Genome editing enables reverse genetics of multicellular development in the choanoflagellate Salpingoeca rosetta
- Howard Hughes Medical Institute and Department of Molecular and Cell Biology, University of California, Berkeley, United States
Figures
Figure 1

Introduction to Salpingoeca rosetta as a simple model for multicellularity and the ancestry of animal cell biology.
(A) Choanoflagellates (blue) are the closest living relatives of animals (red) and last shared a common ancestor (purple) ~800 million years ago (Parfrey et al., 2011). (B) The collar complex, an apical flagellum (f) surrounded by a collar (c) of actin-filled microvilli, typifies choanoflagellates and is uniquely shared between choanoflagellates and animals (Brunet and King, 2017). (C) Wild-type S. rosetta forms multicellular rosette colonies in response to rosette inducing factors (RIFs) secreted by environmental bacteria. In the absence of RIFs (C’), S. rosetta grows as single cells or as a linear chain of cells (star). Upon the addition of RIFs (C’; Alegado et al., 2012; Woznica et al., 2016), S. rosetta develops into spheroidal, multicellular rosettes (arrowhead) through serial cell divisions (Fairclough et al., 2010). (D) The rosetteless C-type lectin gene is necessary for rosette development. A mutation in rosetteless allows normal cell growth as single cells and linear chains in the absence of RIFs (D’) but prevents rosette development in the presence of RIFs (D’; Levin et al., 2014). (E) Wild-type S. rosetta secretes Rosetteless protein from the basal ends of cells into the interior of rosettes. Shown is a representative rosette stained with an antibody to alpha-tubulin to mark cortical microtubules and the apical flagellum of each cell (E’, grey) phalloidin to mark actin-filled microvilli (E’, magenta), and an antibody to Rosetteless protein (E’’’, green). A merge of alpha-tubulin, phalloidin, and Rosetteless staining shows that Rosetteless protein localizes to the interior of rosettes (arrow) where cells meet at their basal ends (E''''; Levin et al., 2014).
Figure 2 with 2 supplements
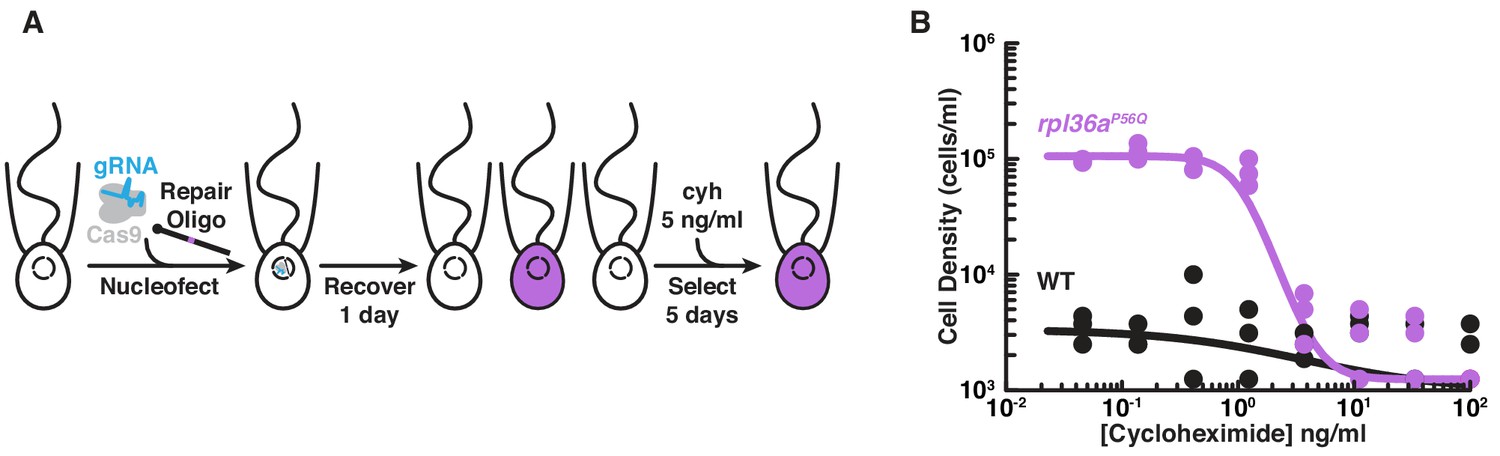
Engineered cycloheximide resistance in S. rosetta provides a proof-of-principle for Cas9-mediated genome editing.
