Use of signals of positive and negative selection to distinguish cancer genes and passenger genes
- Institute of Enzymology, Research Centre for Natural Sciences, Hungary
- Department of Pathogenetics, National Institute of Oncology, Hungary
Figures
Figure 1
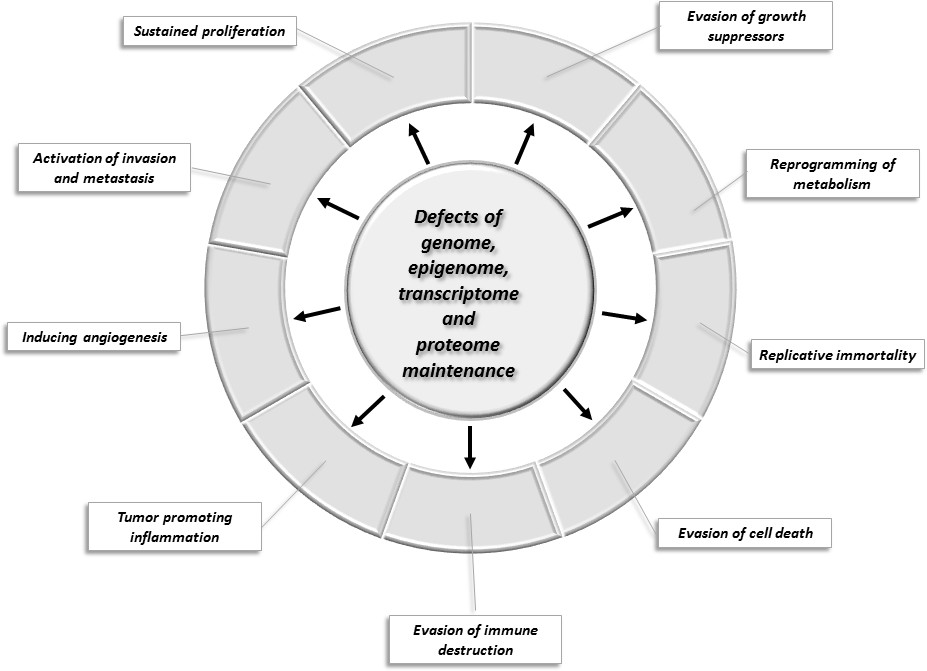
Changes of key cellular processes contributing to carcinogenesis.
The central circle refers to processes involved in the maintenance of the integrity of the genome, epigenome, transcriptome, and proteome: defects in these processes increase the chance that genes and proteins of other cellular pathways (represented by segments of the outer circle) will suffer alterations that favor the acquisition of capabilities that permit the proliferation, survival, and metastasis of tumor cells.
Figure 2
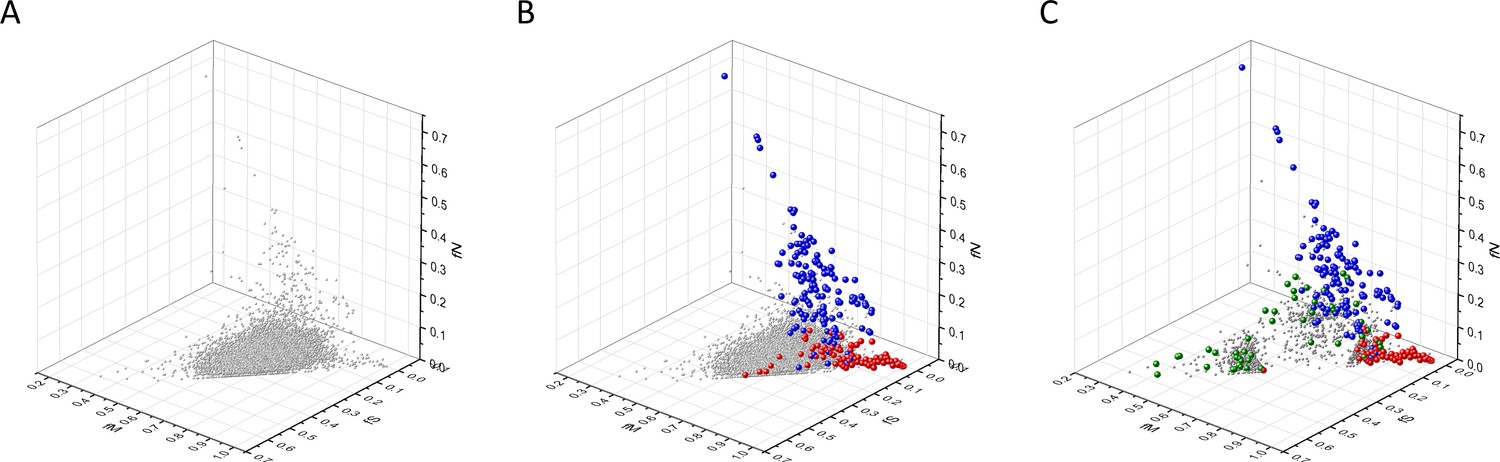
Analyses of fS, fM, and fN parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of 13,803 transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues, including only mutations identified as not single-nucleotide polymorphisms (SNPs). Axes x, y, and z represent the fractions of somatic single-nucleotide substitutions that are assigned to the synonymous (fS), nonsynonymous (fM), and nonsense (fN) categories, respectively. In Panel A, each gray ball represents a human transcript; note that the majority of human genes are present in a dense cluster. Panel B highlights the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls). It is noteworthy that these driver genes separate significantly from the central cluster and from each other: OGs have a significantly larger fraction of nonsynonymous, whereas TSGs have significantly larger fraction of nonsense substitutions. Panel C shows data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 3
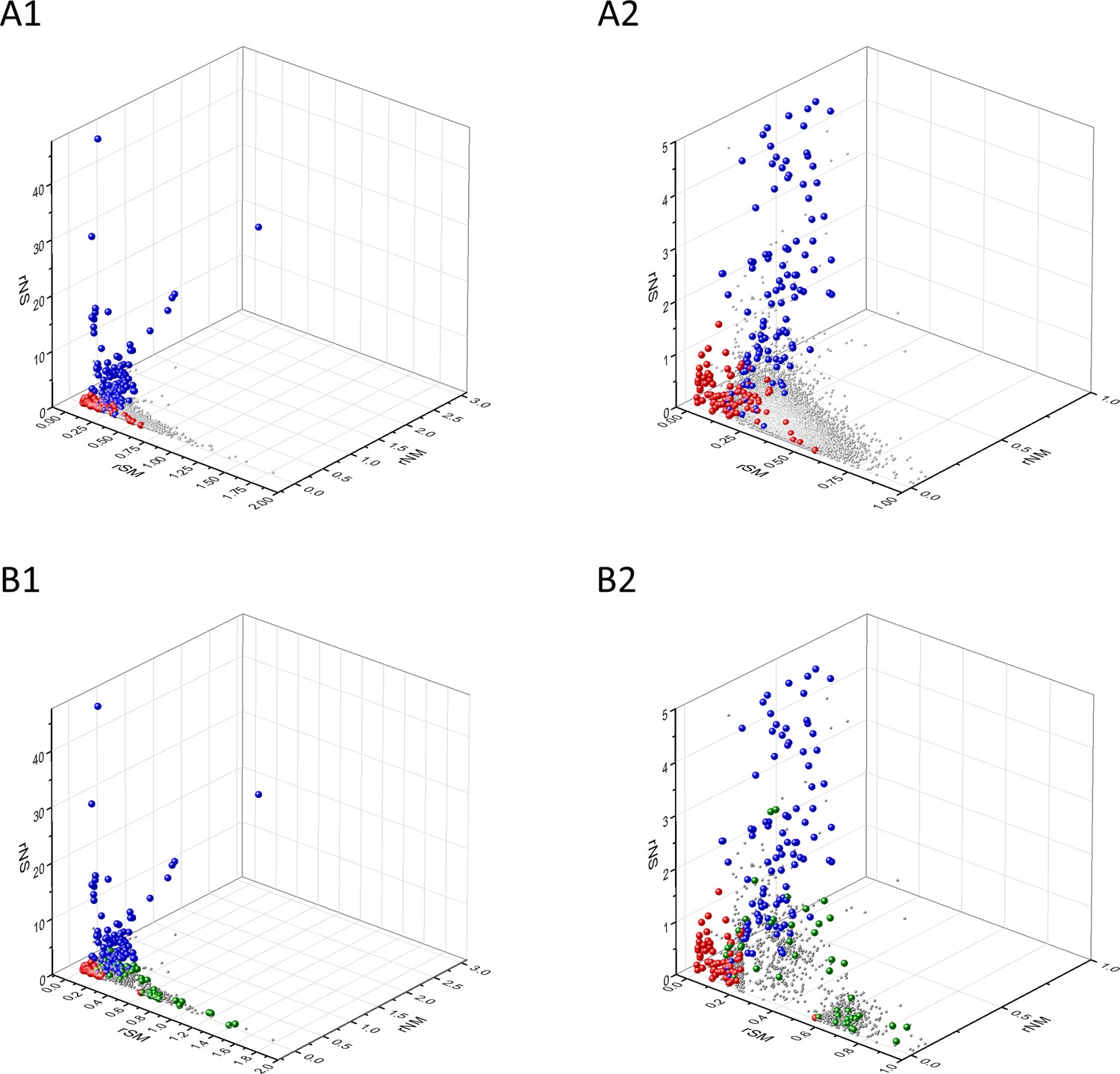
