). Above the critical concentration, the proteins first phase separate into multiple droplet phases (black arrow) which are long-living, metastable structures. However, the growth of these individual droplets to a single large macro-droplet (blue arrow) through coalescence and Ostwald ripening is unlikely at biologically relevant timescales. The multiple metastable micro-droplets can also age into highly cross-linked structures (green arrow) that saturate interactions within themselves and thus cannot grow further. Extremely stable functional interactions or sticky inter-linker interactions could also result in these arrested microphases.
Tables
Table 1
Important simulation variables and order parameters.
| Notation/Terminology | Physical Interpretation | Definition |
|---|---|---|
| Ntot | Total number of polymer chains in the system. Ntot = 400 in our simulations. | – |
| Lclus | Size of the single largest cluster | represented as fraction of Ntot |
| Sclus | Size of the cluster | represented as fraction of Ntot |
| Cmono | Concentration of polymer chains in the simulation box | In units of μM |
| φ | Bulk density of proteins (in their monomeric state) when the individual chains are randomly placed in the simulation box at the start of the simulation. | is the the radius of gyration of proteins when they are randomly positioned in simulation box at the start of the simulation. |
| φclus | Intra-cluster density of polymer chains. | is the radius of gyration of the system of proteins within the cluster. |
| φclus /φ | Normalized intracluster density describing the degree of enrichment of polymer chains within the cluster. | For system-spanning networks, φclus /φ→1. For dense clusters, φclus /φ >> 1. |
| εns | Interaction strength for isotropic, non-specific interactions between linker regions. εns in LD simulations is a pairwise interaction strength between individual beads In kMC simulations, εns is the net non-specific interaction strength between two lattice particles. | – . |
| εsp | Strength of attractive interaction between functional domains (specific interactions). | – |
| φlattice | Bulk density of monomers in the 2D-lattice in kMC simulations. This quantity is analogous to the concentration of monomers on the lattice. | φlattice = Ntot/L2, where L is the size of the 2D square-lattice. |
| λ | Valency of the polymer chain, that is the number of adhesive functional domains per interacting polymer chain. | – |
| Κ | Bending stiffness of the linker regions in LD simulations. A higher value of K is used to model stiffer linkers that prefer a more open configuration. | Κ = 2 kcal/mol, in LD simulations with flexible linkers. |
| η | The viscosity of the medium in LD simulations | η = 10−3Pa.s in LD simulations, unless mentioned otherwise. |
| kdiff | Diffusion rate of free monomers in the 2D-lattice kMC simulations. | – |
| kbond | Rate of formation of specific interactions between neighboring particles in 2D-lattice kMC simulations. | – |
Additional files
-
Source code 1
Langevin Dynamics Simulation LAMMPS Script.
Langevin dynamics simulation script.
- https://cdn.elifesciences.org/articles/56159/elife-56159-code1-v2.zip
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/56159/elife-56159-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Dynamic metastable long-living droplets formed by sticker-spacer proteins
eLife 9:e56159.
https://doi.org/10.7554/eLife.56159




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}