(A) Schematic of Cas9-mediated genome editing to engineer cycloheximide resistance in S. rosetta. Nucleofection was used to deliver SpCas9 (gray) bound to gRNA (cyan), which together form the SpCas9 RNP, and repair oligonucleotides (Repair Oligo; Figure 2—figure supplement 2) to engineer cycloheximide resistance. After recovering cells for one day, successfully edited cells were selected by growth in media supplemented with cycloheximide (cyh), which inhibits the growth of wild-type cells (Figure 2—figure supplement 1) and selects for cycloheximide-resistant cells (purple). (B) A designer cycloheximide-resistant allele (Figure 2—figure supplement 2) allows cell proliferation in the presence of cycloheximide. Wild-type (WT, black dots and line) and rpl36aP56Q (purple dots and line) strains were placed into media supplemented with a range of cycloheximide concentrations (x-axis) at a cell density of 104 cells/ml and then were grown for two days. rpl36aP56Q grew to higher cell densities than the wild-type strain at cycloheximide concentrations < 10 ng/ml. At higher concentrations, cycloheximide inhibited growth of both strains. The dots show cell densities from three independent replicates. The lines show the average from independently fitting a dose inhibition curve to the cell densities from three independent experiments.
Figure 2—figure supplement 1

An approach for selecting cycloheximide resistance in S. rosetta.
(A) Cycloheximide inhibits S. rosetta growth. We seeded each well of a 24-well plate with 0.5 ml of cells at 2 × 104 cells/ml in a 3-fold serial dilution of cycloheximide, including a condition without cycloheximide. Three independent wells were set up for each concentration of cycloheximide for replicate measures. The cell density at each concentration was determined after 48 hr by counting with a hemocytometer. To establish a relationship between growth rate and cycloheximide concentration, the cell density was transformed into relative growth rates (S) using a rearranged form of the logistic equation: , in which the cell density in a given cycloheximide concentration (Pcyh) at time (t = 48 hr) was normalized by the cell density without cyloheximide (P0) and the doubling time without cycloheximide (T = 10 hr) was taken from growth curves of wild-type strains (Figure 3—figure supplement 2). After performing a linear fit of the data, we determined that 5 ng/ml of cycloheximide retards the growth rate two-fold. (B) The cycloheximide resistant mutation rpl36aP56Q disrupts cycloheximde binding to the large ribosomal subunit of yeast (left). A crystal structure of cycloheximide bound to the yeast 80S ribosome (Garreau de Loubresse et al., 2014; PDB 4U3U) shows that the yeast ortholog of Rpl36a (L42A; black) and ribosomal RNA (gray) form the cycloheximide binding pocket. The most critical residues in L42A for cycloheximide binding are Lys55, Pro56, and Phe58 (right). In silico modeling (Goddard et al., 2005) of cycloheximide resistance mutations (Bae et al., 2018) shows that some rotamers of the Pro56Gln substitution (purple) disrupt van der Waals interactions and cause steric clashes. (C) The S. rosetta ortholog of Rpl36a conserves residues that bind cycloheximide in yeast and Chlamydomonas reinhardtii. A sequence alignment (Sievers et al., 2011) of Rpl36a orthologs from S. rosetta, S. cerevisiae (L42A), and C. reinhardtii (RPL41) shows that S. rosetta conserves residues for cycloheximide binding, the most critical of which is Pro56 (purple arrow). (D) The efficiency of genome editing alters the selection of edited alleles. Using growth parameters determined from S. rosetta growth curves (Figure 3—figure supplement 2) and cycloheximide inhibition (Figure 2—figure supplement 1A), we modeled the selection for cycloheximide resistance using the following equation: , where Fcyh is the frequency of the cycloheximide resistant allele, ε is the genome editing efficiency (which also corresponds to the initial frequency of the edited allele), M is the ratio of the carrying capacity to the cell density (which we set as an arbitrarily large number because continuous passaging in the laboratory can keep the cell population far from the carrying capacity), t is the time of growth after starting selection, T is the doubling time in the absence of selection (which is 10 hr, see Fig. S4), and S is the relative growth rate in the presence of selection (which we set to 2, based on the relative growth rate upon adding 5 ng/ml of cycloheximide to cells (panel A). Notably, this model only captures the relative changes in growth upon selection and assumes that the edited allele is insensitive to the drug; the model does not include a term for the rate of cells dying, which we observed to happen after 3 days of selection. Nonetheless, this model helped us determine that after 5 days of growth in a selective media, we could expect to observe genome editing events occurring at frequencies > 0.01%.
Figure 2—figure supplement 2

Engineered cycloheximide resistance establishes genome editing conditions.