Analyses of rSM, rNM, rNS parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of 13,803 transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues, including only mutations identified as not single-nucleotide polymorphisms (SNPs). Axes x, y, and z represent the rSM, rNM, rNS values defined as the ratio of fS/fM, fN/fM, fN/fS, respectively. Each ball represents a human transcript; the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Panels A1, A2 show the distribution of the 13,803 transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The rNS and rNM values of TSGs are higher, whereas the rSM and rNM values of OGs are lower than those of passenger genes. Panels B1, B2 show data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 4

Analyses of rSMN, rMSN, and rNSM parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues, including only mutations identified as not single-nucleotide polymorphisms (SNPs). Axes x, y, and z represent the rSMN, rMSN, and rNSM defined as the ratio of fS/(fM+fN), fM/(fS+fN), and fN/(fS+fM). Each ball represents a human transcript; the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Panels A1, A2 show the distribution of the 13,803 transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The rNSM values of TSGs are higher, their rMSN and rSMN are lower than those of passenger genes (PGs). OGs also separate from PGs in that their rMSN values are higher and their rSMN and rNSM values are lower than those of PGs. Panels B1, B2 show data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 5
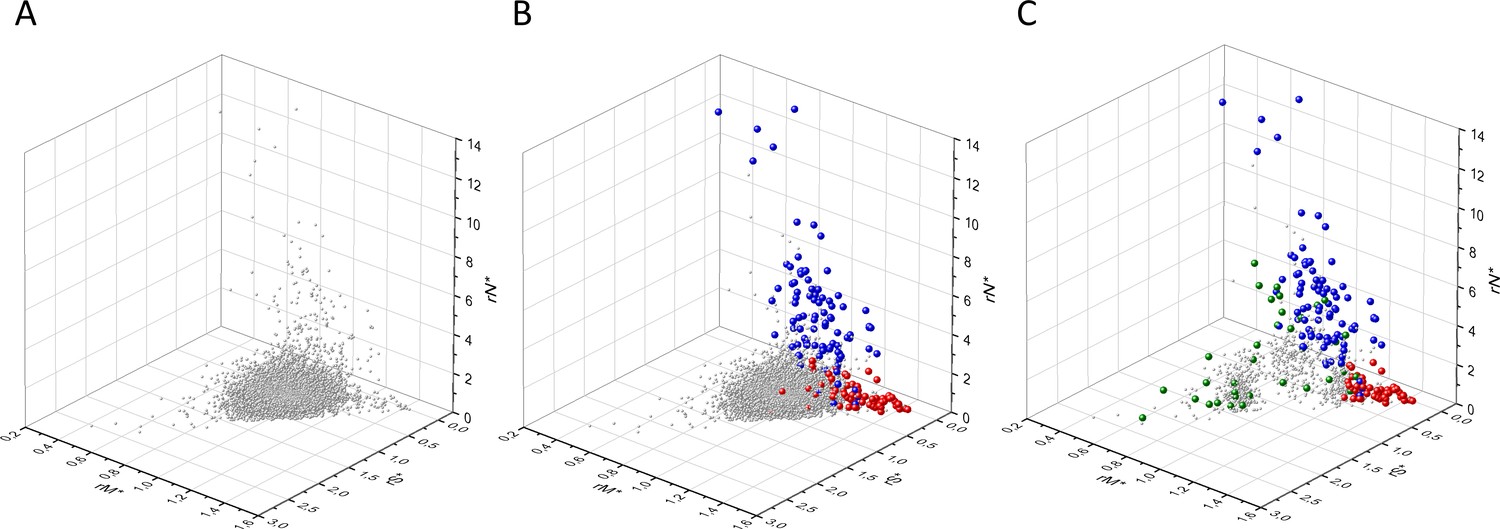
Analyses of rS*, rM*, and rN* parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues. Axes x, y, and z represent rS*, rM*, and rN* values, respectively. In Panel A, each gray ball represents a human transcript; note that the majority of human genes are present in a dense cluster. Panel B highlights the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls). It is noteworthy that these driver genes separate significantly from the central cluster and from each other: OGs have a significantly larger fraction of nonsynonymous, whereas TSGs have significantly larger fraction of nonsense substitutions than expected. Panel C shows data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 6