(A) The design of a cycloheximide resistant allele, rpl36aP56Q, in S. rosetta. The protospacer adjacent motif (PAM, orange) next to the 56th codon of rpl36a (Target, cyan), which is located on the second exon (thick black line labeled 2), provides a suitable site to design a gRNA that targets SpCas9 cleavage (sequence is shown underneath the locus schematic, and carets indicate the target cleavage site). A repair oligonucleotide (black line with knob) introduces a cycloheximide resistant allele, rpl36aP56Q (Mutation, purple), flanked by 100 bases of homologous sequence. The sequence of the edited allele is shown below. (B–D) A comparison of genotypes from populations of unedited cells (B), edited cells (C), and a strain established from a clonal isolate of edited cells (D) shows that cycloheximide selection enriches for rpl36aP56Q. The genotype for each population was determined by amplifying the locus with primers surrounding the editing site (black arrows in panel A) that did not overlap in sequence with the repair oligonucleotide. One of the primers had a T3 primer binding site for Sanger sequencing of amplicons (black arrow with flap). Remarkably, after selection, the wild-type allele was not detected (B). (E) S. rosetta uses repair oligonucleotides with >20 nt homology arms for genome editing. Truncations of repair oligonucleotides encoding the rpl36aP56Q allele were designed in the same orientation as gRNAs (sense, black dots and lines) or the opposite orientation (antisense, gray dots and lines). 24 hr after S. rosetta recovered from transfections with repair templates and SpCas9 RNPs, cycloheximide was added to grow cells in selective media for five days, at which time the cells were harvested for counting cell density and for genotyping. Closed circles indicate that the consensus genotype of the cell population had the rpl36aP56Q allele in Sanger sequencing; whereas, open circles indicate that the cell population had the wild-type allele. E’ and E’’ show two independent trials. Notably, we observed a slight bias for repair oligonucleotides in the sense direction, particularly with shorter homology arms of 20-30 bases. Because repair templates in the sense orientation with 40–80 bases of homologous sequence resulted in the best editing, we performed subsequent optimization with a sense repair oligonucleotide that 50-base homology arms on each side of the double-stranded break. (F) Small quantities of SpCas9 RNPs are sufficient to initiate genome editing. Decreasing concentrations of SpCas9 RNP (SpCas9 was the limiting factor) and a constant amount of repair template were transfected into S. rosetta. After characterizing genome editing outcomes by counting cell density and sequencing the consensus genotype (described in panel E), we found that low concentrations of SpCas9 (20 pmol) were sufficient to introduce the rpl36aP56Q allele. F’ and F’’ show two independent trials. (G) High concentrations of repair oligonucleotides increase genome editing efficiency. A serial dilution of a repair template was delivered into S. rosetta. The cell density and consensus genotypes from these experiments show that all concentrations of repair template can introduce the rpl36aP56Q allele, but the higher cell densities recovered after transfection with increasing concentrations of repair templates