Analyses of rSM*, rNM*, rNS* parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues. Axes x, y, and z represent rSM*, rNM*, rNS* values, respectively. Each ball represents a human transcript; the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Panels A1 and A2 show the distribution of the transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The rNS* and rNM* values of TSGs are higher, whereas the rSM* and rNM* values of OGs are lower than those of passenger genes. Panels B1, B2 show data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 7
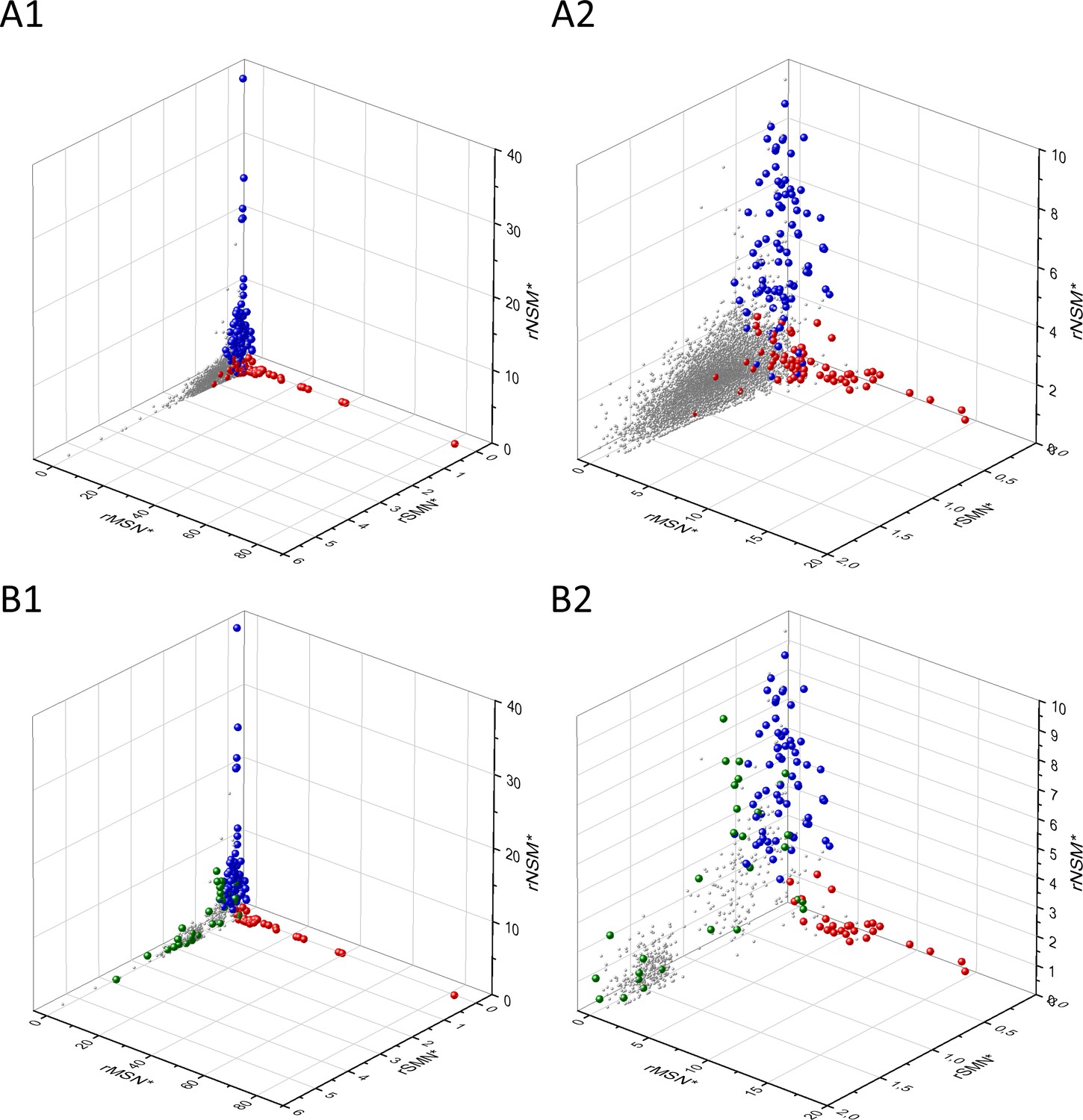
Analyses of rSMN*, rMSN*, and rNSM* parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues. Axes x, y, and z represent the rSMN*, rMSN*, and rNSM* values, respectively. Each ball represents a human transcript; the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Panels A1, A2 show the distribution of the transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The rNSM* values of TSGs are higher, their rMSN* and rSMN* are lower than those of passenger genes (PGs). OGs also separate from PGs in that their rMSN* values are higher and their rSMN* and rNSM* values are lower than those of PGs. Panels B1, B2 show data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Materials and methods). The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Figure 8
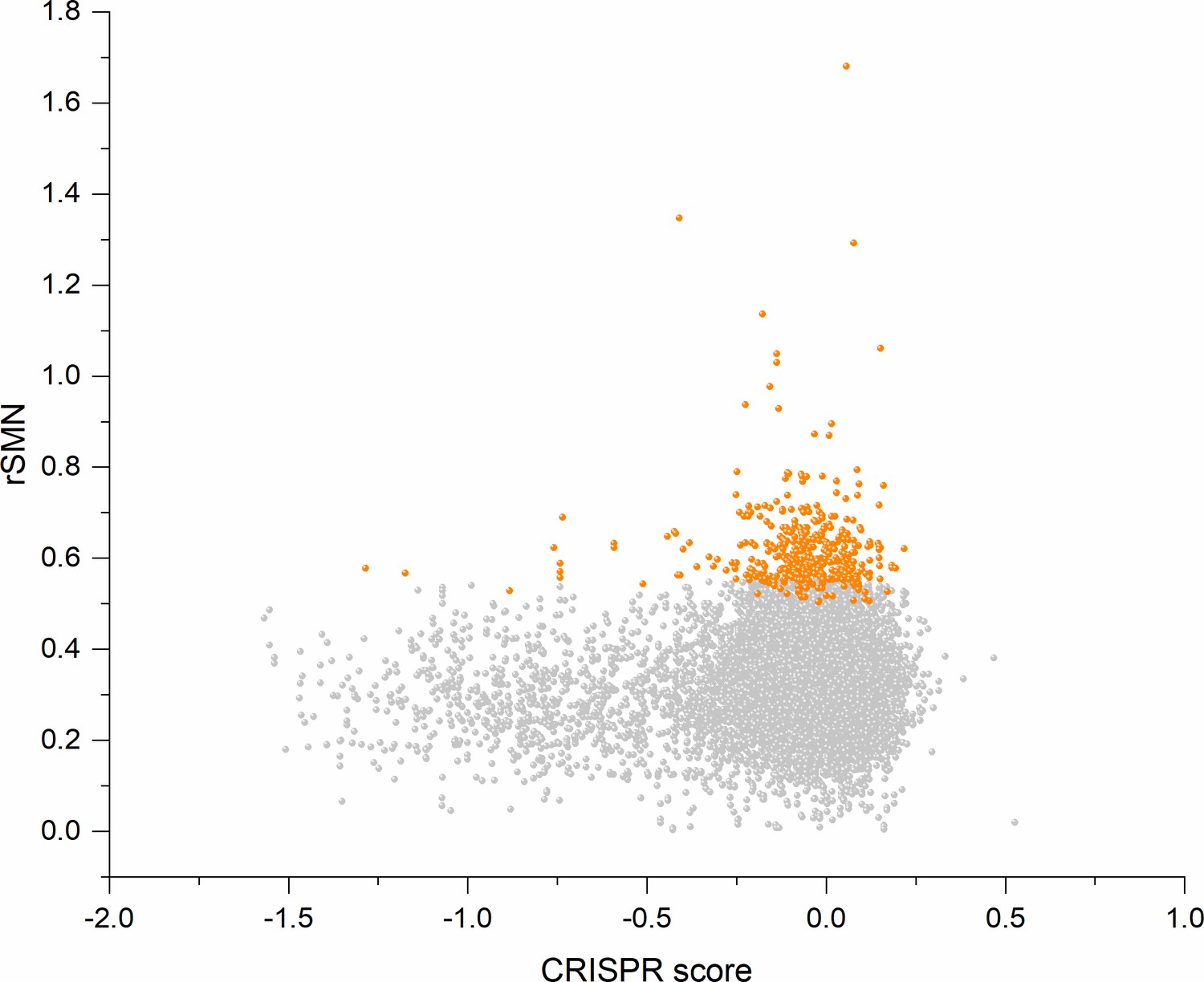
Cell-essentiality scores of human genes and negative selection during tumor evolution.
The figure shows the results of the analysis of transcripts containing at least 100 subtle, confirmed somatic, non-polymorphic mutations from tumor tissues. The abscissa indicates the cell-essentiality score of the genes, the ordinate shows the rSMN parameters of the transcripts. Each ball represents a human transcript. Transcripts showing strongest signals of negative selection (CG_SO2SD rSMN >0.5) are represented by dark orange balls.
Figure 9
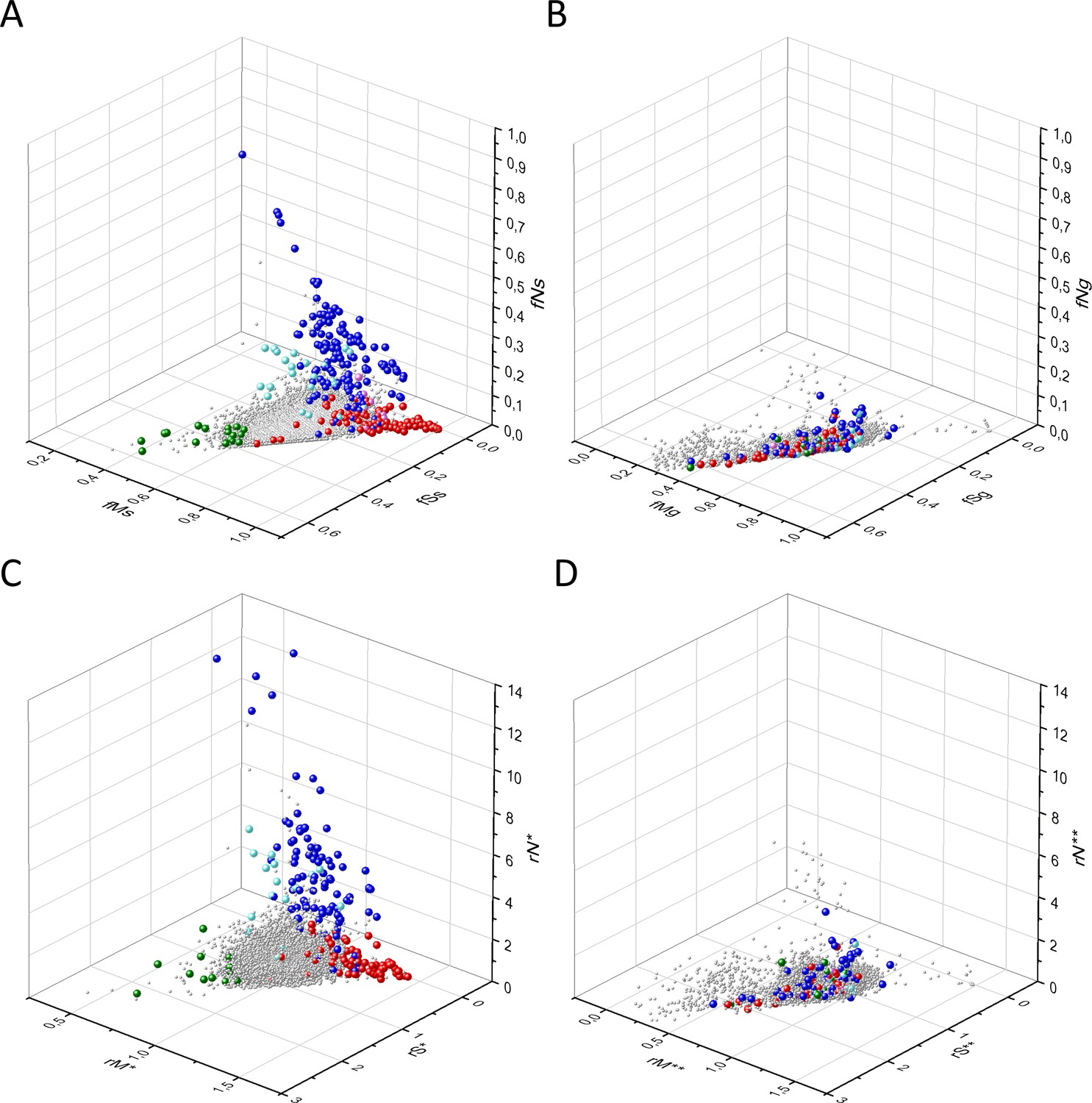
Comparison of the patterns of germline mutations of genes with those of somatic mutations observed during tumor evolution.
Panel A: fS, fM, and fN scores of somatic mutations in cancer, Panel B: fS, fM, and fN scores of germline mutations. Panel C: rS*, rM*, and rN* scores of somatic mutations in cancer, Panel D: rS**, rM**, and rN** scores of germline mutations. Each ball represents a human transcript. The positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Novel proto-oncogenes, tumor suppressors and tumor essential genes identified in the present work are highlighted in magenta, cyan, and green, respectively.
Figure 10
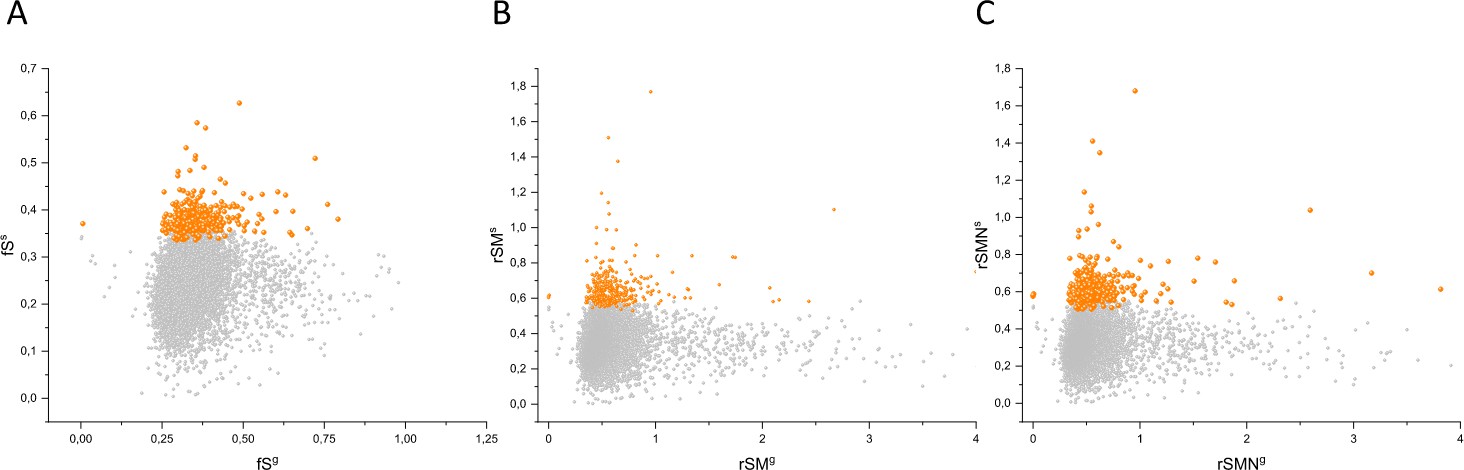
Comparison of fS, rSM, and rSMN scores of genes determined for somatic mutations in tumors with those determined for germline mutations.
The abscissas indicate the fSg (panel A), rSMg (panel B), and rSMNg (panel C) scores of germline mutations of human genes and the ordinates shows the corresponding fSs, rSMs, and rSMNs scores of somatic mutations of tumors for the same genes. Each ball represents a human gene. Transcripts showing the strongest signals of negative selection during tumor evolution (CG_SO2SD rSMN >0.5) are represented by dark orange balls.
Figure 11
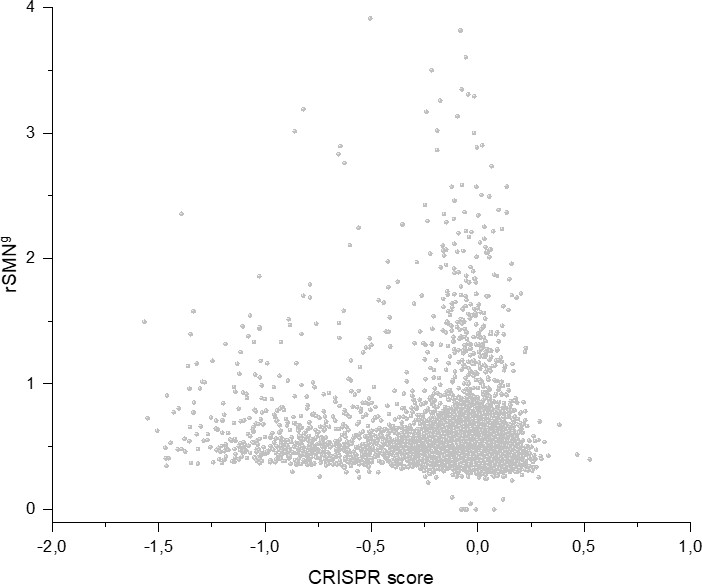
Cell-essentiality scores of human genes and negative selection on single-nucleotide polymorphisms (SNPs).
The figure shows the results of the analysis of transcripts containing at least 100 polymorphic mutations. The abscissa indicates the cell-essentiality score of the genes, the ordinate shows the rSMNg parameters of the transcripts. Each ball represents a human transcript. Note that there is a weak negative correlation (Pearson's r = −0.03662, p<0.05) between the strength of purifying selection of transcripts (rSMNg) and their cell-essentiality scores.