indicate more efficient editing. G’ and G’’ show two independent trials. (H) The addition of gRNAs stimulates genome editing. Genome editing was performed by delivering a repair oligonucleotide with SpCas9 without the addition of any gRNA or with a gRNA that was prepared from in vitro transcriptions (noted as gRNA in figure) or with a synthetic crRNA that was annealed to a synthetic tracrRNA (noted as crRNA). The consensus genotype and cell densities from these experiments show that gRNAs are necessary for editing and that gRNAs from either source were sufficient for editing. The dots show two independent experiments and lines show their average result.
Figure 3 with 2 supplements
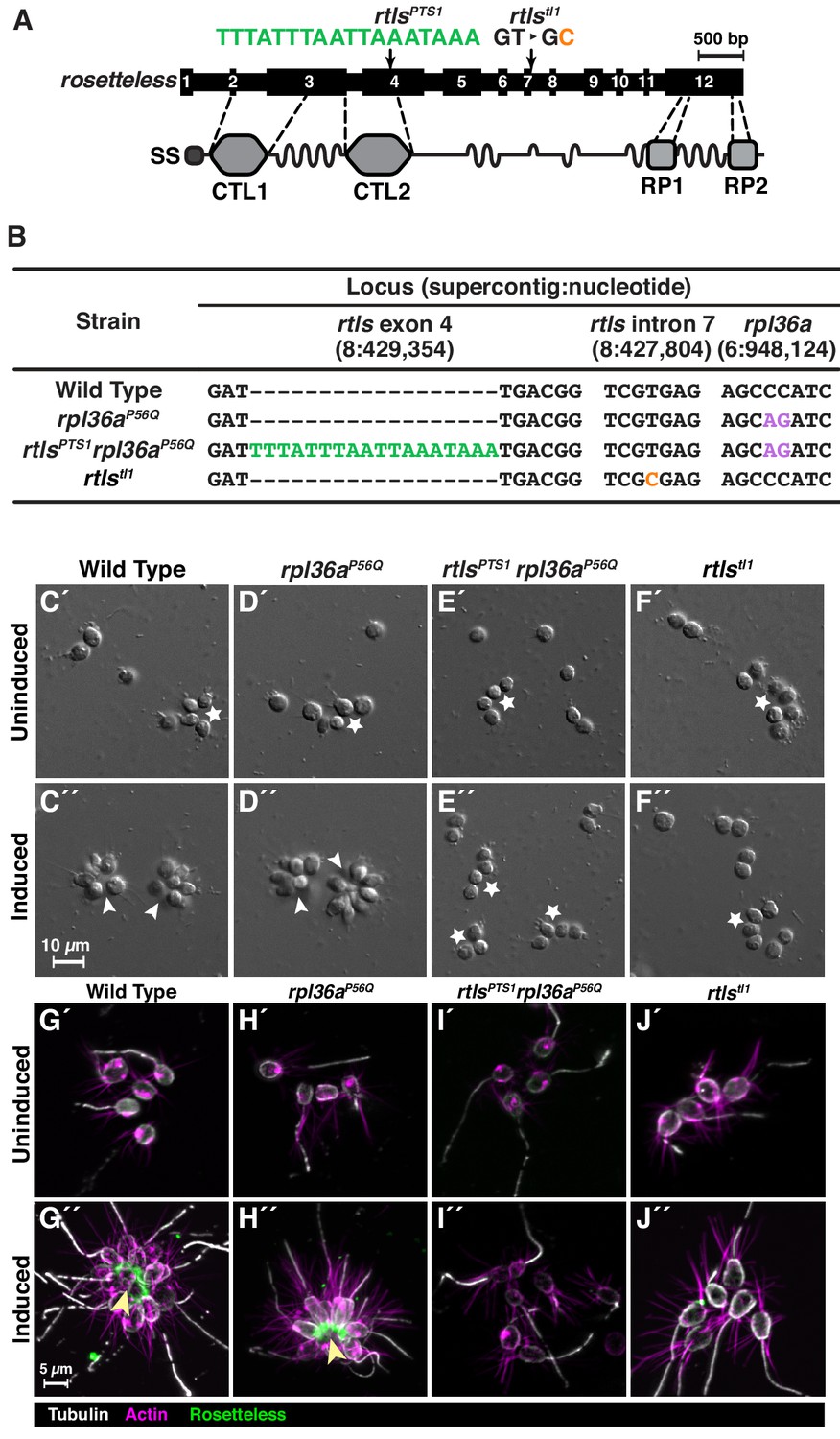
Genome editing of rosetteless enables targeted disruption of multicellular development in S. rosetta.
(A) An engineered mutation in rosetteless introduces a premature termination sequence (PTS) to knockout the expression of rosetteless. The rosetteless gene (exons shown as numbered black boxes, connected by introns) encodes a secreted protein (SS denotes the signal sequence for secretion) with two C-type lectin domains (CTL1 and CTL2) and two carboxy-terminal repeats (RP1 and RP2). A forward genetic screen (Levin et al., 2014) identified a mutation, rtlstl1, in which a T to C transition in the seventh intron disrupts splicing and knocks out rosetteless expression. To increase the likelihood of disrupting rosetteless function with genome editing, we designed the rtlsPTS1 mutation that introduces a PTS (green), with a poly-adenylation sequence and stop codons in each reading frame, into the fourth exon of the gene. (B) The genotypes of strains established from genome-editing confirm that rosetteless and rpl36a incorporated the designed mutations. To enrich for genome-edited cells, SpCas9 RNPs and repair templates for introducing rpl36aP56Q (Figure 2—figure supplement 2) and rtlsPTS1 were simultaneously delivered into S. rosetta. Afterward, cycloheximide resistant cells were clonally isolated and screened for cells that did not develop into rosettes in the presence of RIFs. The genotypes of rtlsPTS1 rpl36aP56Q, and rpl36P56Q confirmed that strains established from genome editing had the rpl36P56Q allele and the strain with the rosetteless phenotype also had the rtlsPTS1 allele. In addition, the wild-type and genome edited strains lacked the T to C transition in the 5’-splice site of intron seven that defined the rtlstl1 allele. (C–F) Phenotypes of genome-edited strains correspond to their respective genotypes. In the absence of RIFs, all strains (C’, D’, E’, and F’) grew as chains (stars) or single cells. Upon the addition of RIFs, the wild-type (C”) and rpl36P56Q strains (D”) formed rosettes (arrowheads). In contrast, rtlsPTS1 rpl36P56Q (E”) and rtlstl1 (F”) did not form rosettes. (G–J) Mutations in rosetteless prevent the secretion of Rosetteless protein at the basal end of cells and into the interior or rosettes. Immunofluorescent staining for Rosetteless (green), alpha tubulin (gray), and actin (magenta) in wild-type (G), rpl36aP56Q (H), rtlsPTS1 rpl36aP56Q (I), and rtlstl1 (J) strains with (G”–J”) and without (G’–J’) rosette induction. Rosetteless localizes in the interior of rosettes (arrow) in wild-type and rpl36aP56Q but not rtlsPTS1 rpl36aP56Q and rtlstl1.