Figure 12
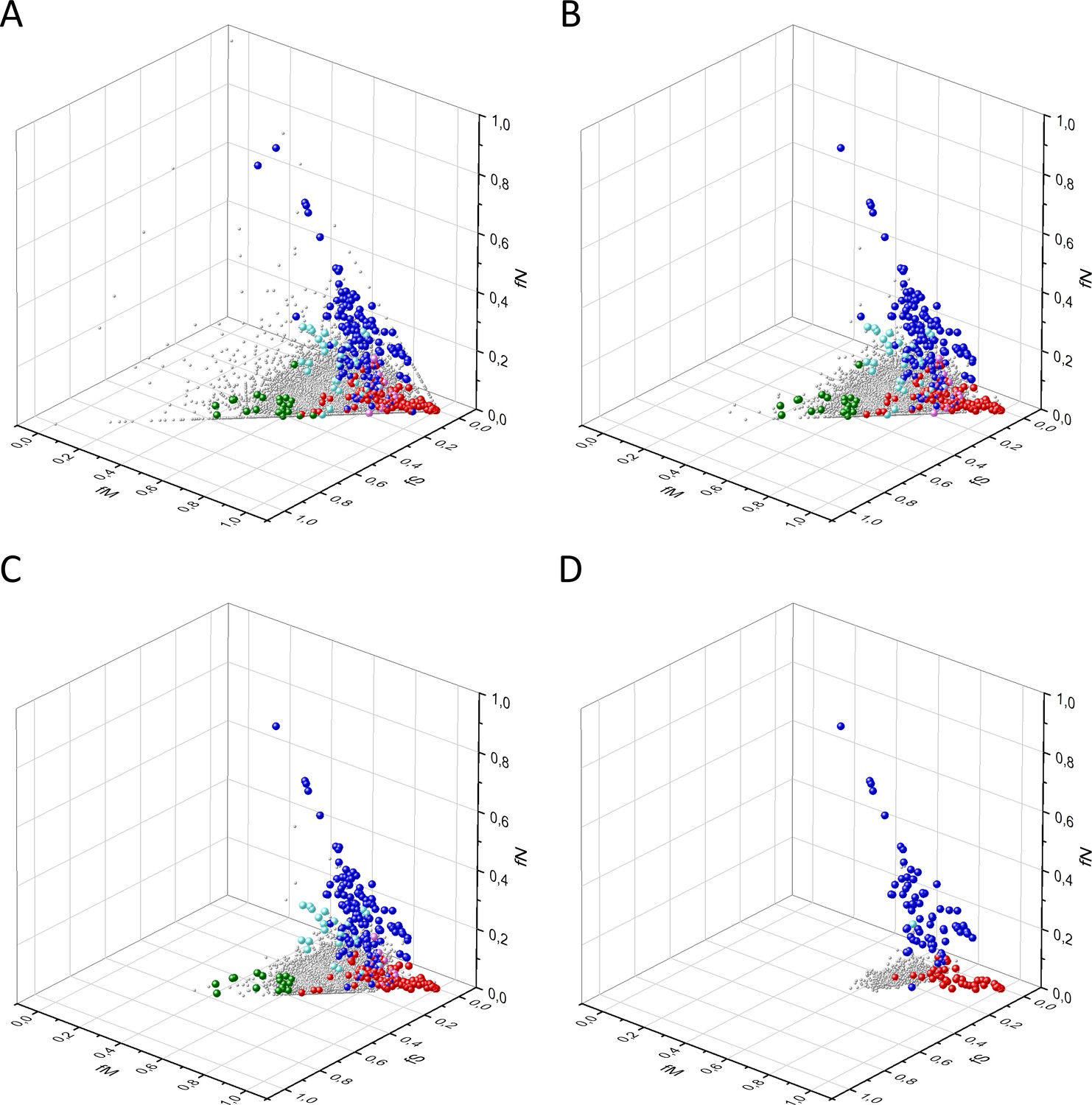
Analyses of fS, fM, and fN parameters of datasets N0, N50, N100, and N500 containing transcripts of human protein-coding genes with at least 0, 50, 100, or 500 somatic substitutions in tumors.
The figure shows the results of the analysis of 29,333, 21,307, 13,803, and 997 transcripts present in datasets N0 (panel A), N50 (panel B), N100 (panel C), and N500 (panel D), respectively. Axes x, y, and z represent the fractions of somatic single nucleotide substitutions that are assigned to the synonymous (fS), nonsynonymous (fM), and nonsense (fN) categories. Each gray ball represents a human transcript. The positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted; novel proto-oncogenes, TSGs, and negatively selected tumor essential genes validated in the present work are represented by large magenta, cyan, and green balls, respectively. It is noteworthy that the requirement of at least 50 somatic mutations per transcript eliminates transcripts where the signal-to-noise ratio is too low to permit detection of signals of selection through the analysis of fS, fM, and fN parameters (compare panel A and panel B). It should also be noted that the requirement of at least 500 somatic mutations per transcript eliminates transcripts of negatively selected genes (compare panel C and panel D), consistent with the view that they tend to be undermutated.
Appendix 2—figure 1
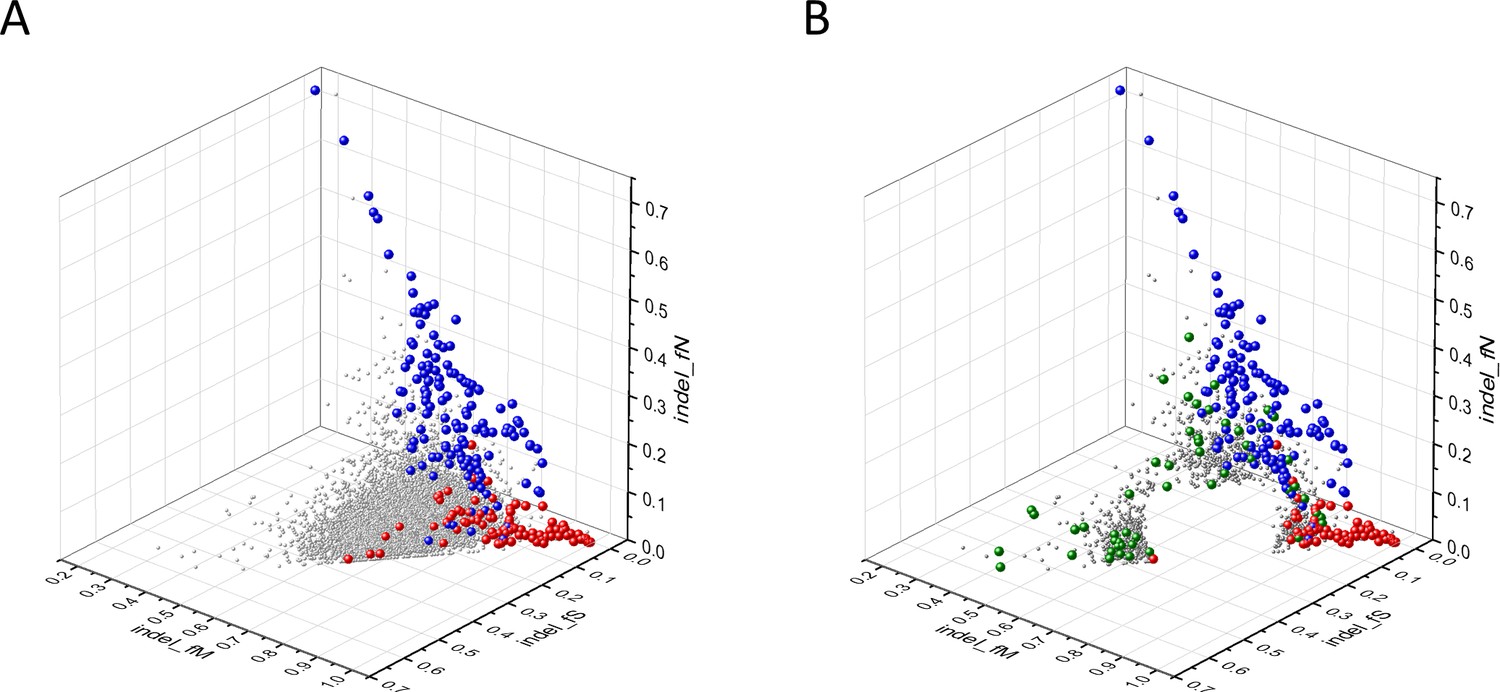
Analyses of indel_fS, indel_fM and indel_fN parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of 13,930 transcripts containing at least 100 subtle, confirmed somatic non-polymorphic mutations from tumor tissues. Axes x, y and z represent the fractions of somatic mutations that are assigned to the indel_fS, indel_fM and indel_fN categories. In Panel A, each ball represents a human transcript; note that the majority of human genes are present in a dense cluster. The positions of transcripts