Figure 3—figure supplement 1
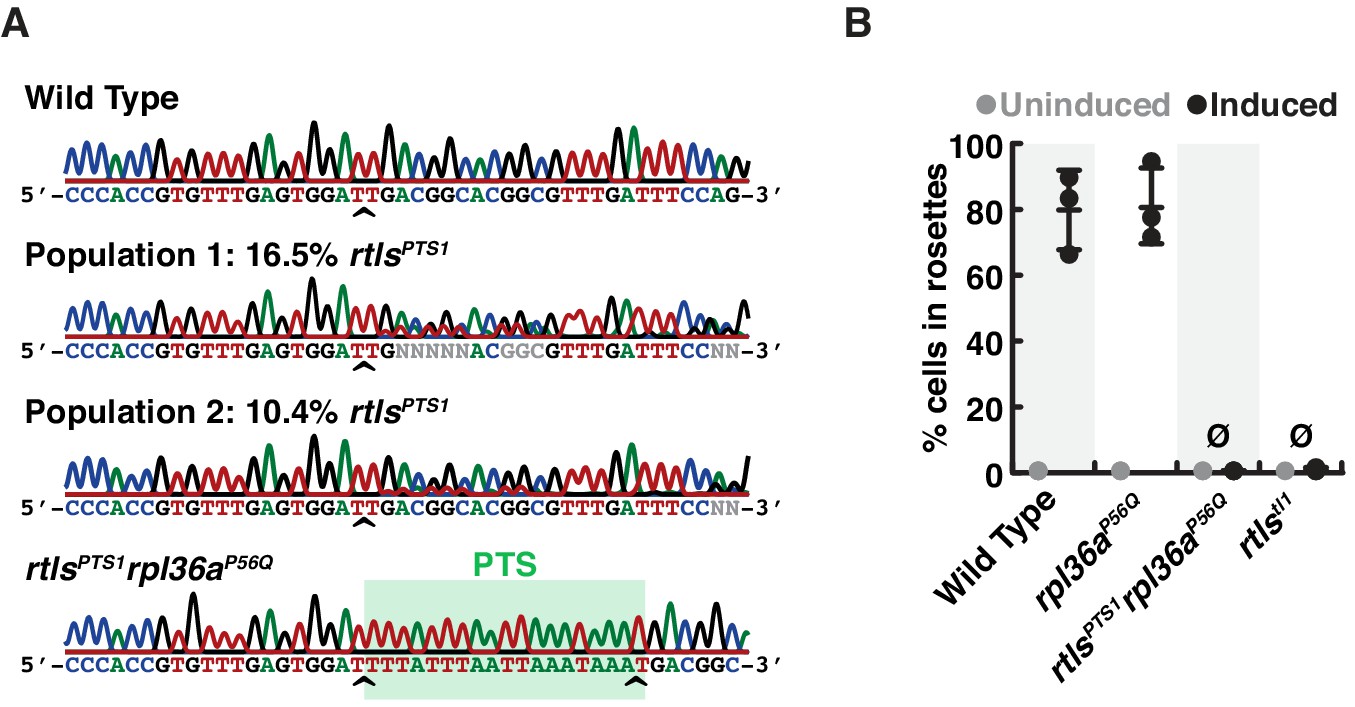
Phenotypes of rosetteless mutants correspond to their genotypes.
(A) The consensus genotype at the site of rosetteless editing in cell populations selected for cycloheximide resistance indicates the presence of the rtlsPTS1 allele. In a wild-type strain (top) and a clonal isolate of rltsPTS1 rpl36aP56Q (bottom), Sanger sequencing at the rtls exon four reveals no heterogeneity in sequence. In populations of cells that had been co-edited to simultaneously engineer rpl36aP56Q and rtlsPTS1 alleles and then selected for cycloheximide resistance, sequence heterogeneity was detected at rtls exon 4 (indicated by N at positions were the base cannot automatically be assigned) and revealed that the rtlsPTS1 allele was present at 16.5% (Population 1) or 10.4% (Population 2) in populations of cells from two independent experiments in which selection for cycloheximide resistant cells was performed after co-editing rpl36aP56Q and rtlsPTS1 alleles. Allele frequency was estimated by unmixing wild-type and rtlsPTS1 alleles in electropherograms from Sanger sequencing (Brinkman et al., 2018). Carets indicate site targeted cleavage by SpCas9. (B) Mutations in rosetteless eliminate rosette development. Rosette development in cell populations (N = 500 cells for each of three independent replicates) shows that wild-type and rpl36aP56Q develop into rosettes in the presence of RIFs while rtlsPTS1 rpl36aP56Q and rtlstl1 do not.
Figure 3—figure supplement 2
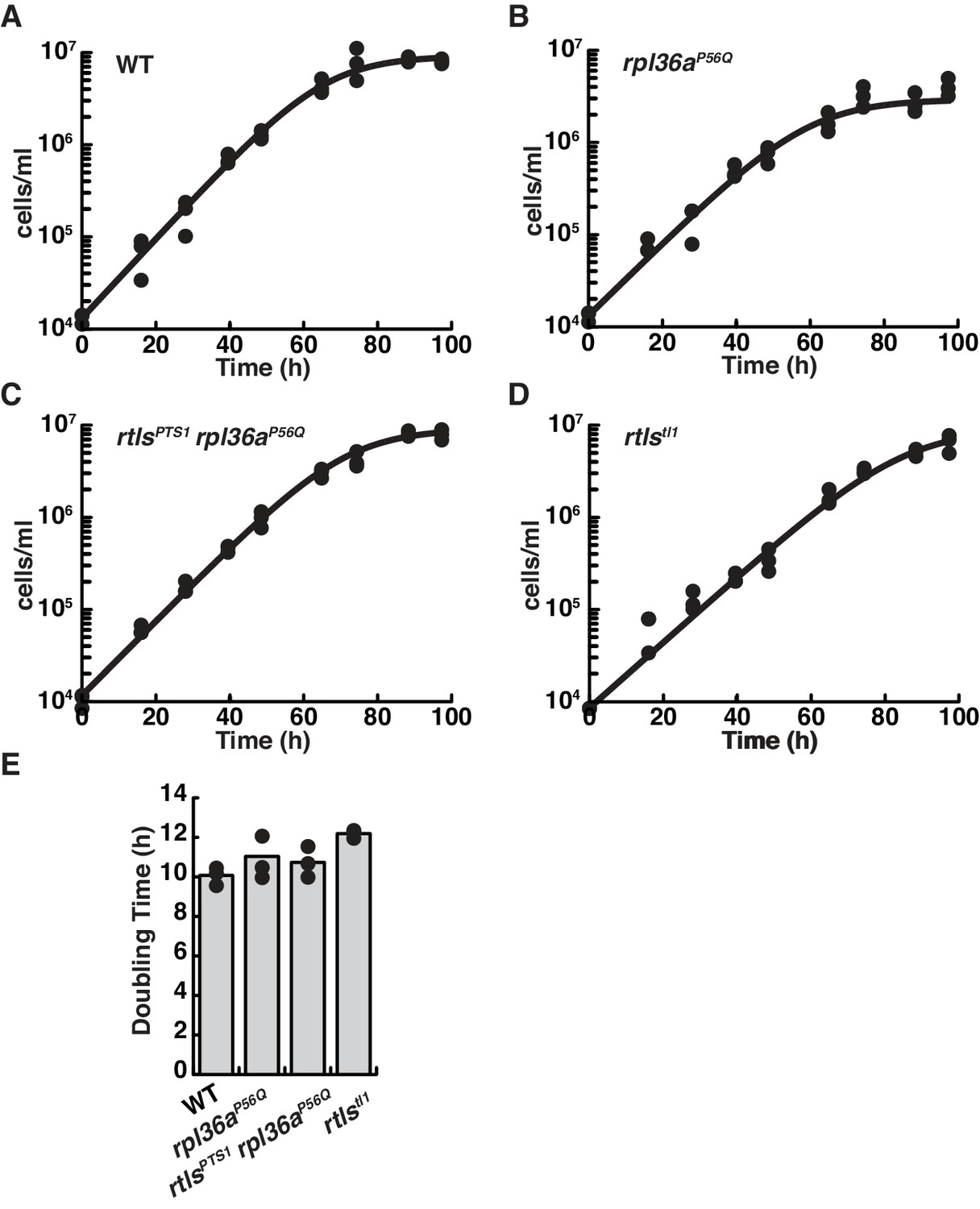
Wild-type and mutant strains proliferate similarly.
Growth curves for wild-type (A), rpl36aP56Q (B), rtlsPTS1 rpl36aP56Q (C), and rtlstl1 (D) show similar rates of proliferation. The growth for each strain was characterized by seeding cells at a density of 1 × 104 cells per ml and determination the cell concentration every ~12 hr. For each time point, triplicate measures were taken. Each replicate growth trajectory was fit with the logistic equation to calculate the doubling time (E). An analysis of variance (ANOVA) between samples showed growth rates were probably the same (p<0.08).