of the genes defined by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. It is noteworthy that these driver genes separate significantly from the central cluster and from each other: OGs have an increased fraction of indel_fM, whereas TSGs have markedly increased fraction of indel_fN. Panel B shows data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Supplementary file 31). The positions of transcripts of the genes identified by Vogelstein et al., 2013 as OGs (large red balls) or TSGs (large blue balls) are highlighted. The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Appendix 2—figure 2
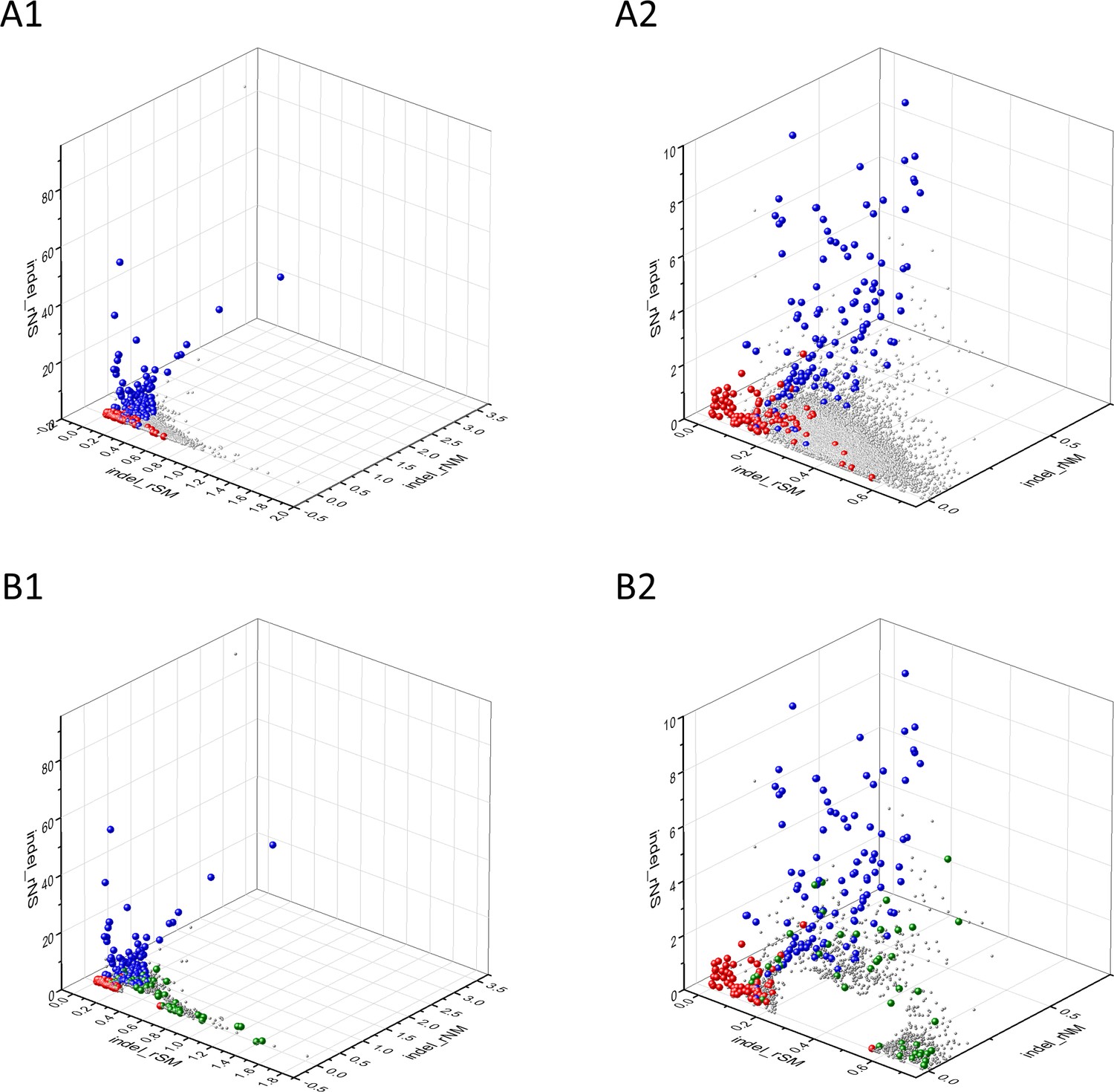
Analyses of indel_rSM, indel_rNM, indel_rNS parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of 13930 transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues, including only mutations identified as not SNPs. Axes x, y, and z represent the indel_rSM, indel_rNM, indel_rNS values defined as the ratio of indel_fS/ indel_fM, indel_fN/ indel_fM, indel_fN/ indel_fS, respectively. Each ball represents a human transcript; the positions of transcripts of the genes identified by Vogelstein et al., 2013 as oncogenes (OGs, large red balls) or tumor suppressor genes (TSGs, large blue balls) are highlighted. Panels A1, A2 show the distribution of the 13,930 transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The rNS and rNM values of TSGs are higher, whereas the rSM and rNM values of OGs are lower than those of passenger genes. Panels B1,B2 show data only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Supplementary file 31). The positions of transcripts of the genes identified by Vogelstein et al., 2013 as OGs (large red balls) or TSGs (large blue balls) are highlighted. The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Appendix 2—figure 3
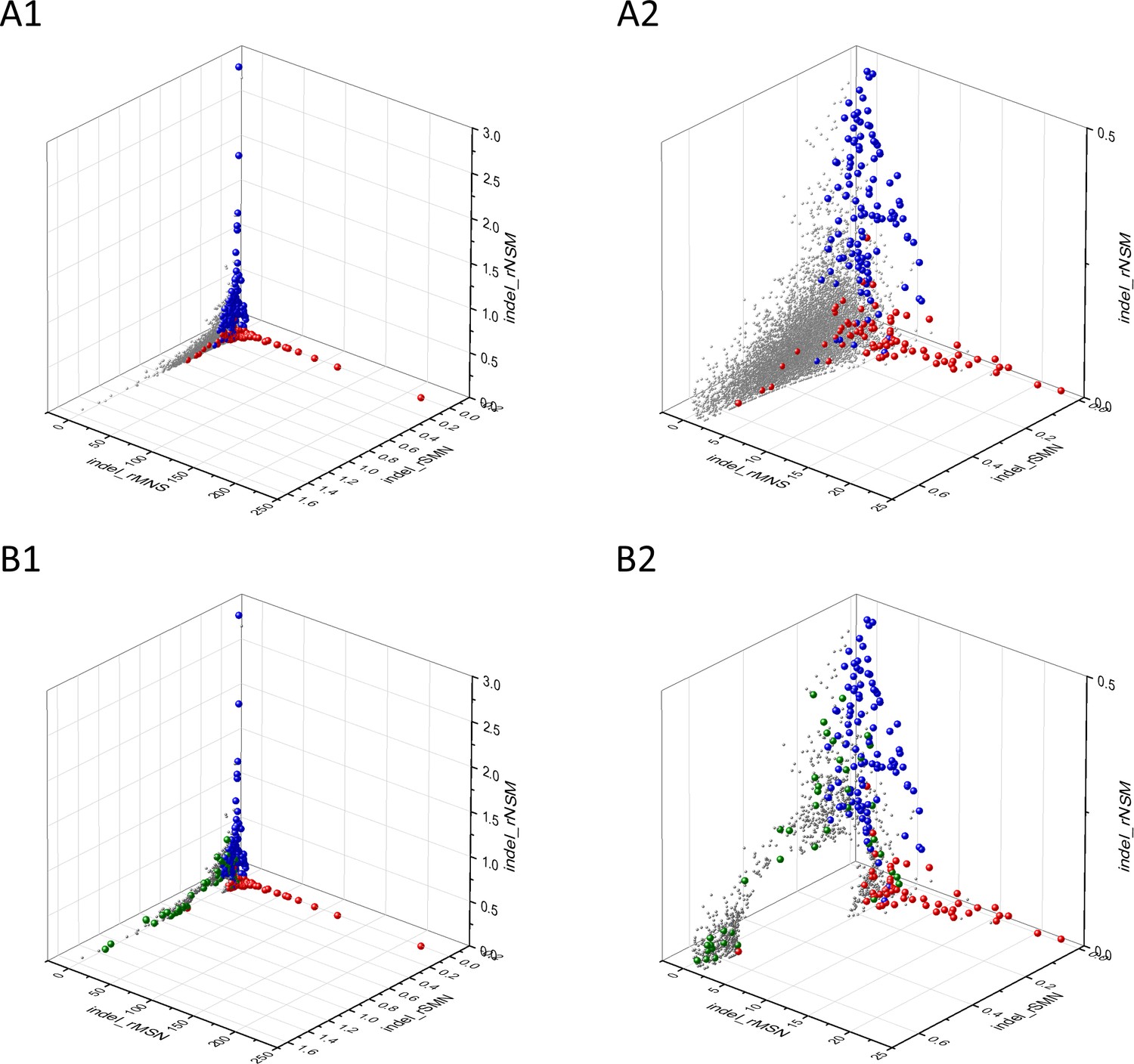
Analyses of indel_rSMN, indel_rMSN and indel_rNSM parameters of human protein-coding genes of tumor tissues.