Figure 4 with 1 supplement

S. rosetta preferentially introduces genome-edited mutations from DNA templates.
(A) Schematic of a gRNA targeting SpCas9 to a genomic locus of interest. A gRNA (cyan, knobs indicate 5’ ends) that encodes a 20 nt targeting sequence from the sense strand of a genomic locus (black) hybridizes with the antisense strand (dark gray). SpCas9 (light gray) introduces a double-stranded break at the genomic locus (carets), 3 bp upstream of a protospacer adjacent motif (PAM, orange). (B) We designed a panel of repair oligonucleotides to test the preferred substrates for repairing double-stranded breaks introduced by SpCas9 at rosetteles exon 4. Oligonucleotide repair templates containing the PTS sequence (green) were delivered as single-stranded DNA in the sense (S) or anti-sense (A) orientations and as a double-stranded template (D) to test which most efficiently templated DNA repair at the SpCas9 cleavage site. (C) SpCas9 stimulated the introduction of PTS mutations from DNA templates. Repair templates with a PTS (from panel B) were delivered in the presence and absence of SpCas9 (+/–). A ~ 450 bp fragment surrounding the rtlsPTS1 cleavage site was amplified from cells that had been transfected the previous day to prepare deep sequencing libraries for quantifying the frequency of PTS insertions (green) or insertions/deletions from error prone editing (black). Each experiment was performed three independent times (dots; mean and standard deviations are shown with lines). The dotted line indicates the limit of detection of the sequencing, based on a 6-base, randomized barcode. Upon transfection with the SpCas9 RNP, 10x more mutations from repair templates (1–2%, green dots) were detected than untemplated insertions or deletions (black dots). In the absence of SpCas9, mutations generated from a double-stranded template, but not single-stranded templates, were rarely (<0.1%) and unreliably (2 of 3 trials) found.
Figure 4—figure supplement 1
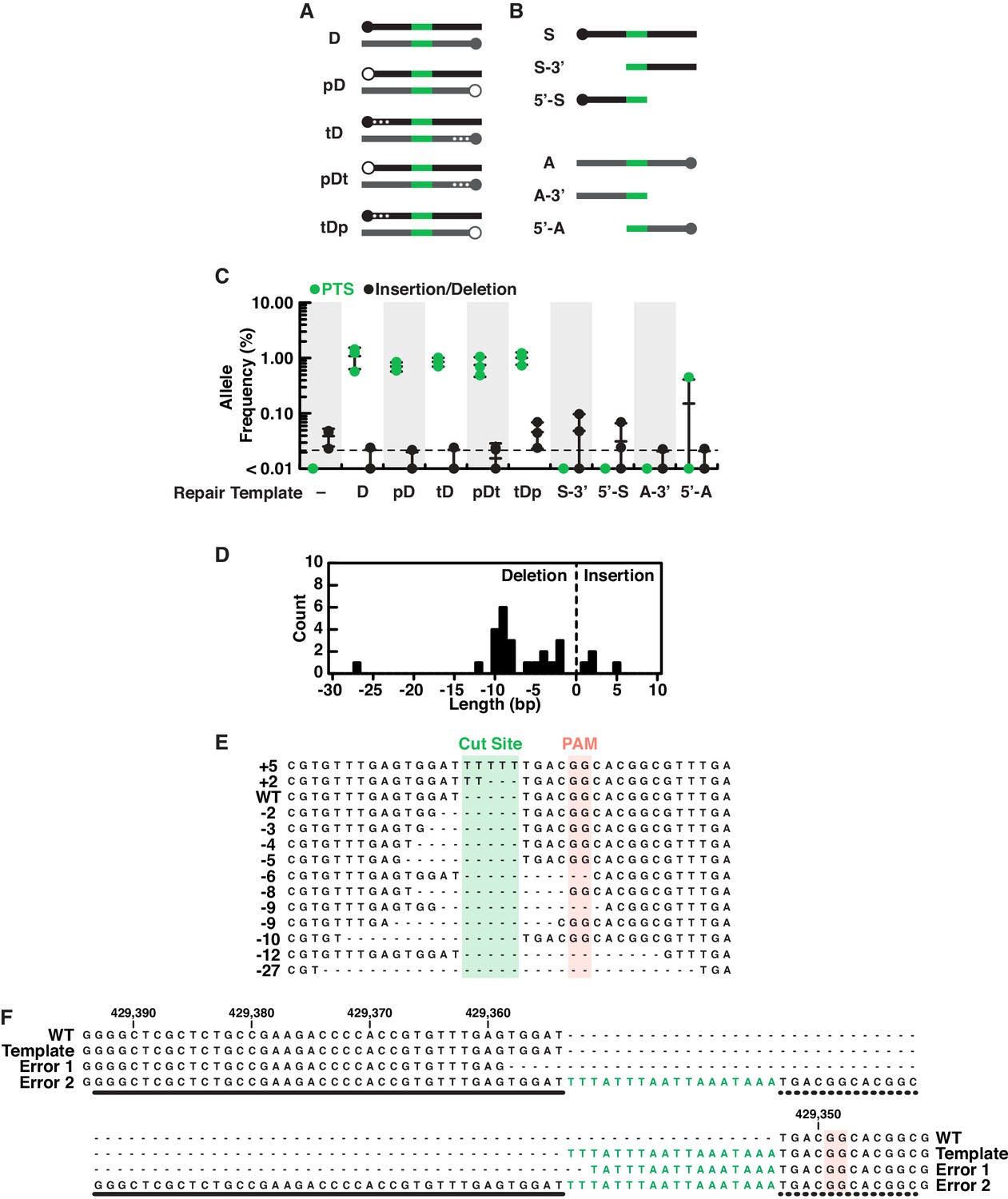
Characterization of editing outcomes at the rosetteless locus with different types of repair templates.
(A) Double-stranded DNA repair templates (black indicates homology arms from the sense strand, gray indicates homology arms from the antisense strand and green is the PTS as in Figure 4) were designed with phosphorylated 5’ ends (indicated with open circles at the 5’ end and a ‘p’ in template names; closed circles indicate unphosphorylated ends) or three phosphorothioate bonds between bases at the 5’ end (indicated with asterisks in diagrams and a ‘t’ in template names). We hypothesized that phosphorylated templates would be more susceptible to nucleases and phosphorothioate bonds less susceptible (Renaud et al., 2016; Yu et al., 2020), altering their utility as repair substrates in vivo, yet S. rosetta used all double stranded templates with similar efficiency (see panel C). (B) We also designed a panel of single-stranded repair templates (colors as in panel A) that lacked 5’ or 3’ arms (Paix et al., 2017) and found that both arms of homology are required for efficient template-mediated genome editing in S. rosetta (see panel C). (C) A comparison of DNA repair templates revealed that S. rosetta efficiently uses double stranded DNA templates during DNA repair and requires both arms of homology for single-stranded DNA templates. Frequencies of alleles containing either the PTS (green) or insertion/deletion mutations (black) are shown for genome editing experiments based on each of the templates described in panels A and B. Genome editing in the presence of double-stranded DNA templates favored template-directed DNA repair. The use of phosphorylated double-stranded DNA templates or double-stranded DNA templates with phosphorothioate bonds (see panel A) did not increase editing efficiency over unmodified double-stranded DNA templates. We also found that removing 5’ or 3’ homology arms from single stranded templates (see panel B) almost completely eliminated efficient editing as compared to single-stranded templates with both homology arms (Figure 4C). Each editing experiment was performed three independent times. (D–E) An aggregate analysis of insertion/deletion mutations identified in deep sequencing of genome editing experiments. (D) A histogram shows the length and frequency of insertion and deletion mutations. (E) A sequence alignment or representative insertion and deletion mutations from each size of insertion/deletion mutations. Notably, the most frequent deletions (8–10 bases) occur at dinucleotide repeats, suggesting that microhomologies may promote deletions after double-stranded breaks. (F) An extreme example of templated repair suggests that S. rosetta may incorporate larger insertions. One mutation identified in deep sequencing shows an 88-base insertion, with the insertion featuring two PTS sequences with an intervening region that has some homology to sequences to the left (thick line) and right (dotted line) of the double stranded break. Although we are unsure of the mechanism that led to this mutation, its presence suggests that large mutations could be incorporated into S. rosetta via genome editing.
Additional files
-
Source code 1
Quantification of DNA repair outcomes.
BASH script for quantifying the frequency of repair outcomes from deep sequencing data that were preprocessed and aligned in a Galaxy server (Afgan et al., 2018).
- https://cdn.elifesciences.org/articles/56193/elife-56193-code1-v3.sc
-
Supplementary file 1
Tables of critical resources.
Table A: Media recipes for making artificial seawater (Hallegraeff et al., 2004; Skelton et al., 2009), high nutrient media (modified from King et al., 2009; Levin and King, 2013; Booth et al., 2018), and low nutrient media. Table B: Oligonucleotide sequences for gRNAs, repair oligonucleotides, and primers that were used to construct and to validate genome edited strains. Table C: S. rosetta strains Genotypes and sources of S. rosetta strains used in this study. Table D: Deep sequencing library primers Sequences for primers (adapted from Lin et al., 2014 used to generate libraries for deep sequencing (Figure 4 and S5)
- https://cdn.elifesciences.org/articles/56193/elife-56193-supp1-v3.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/56193/elife-56193-transrepform-v3.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genome editing enables reverse genetics of multicellular development in the choanoflagellate Salpingoeca rosetta
eLife 9:e56193.
https://doi.org/10.7554/eLife.56193




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}