The figure shows the results of the analysis of 13930 transcripts containing at least 100 subtle, confirmed somatic mutations from tumor tissues. Axes x, y, and z represent paramerters indel_rSMN, indel_rMSN and indel_rNSM defined as the ratio of indel_fS/(indel_fM+indel_fN), indel_fM/(indel_fS+indel_fN) and indel_fN/(indel_fS+indel_fM), respectively. Each ball represents a human transcript; the positions of transcripts of the genes defined by Vogelstein et al., 2013 as oncogenes (OGs, red balls) or tumor suppressor genes (TSGs, blue balls) are highlighted. Panels A1 and A2 show the distribution of the 13,930 transcripts at different magnification. Note that the majority of human genes are present in a dense cluster but known OGs and TSGs separate significantly from the central cluster and from each other. The indel_rNSM values of TSGs are higher, their indel_rMSN and indel_rSMN are lower than those of passenger genes. OGs also separate from passenger genes in that their indel_rMSN values are higher and their indel_rSMN values are lower than those of passenger genes. Panels B1,B2 show data at different magnification only for candidate cancer genes present in the CG_SO2SD_SSI2SD list (see Supplementary file 31). The positions of transcripts of the genes identified by Vogelstein et al., 2013 as OGs (large red balls) or TSGs (large blue balls) are highlighted. The positions of novel cancer gene transcripts validated in the present work are highlighted as large green balls.
Tables
Table 1
Assignment of novel positively or negatively selected cancer genes to key cellular processes of carcinogenesis.
| Hallmarks of cancer | Gene symbol |
|---|---|
| Defects of genome, epigenome, transcriptome, or proteome maintenance | CDK8, FOXG1, IDH3B, MARCH7, MGA, NOVA1, PNCK, RNF128, TGIF1, TNRC6B, TWIST1, ZC3H13, ZFP36L1, ZFP36L2, ZNF750 |
| Sustained proliferation | AURKA, BRD7, ING1, FOXG1, MAPK13, PNCK, PRRT2, RASA1, RIT1, SPRED1, TRIB2, TTK, YAP1, YES1, ZFP36L1, ZFP36L2, ZNF750 |
| Evasion of growth suppressors | |
| Reprogramming of metabolism | BRD7, G6PD, SLC16A1, SLC16A3, SLC2A1, SLC2A8, YAP1, YES1 |
| Replicative immortality | NOVA1 |
| Evasion of cell death | BRD7, ING1, MAPK13, PNCK, PRRT2, TP73, TRIB2, TTK, YAP1, YES1, ZNF750 |
| Evasion of immune destruction | |
| Tumor promoting inflammation | BMP2R, CCR2, CCR5, CX3CR1, MAPK13 |
| Inducing angiogenesis | CCR2 |
| Activation of invasion and metastasis | CCR2, CCR5, CX3CR1, RASA1, TBXA2R |
-
For annotation of novel genes identified in the present study see Appendix 1. The names of negatively selected genes are marked by bold underline.
Additional files
-
Supplementary file 1
Comparison of the lists of genes in datasets CG_SSI2SD_rNSM > 0.125 and CG_SO2SD_rMSN > 3.00 with the lists of cancer genes identified by others (VOG, Vogelstein et al., 2013; TAM, Tamborero et al., 2013; LAW, Lawrence et al., 2014; ABB, Abbott et al., 2015; TOR, Torrente et al., 2016; ZHO, Zhou et al., 2017; MAR, Martincorena et al., 2017; BAI, Bailey et al., 2018; SON, Sondka et al., 2018; ZHA, Zhao et al., 2019a).
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively. Transcripts of CGC genes (SON, Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background. Novel positively or negatively selected cancer genes validated in the present work are highlighted in dark green background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp1-v2.xlsx
-
Supplementary file 2
Comparison of the lists of genes in datasets CG_SSI2SD_rNSM > 0.125 and CG_SO2SD_rMSN > 3.00 with the lists of genes in datasets CG_SO*2SD_rNSM > 3 and CG_SO*2SD_rMSN > 1.50, respectively.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp2-v2.xlsx
-
Supplementary file 3
Comparison of the list of negatively selected genes, CG2SD_rSMN > 0.5 with the lists of negatively selected genes (WEG, ZHOU, ZAPATA, PYATNITSKIY), defined by Zhou et al., 2017, Weghorn and Sunyaev, 2017, Zapata et al., 2018, Pyatnitskiy et al., 2015, respectively as well as the list of genes (De Kegel) identified by De Kegel and Ryan, 2019 as broadly essential genes.
Negatively selected genes discussed in detail in the present work are highlighted in dark green background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp3-v2.xlsx
-
Supplementary file 4
Comparison of the list of genes in dataset CG2SD_rSMN > 0.5 with the list of genes in dataset CG_SO*2SD_rSMN > 1.50.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp4-v2.xlsx
-
Supplementary file 5
SO (Substitution Only) and SSI (Substitutions and Subtle Indel) analyses of somatic mutations of transcripts of human protein coding genes that have at least 100 confirmed somatic, non-polymorphic mutations identified in tumor tissues.
The table also contains lists of passenger genes (PG_SOf_1SD, PG_SOr2_1SD, PG_SOr3_1SD, PG_SSIf_1SD, PG_SSIr2_1SD, PG_SSIr3_1SD) whose parameters deviate from the mean values by ≤1 SD as well as lists of candidate cancer genes (CG_SOf_1SD, CG_SOr2_1SD, CG_SOr3_1SD, CG_SSIf_1SD, CG_SSIr2_1SD, CG_SSIr3_1SD) whose parameters deviate from the mean values by >1 SD. Table also contains lists of candidate cancer genes (CG_SOf_2SD, CG_SOr2_2SD, CG_SOr3_2SD, CG_SSIf_2SD, CG_SSIr2_2SD, CG_SSIr3_2SD) whose parameters deviate from the mean values by >2 SD as well as lists of passenger genes (PG_SOf_2SD, PG_SOr2_2SD, PG_SOr3_2SD, PG_SSIf_2SD, PG_SSIr2_2SD, PG_SSIr3_2SD) whose parameters deviate from the mean values by <2 SD. Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively. Transcripts of CGC (Cancer Gene Census) genes (Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp5-v2.xlsx
-
Supplementary file 6
Numbers and fractions of missense, nonsense, and silent single-nucleotide polymorphisms (SNPs) affecting the coding sequences of the human genes.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively. Transcripts of CGC genes (SON, Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background. Novel positively or negatively selected cancer genes validated in the present work are highlighted in dark green background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp6-v2.xlsx
-
Supplementary file 7
Comparison of fS, rSM, and rSMN scores of genes determined for somatic mutations in tumors with those determined for germline mutations.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp7-v2.xlsx
-
Supplementary file 8
Statistics of transcripts and subtle somatic mutations of human protein coding genes of the different datasets analyzed.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp8-v2.xlsx
-
Supplementary file 9
SO (Substitution Only) and SSI (Substitutions and Subtle Indel) analyses of somatic mutations of transcripts of human protein coding genes.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively. Transcripts of CGC (Cancer Gene Census) genes (Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp9-v2.xlsx
-
Supplementary file 10
Contribution of major types of tumors (‘Tumor Primary site’) to subtle somatic substitutions of the human protein coding genes analyzed.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp10-v2.xlsx
-
Supplementary file 11
Analyses of fS, fM, and fN parameters of transcripts of human protein coding genes that have at least 0 (N0), 50 (N50), 100 (N100), or 500 (N500) somatic substitutions in tumors, respectively.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and light blue backgrounds, respectively. Transcripts of CGC (Cancer Gene Census) genes (Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background. Novel proto-oncogenes, TSGs and negatively selected tumor essential genes validated in the present work are shown in brown, dark blue, and green colors, respectively. For 3D representations of the data, see Figure 12.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp11-v2.xlsx
-
Supplementary file 12
Analyses of fS, fM, and fN parameters of transcripts of human protein coding genes that have at least 0 (N02SD), 50 (N502SD), 100 (N1002SD), or 500 (N5002SD) somatic substitutions in tumors and deviate from average values of fS, fM, and fN by more than 2SD (Sheet ‘CG_SOf_2SD’).
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and light blue backgrounds, respectively. Transcripts of CGC (Cancer Gene Census) genes (SON, Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background. Novel proto-oncogenes, TSGs and negatively selected tumor essential genes (TEGs) validated in the present work are shown in brown, dark blue, and green colors, respectively. Sheet ‘statistics’ contains a summary of the fS, fM, and fN parameters of datasets N0, N50, N100, N500, N02SD, N502SD, N1002SD, N5002SD and indicates the number of known and novel OGs, TSGs and TEGs that are present in the different datasets.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp12-v2.xlsx
-
Supplementary file 13
Negatively selected genes in datasets N0, N50, N100, and N500.
Sheet ‘SO’ lists the genes/transcripts in datasets N0, N50, N100, and N500 that contain transcripts of human protein coding genes with at least 0, 50, 100, or 500 somatic substitutions in tumors, respectively. The lists of negatively selected genes identified by others were taken from the publications of Weghorn and Sunyaev, 2017, Zapata et al., 2018, Zhou et al., 2017 and Pyatnitskiy et al., 2015. Sheet ‘statistics’ indicates the number of negatively selected genes identified by others that are present in the N0, N50, N100, and N500 datasets. Note that only 48%, 64%, 77%, and 89% of the negatively selected genes identified by Weghorn and Sunyaev, 2017, Zapata et al., 2018, Zhou et al., 2017 and Pyatnitskiy et al., 2015, respectively, are present in the dataset N100 that we have analyzed in the present work.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp13-v2.xlsx
-
Supplementary file 14
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that there is no difference in the probability of the substitution classes C>A, C>G, C>T, T>A, T>C, and T>G.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp14-v2.docx
-
Supplementary file 15
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that there is no difference in the probability of the substitution classes C>A, C>G, C>T, T>A, T>C, and T>G.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp15-v2.xlsx
-
Supplementary file 16
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that there is no difference in the probability of the substitution classes C>A, C>G, C>T, T>A, T>C, and T>G.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp16-v2.docx
-
Supplementary file 17
Expected fraction of silent, missense, and nonsense mutations of coding sequences of human protein-coding genes, assuming equal probability of different substitutions classes.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp17-v2.xlsx
-
Supplementary file 18
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only C>A and G>T mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp18-v2.docx
-
Supplementary file 19
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only C>G and G>C mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp19-v2.docx
-
Supplementary file 20
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only C>T and G>A mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp20-v2.docx
-
Supplementary file 21
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only T>A and A>T mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp21-v2.docx
-
Supplementary file 22
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only T>C and A>G mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp22-v2.docx
-
Supplementary file 23
Expected fractions of nonsense, missense, and silent substitutions of various codons in the absence of selection assuming that only T>G and A>C mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp23-v2.docx
-
Supplementary file 24
Expected fractions of nonsense, missense and silent substitutions of various codons in the absence of selection assuming that only C>A or C>G or C>T or T>A or T>C or T>G mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp24-v2.xlsx
-
Supplementary file 25
Expected fractions of nonsense, missense, and silent substitutions in the absence of selection assuming equal codon frequency and that only C>A or C>G or C>T or T>A or T>C or T>G mutations occur.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp25-v2.docx
-
Supplementary file 26
Contributions of C>A, C>G, C>T, T>A, T>C, and T>G mutations to the pattern of Single Base Substitutions in tumors.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp26-v2.xlsx
-
Supplementary file 27
Expected fractions of nonsense (fN*), missense (fM*), and silent (fS*) mutations of human protein-coding genes taking into account the probability of different substitutions classes in tumors.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp27-v2.xlsx
-
Supplementary file 28
Expected fractions of nonsense (fN**), missense (fM**), and silent (fS**) mutations of human protein-coding genes taking into account the probability of different substitutions classes in germline cells.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp28-v2.xlsx
-
Supplementary file 29
Statistics of the results of SO (Substitution Only) and SSI (Substitutions and Subtle Indel) analyses of the data presented in Supplementary file 5.
The column marked 'Expected' indicates the parameters expected if we assume that the structure of the genetic code determines the probability of somatic substitutions.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp29-v2.xlsx
-
Supplementary file 30
Comparison of the results of SO (Substitution Only) and SSI (Substitutions and Subtle Indel) analyses.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp30-v2.xlsx
-
Supplementary file 31
Lists of genes (CG_SOf_2SD, CG_SOr2_2SD, CG_SOr3_2SD, CG_SSIf_2SD, CG_SSIr2_2SD, CG_SSIr3_2SD) whose parameters deviate from the mean values by >2 SD.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively. Transcripts of CGC (Cancer Gene Census) genes (Sondka et al., 2018) that do not correspond to OGs or TSGs of the cancer gene list of Vogelstein et al., 2013 are highlighted by yellow background.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp31-v2.xlsx
-
Supplementary file 32
Observed/expected parameters (rN*, rM*, rS*; rSM*, rNM*, rNS*; rSMN*, rMSN*, and rNSM*) of somatic mutations affecting the coding sequences of the human genes in cancer.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp32-v2.xlsx
-
Supplementary file 33
Observed/expected parameters (rN**, rM**, rS**; rSM**, rNM**, rNS**; rSMN**, rMSN**, and rNSM**) of single-nucleotide polymorphisms (SNPs) affecting the coding sequences of the human genes.
Transcripts of OGs (oncogenes) and TSGs (tumor suppressor genes) of the cancer gene list of Vogelstein et al., 2013 are highlighted by brick red and blue backgrounds, respectively.
- https://cdn.elifesciences.org/articles/59629/elife-59629-supp33-v2.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/59629/elife-59629-transrepform-v2.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Use of signals of positive and negative selection to distinguish cancer genes and passenger genes
eLife 10:e59629.
https://doi.org/10.7554/eLife.59629




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}